

Abstract
The in vitro perfused rectal gland of the dogfish shark (Squalus acanthias) and filter-grown monolayers of primary cultures of shark rectal gland (SRG) epithelial cells were used to analyze the signal transduction pathway by which C-type natriuretic peptide (CNP) stimulates chloride secretion. CNP binds to natriuretic receptors in the basolateral membrane, elevates cellular cGMP, and opens cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels in the apical membrane. CNP-provoked chloride secretion was completely inhibitable by the nonspecific protein kinase inhibitor staurosporine and the PKA inhibitor H89 but insensitive to H8, an inhibitor of type I and II isoforms of cGMP-dependent protein kinase (cGKI and cGKII). CNP-induced secretion could not be mimicked by nonhydrolyzable cGMP analogs added alone or in combination with the protein kinase C activator phorbolester, arguing against a role for cGK or for cGMP-induced PKC signaling. We failed to detect a dogfish ortholog of cGKII by molecular cloning and affinity chromatography. However, inhibitors of the cGMP-inhibitable isoform of phosphodiesterase (PDE3) including milrinone, amrinone, and cilostamide but not inhibitors of other PDE isoenzymes mimicked the effect of CNP on chloride secretion in perfused glands and monolayers. CNP raised cGMP and cAMP levels in the SRG epithelial cells. This rise in cAMP as well as the CNP and amrinone-provoked chloride secretion, but not the rise in cGMP, was almost completely blocked by the Gαi-coupled adenylyl cyclase inhibitor somatostatin, arguing against a role for cGMP cross-activation of PKA in CNP action. These data provide molecular, functional, and pharmacological evidence for a CNP/cGMP/PDE3/cAMP/PKA signaling cascade coupled to CFTR in the SRG.
Keywords: shark rectal gland, cystic fibrosis transmembrane conductance regulator, CFTR, C-type natriuretic peptide, CNP, phosphodiesterase, cyclic GMP
the rectal gland of the spiny dogfish shark, Squalus acanthias, is composed of secretory tubules of a single cell type that are highly specialized for acute, hormone-stimulated chloride secretion (9). The mode of chloride movement across the gland shows many similarities with mammalian chloride secretory tissues such as intestinal crypts and submucosal glands in the airways, including sodium chloride entry at the basolateral side through a Na-K-2Cl cotransporter, Na-K-ATPase, recirculation of potassium through cAMP-sensitive basolateral K channels, and export of chloride through apical cystic fibrosis transmembrane conductance regulator (CFTR) anion channels (15). Shark CFTR (sCFTR) is 72% identical and 83% homologous to human CFTR and has similar biophysical characteristics but a different sensitivity to pharmacological inhibitors (26). Because of its anatomical simplicity and functional specialization, the rectal gland provides a unique model of a native epithelium for investigating the complex signaling pathways involved in CFTR regulation. Novel information gained from these studies is expected to be highly relevant for a better understanding of human CFTR-opathies including cystic fibrosis (CF) caused by mutated, dysfunctional CFTR and secretory diarrhea (SD) characterized by hyperactivation of CFTR in the intestine.
C-type natriuretic peptide (CNP), first identified in nonneuronal tissue by our laboratory in the shark heart (21), is the sole natriuretic peptide in elasmobranchs (sharks, skates, and rays) (13) and functions as the major physiological activator of CFTR-mediated Cl secretion in the shark rectal gland (SRG). CNP activates a CNP-selective receptor guanylyl cyclase at the basolateral membrane (designated NPR-B) (2). Electrophysiological measurements following addition of CNP have been carried out in single perfused rectal gland tubules (12). In contrast to mammalian intestinal epithelial cells, the mechanism by which cyclic GMP (cGMP) activates chloride secretion in SRGs is still ill defined. A major difference with mammalian intestine is the apparent failure of the membrane-permeant and phosphodiesterase (PDE)-resistant cGMP analog 8-bromo-cGMP (8-Br-cGMP) to mimic CNP-provoked chloride secretion in perfused glands (24). In contrast, cGMP-mediated heat-stable enterotoxin (STa), as well as guanylin-induced intestinal chloride secretion, in mammals could be reproduced fully by cGMP analogs (30–31, 33–34).
A key question that remained unresolved is the identity of the cGMP effector protein(s) linking cGMP signaling to the ion transporters in the SRG. In analogy with cGMP signaling in mammalian enterocytes, several mechanisms can be proposed, including 1) cGMP activation of a specific membrane-bound isoform of cGMP-dependent protein kinase (cGK-II); this isoenzyme serves as the major cGMP receptor in the small intestinal epithelium and as a prime target for anti-diarrheal pharmacotherapy (6–7, 14, 33); 2) cGMP inhibition of type III phosphodiesterase (PDE-3), raising cAMP levels (8, 31); 3) cGMP cross-activation of cAMP-dependent protein kinase (PK-A) (10, 33); and 4) direct interaction of cGMP with a cyclic nucleotide-binding domain in CFTR (19, 27). Mechanism 3 could explain the successful reconstitution of CNP/cGMP activation of CFTR in the Xenopus oocyte model (2) but requires excessive accumulation of cGMP in the rectal gland (up to submillimolar levels) for which experimental evidence is presently lacking. Therefore, we chose to focus on mechanisms 1 and 2.
In the current study, cGMP inhibition of PDE3 emerged as the major and perhaps sole signaling mechanism accounting for CNP-provoked CFTR-mediated chloride secretion in the SRG whereas molecular cloning, affinity purification, and functional studies failed to provide evidence for the operation of a CNP/cGMP/cGKII/CFTR pathway.
MATERIALS AND METHODS
Reagents.
All reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO).
In vitro perfusion of SRGs.
Rectal glands were obtained from male and female dogfish sharks, Squalus acanthias, weighing 2–4 kg, which were caught by gill nets in Frenchman's Bay, ME, and kept in a 5,000 gallon circulating seawater system at the Mount Desert Island Biological Laboratory (MDIBL). Animal care and use at the MDIBL is guided by an institutional assurance (A-3562–01) approved by the National Institutes of Health Office of Laboratory Animal Welfare. Sharks were killed by pithing the brain and spinal cord (MDIBL Institutional Animal Care and Use Committee Approved Protocol No. 10-03). Rectal glands were excised, and cannulae were inserted into the artery, vein, and duct. The rectal glands were then placed in a glass perfusion chamber maintained at 15°C with running seawater and perfused with elasmobranch Ringer's solution equilibrated to pH 7.5 by bubbling with 99% oxygen and 1% CO2 as previously described (15). All glands were perfused first for 30 min in the absence of agonists to attain basal levels of secretion. Measurements of duct flow and chloride secretion were made at 1-min intervals after the addition of secretagogues or specific inhibitors of phosphodiesterase enzymes. Results are expressed as microequivalents of chloride secreted per hour per gram wet weight (μeq·h−1·g−1) ± SE.
Short-circuit current measurements.
To further pursue the mechanism of CNP-induced secretion we performed electrical measurements of transepithelial chloride transport in primary cultures of SRG epithelial cells in Ussing chambers. SRG tubular epithelial cells were isolated and cultured on collagen-coated CoStar Transwell filters for 10-25 days using techniques previously described (35). Confluent monolayers were mounted in Ussing chambers (P2300; Physiological Instruments, San Diego, CA) and bathed with a solution containing 270 mM NaCl, 6 mM KCl, 3 mM MgCl2, 5 mM CaCl2, 20 mM NaHCO3, 350 mM urea, and 5 mM glucose, at pH 7.5. The chamber was kept at 20°C and was constantly gassed with 95% O2-5% CO2. Voltage clamping and data acquisition were performed using the VCC MC-6 Multi-Channel Voltage Clamp and software designed and constructed by Physiological Instruments (San Diego, CA). Hormones, antagonists, and other reagents were added to both the mucosal and the serosal side. Data shown are short-circuit current (Isc) tracings reflecting CFTR-mediated Cl− secretion (15, 26) and are representative of three to eight identical experiments.
Cyclic nucleotide measurements.
Following incubation under basal conditions or in the presence of secretagogues, filter-grown monolayers of SRG cells were lysed for 10 min with 0.1 M HCl, centrifuged at 600 g at room temperature, and dried down before reconstitution in assay buffer. cGMP levels were determined using the acetylated version of the cGMP ELISA kit (ADI-900-164; ENZO Life Sciences, Farmingdale, NY). cAMP was detected using a cAMP ELISA kit (ADI-900-163, ENZO Life Sciences). Perfused rectal glands were snap frozen in liquid nitrogen with Wallenberg clamps, and the frozen tissue was pulverized in a porcelain mortar, weighed, and homogenized in 10 vol of cold 5% TCA in a glass-Teflon tissue grinder. Following centrifugation at 600 g for 10 min, the supernatants were extracted with 3 vol of water-saturated ether, and the aqueous extracts were dried and run directly for cAMP determinations. cAMP and cGMP levels are expressed relative to cell protein content, which was determined on monolayers or perfused glands (DC protein assay kit; Bio-Rad, Hercules, CA).
Molecular cloning of cGMP-dependent protein kinases.
Primers were designed based on consensus sequences made by alignment of all proven and putative cGKI and cGKII sequences known to date. Selected cGKII sequences were from human (NM_006259), rhesus monkey (XM_001084948), chimpanzee (XM_517194), cow (XM_612707), dog (XM_544949), rat (Z36276), mouse (NM_008926), opossum (XM_001364891), frog (NM_001092130), platypus (XM_001506960 and XM_001513120), zebrafish (NM_001105279 and XM_679108), pufferfish (CAAE01015003), beetle (XM_963625), and California sea hare (AY362340). Putative cGKI sequences were from human (NM_001098512 cGKIα, NM_006258 cGKIβ), chimpanzee (XM_001162858, cGKIβ), cow (Y08961, cGKIβ), mouse (AF084547, cGKIβ), rhesus monkey (XM_001099261, cGKIα), wild boar (NM_001044574, cGKIα), rabbit (NM_001082042, cGKIα), rat (NM_001105731, cGKIα), chicken (XR_027075), opossum (XM_001374648), zebrafish (XM_689701 and BC054581), and the Japanese killifish (AB164051). All alignments were made using Genious software.
The first 10 forward and 8 reverse primers listed in Table 1 were initially selected for amplification of a potential shark cGKII ortholog. Numbering is relative to the coding region of human cGKII. Sequences were selected partly manually and partly using the primer3 module of Genious. cGKI-specific primers (Fw11–14 and Rv9–10) were designed in a similar way. Primers Fw15 and Rv11–12 were aimed to amplify any cGMP-dependent kinase and were designed from the most conserved parts of the protein sequences, namely the cGMP binding domain and the kinase domain. After two putative cGKI fragments were amplified by these methods, the remaining parts of the open reading frames were amplified using various combinations of the aforementioned primers and sequences identical to those from the known fragments of the S. acanthus cGKI sequences (primers Fw15–18 and Rv13–19; Table 1).
Table 1.
Primer sequences used for cloning and sequencing of Squalus acantias cGMP-dependent protein kinase
| Primer | Target | Sequence | Target |
|---|---|---|---|
| Fw1 | cGKII | ATGGGAAATGGTTCAGTGAAACCCAAAC | cGKII 1–28 |
| Fw2 | cGKII | CAAGTGCATCCAGCTGAACAAGCTGCAGGA | cGKII 222–251 |
| Fw3 | cGKII | TCCAGCTNAACAARCTKCARGA | cGKII 230–251 |
| Fw4 | cGKII | AAGAAGCTCATTACAGATGCCCTKAATAANAAYCAGT | cGKII 466–502 |
| Fw5 | cGKII | ACCAYNTCTWTGTGYTNGCAGAKGG | cGKII 608–632 |
| Fw6 | cGKII | ATYMGAGARGGNGARGAAGGARRYACCTT | cGKII 940–968 |
| Fw7 | cGKII | TTTGCYATGAARTGYATNARGAARAAGCACATHGTKGA | cGKII 1435–1472 |
| Fw8 | cGKII | TTCAAGGAYAATAAGTATGTRTACATG | cGKII 1558–1584 |
| Fw9 | cGKII | TGGAGGCCTGYYTRGGTGGRGARCTCTGGA | cGKII 1589–1618 |
| Fw10 | cGKII | ACATGGACATTCTGTGGGACTCC | cGKII 1819–1841 |
| Rv1 | cGKII | TCCTGCAGCTTGTTCAGCTGGATGCACTTG | cGKII 222–251 |
| Rv2 | cGKII | ACTGRTTNTTATTMAGGGCATCTGTAATGAGCTTCTT | cGKII 466–502 |
| Rv3 | cGKII | ACAAAGATATGGTTTCCNGGTTCTCC | cGKII 595–620 |
| Rv4 | cGKII | TGGAANACYTCTCKATCBAGNGCCCA | cGKII 763–788 |
| Rv5 | cGKII | TCMACDATGTGCTTYTTCYTNATRCAYTTCATRGCAAA | cGKII 1435–1472 |
| Rv6 | cGKII | TCATCRAARCTNCCYCTGTCYCTTARWATRSTCCA | cGKII 1615–1649 |
| Rv7 | cGKII | ACATACTCTGGAGTCCCACAGAATGTCCA | cGKII 1822–1850 |
| Rv8 | cGKII | AGAAGTCTTTRTCCCAGCCTGACANYTCATC | cGKII 2262–2287 |
| Fw11 | cGKIα | ATGAGYGANCTAGANGAAGACTTYGCSAAGATT | cGKIα 1–33 |
| Fw12 | cGKIβ | ATGGGCACCYTGCGNGAYYTNCAGTACGCGCTCCA | cGKIβ 1–34 |
| Fw13 | cGKI | CTNATMAAGGARGCYATCYTKGAYAATGACTTYATGAA | cGKIα/β |
| Fw14 | cGKI | GATGCYACSACTCGKTTYTACACKGCCTGTGTG | cGKIα/β |
| Rv9 | cGKI | GAAGCTCAGACAATCAGAAGGGCTCC | cGKIα/β |
| Rv10 | cGKI | TTGTAMGTTTTCATKGGRTCGGRRCCTGAGAACGG | cGKIα/β |
| Fw15 | cGKI and II | GGNGARCTGGCCATYYTNTAYAAYTGYACNMGRACNGC | cGMP binding domain |
| Rv11 | cGKI and II | TTNACNAGYTCNACNCKNCCRAANCCWCCSACNCCYARWGT | Kinase domain |
| Rv12 | cGKI and II | CCYTTRTTNAKDATNAYYTCNGGNGCCATRTAYTCNGGWGTNCC | Kinase domain |
| Fw17 | S. acanthias cGKI | ACAGTCAAAGCCCTCGCCAACATC | 224–247 |
| Fw16 | S. acanthias cGKI | GAGCGTACAGATTTCAATGTCTG | 688–710 |
| Fw15 | S. acanthias cGKI | GCCGGAGAACATAATCCTCA | 1080–1099 |
| Fw18 | S. acanthias cGKI | GAGGCTCTATAGGACATTCAAG | 1431–1452 |
| Rv13 | S. acanthias cGKI | GGATTGTCCCTGCACAGTTT | 1399–1418 |
| Rv14 | S. acanthias cGKI | CATCACCCTCTTTGATGATGCA | 70–91 |
| Rv18 | S. acanthias cGKI | CCTGGACAGAGAACTGGGAAGCT | 458–480 |
| Rv15 | S. acanthias cGKI | ATCGAAAGAGCCCCTGTCCCTCAG | 993–970 |
| Rv16 | S. acanthias cGKI | TCTGTATCTGTCCCTGAGAATAGTC | 1026–1050 |
| Rv19 | S. acanthias cGKI | CTTAGCATAGCCCCGGTGATCA | 1185–1206 |
| Rv17 | S. acanthias cGKI | AGGACCAGTAATCTGCGGAGC | 1232–1252 |
| Fw19 | NPR-C | GACGGACGAACGTCATTTTT | 540–559 |
| Rv18 | NPR-C | TCCAGATCAGCACTCGACAC | 985–1004 |
Positions for cGMP-dependent protein kinase II (cGKII) are relative to the start of the open reading frame of human cGKII. Positions in S. acanthias-specific primers refer to the start of the open reading frame or the start of the known sequence. NPR-C, natriuretic peptide receptor C type.
Amplification was performed using total rectal gland RNA. Fifty nanograms of RNA were reverse transcribed in a volume of 20 μl using 200 units Sigma Superscript II reverse transcriptase using standard conditions as recommended by the manufacturer. After RNAse H treatment for 15 min at 37°C, samples were diluted to 50 μl and PCR reactions were performed on 2 μl of cDNA using Sigma Redtaq polymerase. Alternatively, 100 ng of a SRG cDNA library were used as input in the PCR reactions (for a description of the cDNA library preparation procedure, see ref. 2). The PCR conditions were denaturation 94°C for 4 min followed by 30 cycles (94°C for 30 s, annealing at 50°C for 30 s, extension at 72°C for 2 min) followed by a further extension at 72°C for 5 min. Picogram quantities of plasmids containing human or mouse cGKI and cGKII were used to optimize PCR conditions and check for successful amplification. Amplified products were analyzed on a 1% agarose gel. If by-products were present, products were cut from gel and reamplified if necessary. Sequencing was performed using BigDyedideoxy terminators (Perkin Elmer) and an ABI automated sequencer.
Statistical analysis.
Data are presented as mean values ± SE. Statistical significance of differences between means was assessed using the Student's t-test (2 sided), and accepted at values of *P < 0.05, **P < 0.01, or ***P < 0.001.
RESULTS
Partial cloning of cGMP-dependent protein kinases and identification by protein purification.
In an attempt to identify the cGMP target protein(s) linking cGMP signaling to chloride secretion in the SRG, we first searched for the expression of a dogfish ortholog of cGKII in this tissue by a combination of molecular cloning and protein purification experiments.
To clone a putative cGK encoding gene, degenerate primer sets were designed from 14 species known to express a cGKII sequence, including two species of teleosts (Danio rerio and Tetraodon nigroviridis; Table 1). However, with the use of these primers no cGKII homolog was detected after RT-PCR amplification of cDNA isolated from the rectal gland, although most primer combinations were able to amplify picogram quantities of a plasmid containing the mouse cGKII gene. One primer combination (Fw9 and Rv7) specifically amplified a ∼260-bp fragment. Sequence analysis showed much stronger homology to cGKI than to cGKII. Subsequently, long and highly degenerate primers were designed to the most conserved sequences in both cGKI and cGKII, i.e., the cGMP binding sites and part of the kinase domain consensus sequence (Table 1). With the use these primers, fragments from two different cGKI isoforms were amplified from the rectal gland. Attempts to amplify the 5′-end of these genes using degenerate primers specific for cGKIα or cGKIβ failed, suggesting that the NH2-terminal part of these proteins differs strongly from those in mammals. Again, no cGKII homolog was detected.
With the use of degenerate primers to amplify a sequence downstream of the 5′ end (starting at exon 2 from the human gene), larger fragments of both S. acanthias cGKI isoforms were obtained, both starting at bp317 of the human cGKIα coding sequence. The sequences of the final amplification products of 1448 and 1654 bp were deposited in the GenBank (Accession Nos. FJ624866 and FJ624865, respectively). One of the two isoforms, which is partially identical to a previously identified EST clone (GenBank Accession No. 452242), possibly a pseudogene, was shown to contain a stop codon at position aa 173 (relative to human cGKIα), which lies outside the part of the EST clone sequenced previously. The other cGKI gene fragment was highly homologous to cGKI from various species. The predicted protein sequence is 94% identical to human cGKI and 90% identical to zebrafish. In particular the cGMP binding domains and the catalytic domain are highly conserved, suggesting that this gene encodes an active cGMP-dependent protein kinase.
In parallel we attempted to detect cGK protein in lysates of cultured SRG epithelial cells by cGMP- or cAMP-agarose affinity-chromatography followed by cGMP-triggered autophosphorylation in the presence of [γ-32P]ATP, SDS-PAGE, and autoradiography or Western blotting using cGKII-or cGKI-specific antibodies (cf. refs. 6–7 for Methods). Whereas trace amounts of recombinant human cGKII mixed with the SRG cell extracts were readily detected, endogenous shark cGKII could not be identified by this approach (results not shown). In conclusion, both molecular cloning and protein isolation approaches were unable to provide evidence for the expression of a type II ortholog of mammalian cGKs in SRG epithelial cells, arguing strongly against a key role of cGKII as a regulator of CNP/cGMP-activated chloride secretion in this cell type. In contrast, cGKI is expressed in the SRG gland but does not qualify as a regulator of CFTR-mediated chloride secretion because 1) it could not substitute for cGKII as an activator of CFTR in mammalian cells (11, 34), and 2) preliminary experiments indicated that cGKI is expressed in vascular smooth muscle and neuronal cells of the SRG, not in SRG epithelial cells (data not shown).
Functional studies.
In agreement with our previous studies (2, 15), saturating concentrations of the NPR-B/guanylyl cyclase activator CNP (10 nM) stimulated chloride secretion in the perfused SRG to a similar extent as saturating concentrations of the adenylyl cyclase activator forskolin (1 μM) in combination with the nonspecific PDE inhibitor IBMX (Fig. 1A). To ascertain that the CNP effect was of nonneuronal origin, filter-grown confluent monolayers of primary cultures of SRG epithelial cells that are devoid of neural elements (35) were exposed to CNP and transepithelial chloride secretion was monitored by Isc measurements in Ussing chambers. As shown in Fig. 1B, CNP provoked a large increase in Isc that was further enhanced by forskolin, shown previously to trigger cAMP/PKA-mediated phosphorylation, membrane insertion of CFTR, and activation of the CFTR chloride channel in SRG epithelial cells (15). In support of this concept, the Isc response to CNP and forskolin was almost fully blocked by a combination of bumetanide, a specific inhibitor of the basolateral Na+-K+-2Cl− cotransporter NKCC, and the general K+ channel inhibitor Ba2+ (Fig. 1B), the latter strongly reducing the electrochemical driving force for CFTR-mediated Cl− exit at the apical membrane.
Fig. 1.
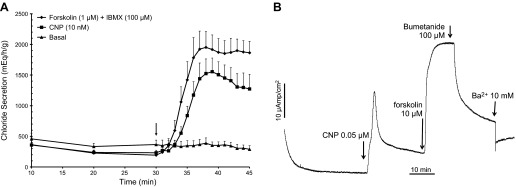
A: perfused shark rectal glands (SRGs). Glands were perfused with shark Ringer's solution for 30 min under basal conditions with measurements of chloride secretion every 10 min. At 30 min (arrow), 1-min readings were begun and glands were perfused with Ringer's solution containing either forskolin (1 μM + IBMX 100 μM; n = 13) or C-type natriuretic peptide (CNP; 10 nM; n = 10) or basal Ringer's only was continued as a control (n = 10). Values are mean chloride secretion (μeq·h−1·g−1) ± SE. B: representative short circuit current (Isc) experiment in primary culture monolayers of SRG tubular epithelial cells demonstrating the response to CNP (0.05 μM), followed by forskolin (10 μM). Isc was then inhibited by the addition of bumetanide (100 μM) and BaCl2 (10 mM). This experiment is representative of 4 similar experiments.
Previous studies in our laboratory have identified, cloned, and characterized an ancestral receptor guanylyl cyclase (designated NPR-B or GC-B) as the effector of CNP in the SRG (2), suggesting that cGMP elevation is the key signaling event coupling CNP to CFTR channel opening. Surprisingly, however, CNP-induced Cl− secretion could not be mimicked by any of the membrane-permeant and phosphodiesterase (PDE)-resistant cGMP analogs, including the general activator of cGMP-dependent protein kinases (cGK) 8-Br-cGMP (Fig. 2, A and B), the cGK type II-specific analog 8-pCPT-cGMP (Fig. 2B), and the cGKI-specific analog 8-Br-PET-cGMP (not shown). In contrast, the PKA-specific analog 6-MB-cAMP caused a full activation of chloride secretion, mimicking the effect of forskolin (Fig. 2B). A similar insensitivity to 8-Br-cGMP alone has been reported previously in SRG perfusion studies (24). In this previous report the combination of 8-Br-cGMP and activation of protein kinase C (PKC) by the phorbolester PMA mimicked the effect of CNP on chloride secretion in the perfused gland. In the present study, however, no evidence could be found to support a synergistic effect of PMA and 8-Br-cGMP on SRG chloride secretion, neither in the perfused gland (Fig. 2A) nor in cultured cells monitored in the Ussing chamber (Fig. 2B).
Fig. 2.
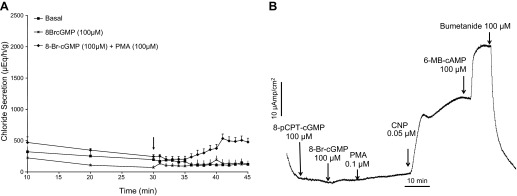
A: perfused SRGs. Glands were perfused with shark Ringer's solution for 30 min under basal conditions with measurements of chloride secretion every 10 min. At 30 min (arrow), 1-min readings were begun and glands were perfused with Ringer's solution containing either 8-bromo-cGMP (8 Br-cGMP; 100 μM; n = 4) or 8 Br-cGMP + PMA (100 μM; n = 3) or basal Ringer's only was continued as a control (n = 12). Whereas 8-Br-cGMP alone did not increase chloride secretion above basal levels, 8 Br-cGMP + PMA slightly increased chloride secretion from min 41–45 (P < 0.05) Values are mean chloride secretion (μeq·h−1·g−1) ± SE. B: representative Isc experiment in primary culture monolayers of SRG tubular epithelial cells demonstrating a lack of response to 8 Br-cGMP and PMA, whereas CNP (0.05 μM) and 6 MB-cAMP markedly increased Isc, which was inhibited to baseline by the addition of bumetanide (100 μM). The experiment is representative of 4 similar experiments.
Our failure to mimic CNP-induced chloride secretion in the SRG by cGMP analogs, despite their known action as potent activators of cGKs in mammalian epithelial tissues (30–31, 33–34), and our failure to identify a shark ortholog of the CFTR-activating enzyme cGKII in the rectal gland, argue strongly against a role for cGKs in the CNP-CFTR signaling pathway. Subsequent protein kinase inhibition experiments likewise pointed to a role for PKA rather than cGK in CNP-provoked chloride secretion in SRG epithelial cells (Fig. 3). The relatively selective cGK inhibitor H-8, tested at a concentration that caused inhibition of cGMP-, but not of cAMP-, provoked chloride secretion in intestinal epithelium (30), was unable to block the CNP/cGMP- and forskolin/cAMP-induced Isc response in SRG epithelial cells (Fig. 3). In contrast, preexposure of the cells for 30 min to H-89, a relatively selective inhibitor of PKA but a poor inhibitor of PKC and many other protein kinases (16), as well as to the broad spectrum kinase inhibitor staurosporine, severely blunted both the CNP- and forskolin-provoked chloride secretion (Fig. 3). These findings strongly suggest that CNP acts through a PKA rather than a cGK- or PKC-dependent pathway and are in apparent conflict with a previous model postulated solely on the basis of SRG perfusion studies (24).
Fig. 3.

Bar graph of Isc experiments in primary culture monolayers of SRG tubular epithelial cells. Both CNP-and forskolin-induced chloride secretion were inhibited to a similar extent by the nonspecific protein kinase inhibitor staurosporine (1 μM) and the specific PKA inhibitor H-89 (50 μM) but were insensitive to the inhibitor of both isoforms of cGMP-dependent protein kinase H-8 (100 μM). Filter-grown monolayers of SRG cells were preexposed to the PK inhibitors (added to the mucosal and serosal bath of the Ussing chamber) for 30 min before the addition of the secretagogues. Values are percentage inhibition of the CNP (0.5 μM) or forskolin (1 μM) induced Isc response averaged from 4 independent experiments ± SE. **P < 0.01, compared with the Isc response without inhibitor; n.s, not significantly different from the Isc response without inhibitor.
Two potential mechanisms could explain how CNP, following activation of guanylyl cyclase and cGMP generation, could activate PKA, i.e., through cGMP cross-activation of PKA or through cGMP inhibition of PDE3. The latter action is expected to result in a reduced breakdown of cAMP and a local increase in cAMP levels. The effects of specific inhibitors of PDE isoforms were thus pursued in 55 perfusion experiments. In support of the second mechanism, only the specific PDE3 inhibitors cilostamide, amrinone, and milrinone and the nonspecific PDE inhibitor IBMX were found to almost fully mimic the effect of CNP on epithelial chloride secretion in the perfused SRG (Figs. 4A and 5).
Fig. 4.
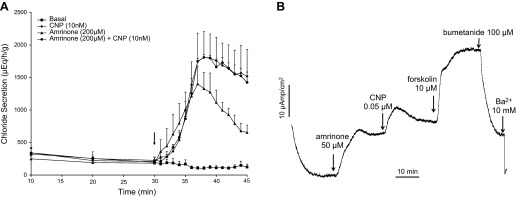
A: perfused SRGs. Glands were perfused with shark Ringer's solution for 30 min under basal conditions with measurements of chloride secretion every 10 min. At 30 min (arrow), 1-min readings were begun and glands were perfused with Ringer's solution containing either CNP (10 nM, n = 5), the PDE-3 inhibitor amrinone (200 μM, n = 4), CNP (10 nM + amrinone 200 μM n = 4), or control basal Ringer's only (n = 12). The chloride secretory response to CNP vs. CNP + amrinone were identical and not additive. Values are mean chloride secretion (μeq·h−1·g−1) ± SE. B: representative Isc experiment in primary culture monolayers of SRG tubular epithelial cells demonstrating an increase in Isc with amrinone, CNP, and forskolin, which was inhibited (9) by bumetanide and BaCl2. The experiment is representative of 4 similar experiments.
Fig. 5.

Perfused SRGs. Mean maximum chloride secretion (μeq·h−1·g−1 ± SE) with nonspecific (IBMX) and isotype-specific PDE inhibitors. Only IBMX, a nonspecific PDE inhibitor, and PDE-3 specific inhibitors (cilostamide, amrinone, and milrinone) increased chloride secretion above basal values. Total n = 55 perfusions, n = 3–19 per group. ***P < 0.001, **P < 0.01, *P < 0.05, all compared with basal.
In contrast to PDE3 inhibitors, none of the inhibitors of other PDE isoforms, including vinpocetine (PDE1-specific), EHNA (PDE2-specific), rolipram (PDE4-specific), zaprinast (PDE5/6/9/11-specific), and dipyridamole (PDE5/6/7-specific) were capable of stimulating gland secretion at concentrations far above their IC50 (Fig. 5). Amrinone was likewise able to mimic the effect of CNP on SRG chloride secretion in cultured cells (Fig. 4B) whereas the addition of amrinone on top of CNP had no further effect on the Isc (results not shown; cf. Fig. 6).
Fig. 6.

Representative Isc experiment in which the basolateral membrane was permeabilized with nystatin and a mucosal to serosal chloride gradient was established by replacing all basolateral Na and K salts with Na and K gluconate. The experiment demonstrates that CNP, amrinone, and forskolin stimulate chloride transport by the opening of anion channels in the apical membrane under conditions of a clamped electrochemical gradient of Cl− across this membrane. The experiment is representative of 3 similar experiments.
The Ussing chamber Isc tracing shown in Fig. 6 was obtained following nystatin permeabilization of the basolateral membrane and after imposing a mucosal-to-serosal Cl− gradient. These conditions allow macroscopic measurements of CFTR-mediated Cl− currents across the apical membrane unaffected by potential rate-limiting ion movements across the basolateral membrane. The outcome proves unambiguously that both CNP and forskolin are capable of enhancing CFTR-mediated Cl− movement across the apical membrane at a fixed electrochemical gradient, i.e., through recruitment and activation of the CFTR Cl− channel itself rather than through effects on other channels or transporters (e.g., basolateral K+ channels) that modulate the driving force for Cl− exit through the CFTR channel.
The experiments above demonstrate clearly that CNP is capable of recruiting and activating the CFTR channel in the apical membrane of SRG epithelial cells through a guanylyl cyclase-cGMP-PKA pathway that is nonadditive with a PDE3-cAMP-PKA inhibition pathway. However, they do not firmly rule out a possible role of cGMP cross-activation of PKA as an alternative or parallel mode of action of CNP. To discriminate between these pathways, we exploited the ability of the peptide hormone somatostatin to bind to somatostatin receptors in the basolateral membrane and to inhibit adenylyl cyclase through a Gi protein-mediated interaction (28). As illustrated in Figs. 7A and 8A, addition of somatostatin to the perfused SRG or to monolayers of epithelial cells in the Ussing chamber caused a complete inhibition of the Cl− secretory response to CNP, whereas the response to supramaximal levels of forskolin (presumably bypassing Gi inhibition) remained unaltered (Fig. 8A). A similar almost complete inhibition by somatostatin was observed for amrinone-induced Cl− secretion (Fig. 8A). In line with the mechanism of action proposed, CNP and amrinone were found able to elevate both cAMP and cGMP levels in perfused glands (Fig. 7B and Ref. 2) and in cultured SRG epithelial cells (Fig. 8, B and C), whereas somatostatin strongly reduced the cAMP increase but not the cGMP increase (Figs. 7B and 8, B and C). These experiments confirm the ability of somatostatin to inhibit the basal activity of adenylyl cyclase and to counteract cAMP elevation by PDE3 inhibitors and CNP in the absence of any significant inhibitory effect on CNP-triggered GC-B activation and subsequent cGMP formation. They virtually rule out a significant contribution of the cGMP-PKA cross-activation pathway to the CNP-induced Cl− secretory response that was completely blocked by somatostatin whereas cGMP signaling remained unaltered.
Fig. 7.
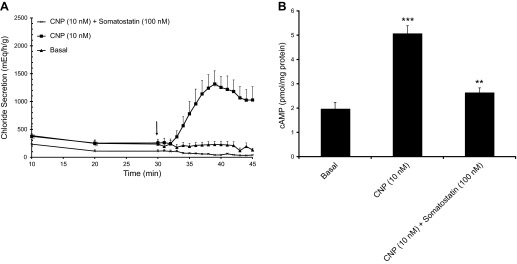
A: perfused SRGs. Glands were perfused with shark Ringer's solution for 30 min under basal conditions with measurements of chloride secretion every 10 min. At 30 min (arrow), 1-min readings were begun and glands were perfused with Ringer's solution containing either CNP (10 nM, n = 5), or CNP 10 nM + somatostatin (100 nM, n = 5), compared with basal (n = 4). B: tissue cAMP content (pmol/mg protein) at the 45-min time point in these glands. ***P < 0.001, comparing CNP to basal; **P < 0.01, comparing CNP + somatostatin to CNP.
Fig. 8.

A: representative Isc experiment in primary culture monolayers of SRG tubular epithelial cells demonstrating complete inhibition of CNP-and amrinone-induced chloride secretion by somatostatin (0.1 μM). The experiment is representative of 4 similar experiments. B and C: intracellular cAMP (B) and cGMP (C) levels in filter-grown SRG epithelial cells before and after addition of secretagogues (CNP, 0.05 μM; amrinone, 50 μM) in the absence or presence of somatostatin (0.1 μM). Cells were harvested and lysed after reaching the plateau phase of the Isc response (cf. A), and cyclic nucleotide assays were performed as described in materials and methods. n = 4 per condition. **P < 0.01, comparing cAMP content with CNP + somatostatin to CNP alone and comparing amrinone + somatostatin to amrinone alone (B); n.s., not significant comparing cGMP content with CNP alone with CNP + somatostatin and comparing amrinone alone with amrinone + somatostatin (C).
DISCUSSION
Sharks maintain body fluid homeostasis in the hypertonic sea by the secretion of NaCl in tubules of the rectal gland, an osmoregulatory organ positioned as an appendage of the gastrointestinal tract. In contrast to mammals, employing atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) as cardiac natriuretic peptides that trigger natriuresis in the kidney, CNP, stored as pro-CNP in the shark heart, is the major physiological activator of CFTR-mediated Cl− secretion in the SRG (2, 21, 25). Its effectiveness relies on direct activation of a NPR-B/GC-B receptor guanylyl cyclase resulting in cGMP signaling (2). However, previous studies in the SRG failed to define the molecular mechanism by which cGMP activates CFTR in the gland tubular cells.
The present study, using both isolated perfused glands and filter-grown monolayers of tubular cells in primary culture as a model, identifies the cGMP/PDE3/cAMP/PKA pathway as the prime signaling cascade linking cGMP elevation with CFTR activation in the apical membrane (Fig. 9, pathway 1). This conclusion is based on two major lines of evidence: the exclusion of a role for other potential cGMP-linked pathways (Fig. 9, pathways 2–4), and the demonstration that pharmacological inhibitors of PDE3 are able to mimic CNP-induced secretion in both models in a nonadditive fashion.
Fig. 9.

Hypothetical model of CNP action in the SRG. CNP binds to and activates a CNP-selective receptor guanylyl cyclase at the basolateral membrane (designated NPR-B), resulting in cGMP elevation. Endogenous cGMP, but not membrane-permeable and nonhydrolyzable cGMP analogs, compete with cAMP for degradation by a type 3 phosphodiesterase (PDE3A) situated in the apical membrane, resulting in a local rise of cAMP levels. Cyclic AMP in the apical compartment, through activation of cAMP-dependent protein kinase (PKA), causes multisite phosphorylation (Pn) of the regulatory (R) domain of cystic fibrosis transmembrane conductance regulator (CFTR) and opening of the CFTR chloride channel (pathway 1, solid lines). Activation of CFTR by CNP is dependent on basal cAMP production by adenylyl cyclase (AC), which is inhibited by somatostatin through a specific receptor coupled to the inhibitory G protein Gαi. The operation of 3 other potential cGMP signaling pathways (dotted lines), i.e., cGMP activation of the type II isoform of cGMP-dependent protein kinase (cGKII) anchored through myristoyl groups to the apical membrane (pathway 2), cGMP cross-activation of PKA (pathway 3), and direct allosteric activation of CFTR by cGMP (pathway 4), is virtually excluded by the experiments reported here, identifying pathway 1 as the predominant, if not sole signaling pathway operating in the SRG epithelial cells. We emphasize that the positioning of PDE3A in the apical membrane in close vicinity to CFTR is entirely hypothetical and based on analogy with a recent PDE3-CFTR signaling model in human airway epithelial cells (18).
Our initial hypothesis considered a role for a shark ortholog of the mammalian type II cGK as the link between CNP-cGMP signaling and CFTR activation. This homology model was based on the colocalization of cGKII and CFTR in the apical membrane of intestinal epithelial cells and the ability of cGKII, but not of cGKI, to activate the CFTR Cl− channel through multisite phosphorylation of the regulatory (R) domain (6–7, 11, 14, 33–34) (Fig. 9, pathway 2). However, a number of findings in the present study argue strongly against the operation of a similar pathway in the SRG:
1) Our failure to detect a cGKII sequence after RT-PCR amplification of cDNA isolated from the SRG, using sets of degenerate primers directed to part of the cGKII gene shared by all 15 species known to express a cGKII sequence, including the teleosts Danio rerio and Tetraodon nigroviridis, and two invertebrates, insect and mollusc. In contrast, the use of degenerate primers designed to the most conserved sequences among the cGKI and cGKII isoforms allowed us to identify and partially clone a shark cGKI gene that at the protein level was 94% identical to human cGKI and 90% identical to zebrafish (GenBank Accession No. FJ624865). However, this soluble cGKI isoform, in contrast to cGKII, lacks a myristoyl anchor for attachment to the apical membrane and is unable to activate CFTR (11, 32, 34). Moreover preliminary experiments pointed to its expression in smooth muscle and neuronal cell types rather than in epithelial cells of the SRG, as documented previously for mammalian intestine (14, 33).
2) Our failure to detect cGKII protein in lysates of cultured SRG epithelial cells by cGMP-or cAMP-agarose affinity chromatography and autophosphorylation in the presence of [γ-32P]ATP or by Western blotting using cGKII-specific antibodies.
3) The inability of membrane-permeable, hydrolysis-resistant cGMP analogs including the relatively specific cGKII activator 8-pCPT-cGMP to mimic CNP-provoked CFTR activation and Cl− secretion in the SRG epithelial cells in contrast to cAMP analogs. Likewise the combined addition of a cGMP analog and PMA, a specific activator of PKC, failed to stimulate Cl− secretion, both in the perfused gland and in the cultured tubular cells. Therefore, a synergistic interaction between cGMP and PKC signaling pathways converging at CFTR in the perfused SRG reported earlier (24) could not be confirmed in the present study and is not known to operate in any mammalian epithelial cell type, including cells from the gastrointestinal tract. The reason for these conflicting results is unclear.
4) The profound effect of the relatively specific PKA inhibitor H89 and of the nonspecific kinase inhibitor staurosporine on SRG Cl− secretion, in contrast to a lack of effect of the relatively specific cGK inhibitor H8, also point to a role of PKA rather than PKG in the CNP signaling cascade.
5) The ability of the inhibitory modulator of SRG Cl− secretion somatostatin to counteract the CNP-induced rise in cAMP levels but not the concomittant elevation of cGMP. If CNP would act through cGMP/cGKII signaling, somatostatin would not be expected to cause the complete inhibition of CNP-induced Cl− secretion observed in this study.
Collectively, these results argue strongly against a role of cGKII as a CFTR activator in the SRG. Additional cloning and functional studies are needed to define a possible role of shark cGKII in other epithelial tissues, in particular the gastrointestinal tract, and to clarify its role in CFTR-expressing epithelia from noncartilaginous fishes, e.g., the gills of teleosts (23).
Two other potential modes of action of cGMP, i.e., cGMP cross-activation of PKA (Fig. 9, pathway 3) and direct allosteric activation of CFTR by interaction with a cGMP binding site located at the NH2-terminal end of NBD1 (19, 27) (Fig. 9, pathway 4), could also be virtually ruled out because both mechanisms would be in conflict with our finding of somatostatin inhibition of CNP-induced Cl− secretion occurring at maximally elevated cGMP levels. Moreover, these mechanisms would not explain the inability of membrane-permeable cGMP analogs to provoke CFTR activation in the SRG.
Instead the data strongly support a key role for the type 3 isoform of PDE3 in CNP action in the SRG. First, it was found that three specific inhibitors of PDE3, i.e., cilostamide, milrinone, and amrinone, and a nonspecific PDE inhibitor, IBMX, were each able to mimic the prosecretory effect of CNP in a nonadditive fashion. In contrast, none of the inhibitors of other mammalian PDE isoforms, i.e., PDE1, 2, 4–7, and 11 (3), appeared able to elicit CFTR-mediated Cl− secretion in the SRG. Second, CNP elevated not only cGMP but also cAMP levels in the perfused gland and in cultured tubular cells, most plausibly through competitive inhibition of PDE3-catalyzed hydrolysis of cAMP by cGMP. The failure of PDE-resistant cGMP analogs to mimic CNP action can be readily explained by their inability to interact with PDE3, in contrast to cGMP itself (1, 5). A similar mechanism has been postulated previously for the action of the cGMP agonist Escherichia coli heat-stable enterotoxin (STa) in mouse proximal colon, showing residual STa stimulation of CFTR-mediated chloride secretion in cGKII−/− mice (mimicked by amrinone) under conditions in which 8-Br-cGMP had completely lost its effect (31). Finally, the parallel inhibition of CNP- and amrinone-provoked Cl− secretion and cAMP but not cGMP elevation by somatostatin, the pronounced synergism between CNP/cGMP- and VIP/cAMP-induced secretion, and the inhibition of CNP-induced secretion by the PKA inhibitor H-89 further support the CNP/cGMP/cAMP/PKA model (Fig. 9, pathway 1).
These data provide strong functional evidence for the expression of a shark ortholog of PDE3 in SRG epithelia. Molecular cloning experiments using degenerate primers provided preliminary evidence for the expression of the PDE3A rather than the PDE3B isoform in the SRG (results not shown). Interestingly, although the catalytic domains of PDE3A and 3B are very similar, PDE3A is more sensitive to cGMP inhibition than PDE3B (8). Moreover, the NH2-terminal portion of PDE3A contains six helical transmembrane segments involved in anchoring the enzyme to the apical membrane of polarized epithelial cells (8). Using the Calu-3 human airway epithelial cell line as a model, Penmatsa et al. (18) have recently demonstrated that PDE3A physically and functionally interacts with CFTR in the apical membrane and that PDE3 inhibition results in compartmentalized cAMP formation that serves to promote CFTR activation within a CFTR/PDE3A/PKA macromolecular complex and augments CFTR-dependent secretion. Clearly additional studies including full-length cloning of the shark ortholog of PDE3 are needed to verify the occurrence of a similar CFTR/PDE3 signaling complex in the SRG.
In summary, this study provides strong evidence for the operation of a CNP/cGMP/PDE3/cAMP/PKA signaling cascade as the major if not sole CFTR-activating mechanism in the SRG. This mode of cGMP-triggered CFTR activation, using cAMP/PKA rather than a membrane-bound isoform of cGK as the key signal transducer, is highly conserved evolutionary from sharks to mammals. Most plausibly, a CFTR activating cGK isoform functioned first as an inhibitor of salt absorption in epithelial cells, as suggested by ANF/cGMP studies in the intestine of teleosts (17, 20, 29) and studies of guanylin and STa inhibition of the apical sodium-hydrogen exchanger NHE3 in mammalian intestine (4, 31). Its ability to phosphorylate and activate CFTR may have evolved later as an additional and redundant mechanism to combine its antiabsorptive with a prosecretory action and to facilitate the regulation of epithelial salt transport by cGMP-linked luminocrinic peptides such as guanylin and uroguanylin, acting through GC-C rather than GC-B (22, 33).
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-34208 and National Institute of Environmental Health Sciences Grant 5-P30-ES-03828 (Center for Membrane Toxicology Studies; to J. N. Forrest), National Science Foundation Grant DBI-0139190 (Research Experience for Undergraduates site at MDIBL), and an MDIBL New Investigator Award (to H. R. De Jonge).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.R.d.J. and J.N.F. conception and design of research; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., R.M.V., and J.N.F. performed experiments; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., R.M.V., and J.N.F. analyzed data; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., and J.N.F. interpreted results of experiments; H.R.d.J., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., R.M.V., and J.N.F. prepared figures; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., and J.N.F. drafted manuscript; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., R.M.V., and J.N.F. edited and revised manuscript; H.R.d.J., B.C.T., B.M.H., D.J.P., C.A.K., M.H.K., A.M.M., M.T.M., R.M.V., and J.N.F. approved final version of manuscript.
REFERENCES
- 1.Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res 93: 406–413, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Aller SG, Lombardo ID, Bhanot S, Forrest JN., Jr Cloning, characterization, and functional expression of a CNP receptor regulating CFTR in the shark rectal gland. Am J Physiol Cell Physiol 276: C442–C449, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Cha B, Kim JH, Hut H, Hogema BM, Nadarja J, Zizak M, Cavet M, Lee-Kwon W, Lohmann SM, Smolenski A, Tse CM, Yun C, de Jonge HR, Donowitz M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem 280: 16642–16650, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481–511, 2007 [DOI] [PubMed] [Google Scholar]
- 6.De Jonge HR. cGMP-dependent phosphorylation and ion transport in intestinal microvilli. In: Cold Spring Harbor Conferences on Cell Proliferation-Protein Phosphorylation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratories, 1981, p. 1313–1332 [Google Scholar]
- 7.de Jonge HR. Cyclic GMP-dependent protein kinase in intestinal brushborders. Adv Cyclic Nucleotide Res 14: 315–333, 1981 [PubMed] [Google Scholar]
- 8.Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J Biol Chem 272: 6823–6826, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Forrest JN., Jr Cellular and molecular biology of chloride secretion in the shark rectal gland: regulation by adenosine receptors. Kidney Int 49: 1557–1562, 1996 [DOI] [PubMed] [Google Scholar]
- 10.Forte LR, Thorne PK, Eber SL, Krause WJ, Freeman RH, Francis SH, Corbin JD. Stimulation of intestinal Cl− transport by heat-stable enterotoxin: activation of cAMP-dependent protein kinase by cGMP. Am J Physiol Cell Physiol 263: C607–C615, 1992 [DOI] [PubMed] [Google Scholar]
- 11.French PJ, Bijman J, Edixhoven M, Vaandrager AB, Scholte BJ, Lohmann SM, Nairn AC, de Jonge HR. Isotype-specific activation of cystic fibrosis transmembrane conductance regulator-chloride channels by cGMP-dependent protein kinase II. J Biol Chem 270: 26626–26631, 1995 [DOI] [PubMed] [Google Scholar]
- 12.Greger R, Bleich M, Warth R, Thiele I, Forrest JN. The cellular mechanisms of Cl− secretion induced by C-type natriuretic peptide (CNP). Experiments on isolated in vitro perfused rectal gland tubules of Squalus acanthias. Pflügers Arch 438: 15–22, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Hyodo S, Kawakoshi A, Bartolo RC, Takei Y, Toop T, Donald JA. Extremely high conservation in the untranslated region as well as the coding region of CNP mRNAs throughout elasmobranch species. Gen Comp Endocrinol 148: 181–186, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Jarchau T, Hausler C, Markert T, Pohler D, Vanderkerckhove J, De Jonge HR, Lohmann SM, Walter U. Cloning, expression, and in situ localization of rat intestinal cGMP-dependent protein kinase II. Proc Natl Acad Sci USA 91: 9426–9430, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehrich RW, Aller SG, Webster P, Marino CR, Forrest JN., Jr Vasoactive intestinal peptide, forskolin, and genistein increase apical CFTR trafficking in the rectal gland of the spiny dogfish, Squalus acanthias. Acute regulation of CFTR trafficking in an intact epithelium. J Clin Invest 101: 737–745, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev 24: 261–274, 2006 [DOI] [PubMed] [Google Scholar]
- 17.O'Grady SM, Field M, Nash NT, Rao MC. Atrial natriuretic factor inhibits Na-K-Cl cotransport in teleost intestine. Am J Physiol Cell Physiol 249: C531–C534, 1985 [DOI] [PubMed] [Google Scholar]
- 18.Penmatsa H, Zhang W, Yarlagadda S, Li C, Conoley VG, Yue J, Bahouth SW, Buddington RK, Zhang G, Nelson DJ, Sonecha MD, Manganiello V, Wine JJ, Naren AP. Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell 21: 1097–1110, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira MM, Parker J, Stratford FL, McPherson M, Dormer RL. Activation mechanisms for the cystic fibrosis transmembrane conductance regulator protein involve direct binding of cAMP. Biochem J 405: 181–189, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao MC, Nash NT, Field M. Differing effects of cGMP and cAMP on ion transport across flounder intestine. Am J Physiol Cell Physiol 246: C167–C171, 1984 [DOI] [PubMed] [Google Scholar]
- 21.Schofield JP, Jones DS, Forrest JN., Jr Identification of C-type natriuretic peptide in heart of spiny dogfish shark (Squalus acanthias). Am J Physiol Renal Fluid Electrolyte Physiol 261: F734–F739, 1991 [DOI] [PubMed] [Google Scholar]
- 22.Schwabe K, Cetin Y. Guanylin and functional coupling proteins in the hepatobiliary system of rat and guinea pig. Histochem Cell Biol 137: 589–597, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Shaw JR, Bomberger JM, VanderHeide J, LaCasse T, Stanton S, Coutermarsh B, Barnaby R, Stanton BA. Arsenic inhibits SGK1 activation of CFTR Cl- channels in the gill of killifish, Fundulus heteroclitus. Aquat Toxicol (Amst) 98: 157–164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva P, Solomon RJ, Epstein FH. Mode of activation of salt secretion by C-type natriuretic peptide in the shark rectal gland. Am J Physiol Regul Integr Comp Physiol 277: R1725–R1732, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Solomon R, Protter A, McEnroe G, Porter JG, Silva P. C-type natriuretic peptides stimulate chloride secretion in the rectal gland of Squalus acanthias. Am J Physiol Regul Integr Comp Physiol 262: R707–R711, 1992 [DOI] [PubMed] [Google Scholar]
- 26.Stahl M, Stahl K, Brubacher MB, Forrest JN., Jr Divergent CFTR orthologs respond differently to the channel inhibitors CFTRinh-172, glibenclamide, and GlyH-101. Am J Physiol Cell Physiol 302: C67–C76, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan SK, Agellon LB, Schick R. Identification and partial characterization of a domain in CFTR that may bind cyclic nucleotides directly. Curr Biol 5: 1159–1167, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol 34: 228–252, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Toskulkao C, Rao MC. Identification of a 50-kDa Ca-, cAMP-, and cGMP-dependent epithelial phosphoprotein as a cAMP regulatory protein. Am J Physiol Cell Physiol 258: C889–C901, 1990 [DOI] [PubMed] [Google Scholar]
- 30.Vaandrager AB, Bot AG, De Jonge HR. Guanosine 3′,5′-cyclic monophosphate-dependent protein kinase II mediates heat-stable enterotoxin-provoked chloride secretion in rat intestine. Gastroenterology 112: 437–443, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Vaandrager AB, Bot AG, Ruth P, Pfeifer A, Hofmann F, De Jonge HR. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 118: 108–114, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Vaandrager AB, Ehlert EM, Jarchau T, Lohmann SM, de Jonge HR. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J Biol Chem 271: 7025–7029, 1996 [DOI] [PubMed] [Google Scholar]
- 33.Vaandrager AB, Hogema BM, de Jonge HR. Molecular properties and biological functions of cGMP-dependent protein kinase II. Front Biosci 10: 2150–2164, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Vaandrager AB, Tilly BC, Smolenski A, Schneider-Rasp S, Bot AG, Edixhoven M, Scholte BJ, Jarchau T, Walter U, Lohmann SM, Poller WC, de Jonge HR. cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl- channels co-expressed with cGMP-dependent protein kinase type II but not type Ibeta. J Biol Chem 272: 4195–4200, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Valentich JD, Forrest JN., Jr Cl− secretion by cultured shark rectal gland cells. I. Transepithelial transport. Am J Physiol Cell Physiol 260: C813–C823, 1991 [DOI] [PubMed] [Google Scholar]