

Abstract
Due to evolutionary pressure, there are many complex interactions at the interface between pathogens and eukaryotic host cells wherein host cells attempt to clear invading microorganisms and pathogens counter these mechanisms to colonize and invade host tissues. One striking observation from studies focused on this interface is that pathogens have multiple mechanisms to modulate and disrupt normal cellular physiology to establish replication niches and avoid clearance. The precision by which pathogens exert their effects on host cells makes them excellent tools to answer questions about cell physiology of eukaryotic cells. Furthermore, an understanding of these mechanisms at the host-pathogen interface will benefit our understanding of how pathogens cause disease. In this review, we describe a few examples of how pathogens disrupt normal cellular physiology and protein trafficking at epithelial cell barriers to underscore how pathogens modulate cellular processes to cause disease and how this knowledge has been utilized to learn about cellular physiology.
Keywords: epithelium, endocytosis, exocytosis, microbe, pathogen
disease-causing pathogens have been studied with the intention of eradicating infections involving these microorganisms and improving human health. From these studies, it is evident that host cells have developed numerous mechanisms for dealing with infectious agents and pathogens have correspondingly established elaborate ways of manipulating host cells for their benefit. As a consequence, a new discipline called cellular microbiology emerged almost 20 years ago and incorporates aspects of cell biology and microbiology to study the interface between hosts and pathogens.
A major focus of this field was initially aimed at utilizing pathogens to answer questions about cell physiology (20, 29). For example, tetanus and botulinum neurotoxins, produced by Clostridium tetani and Clostridium botulinum, respectively, are ideal tools for studying essential aspects of neuronal physiology and cell biology due to their ability to specifically cleave soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) family member proteins (VAMP/synaptobrevin, SNAP-25, and syntaxin 1) at the plasma membrane of neurons and subsequently prevent fusion of synaptic vesicles with neuronal membranes (52). In similar regard, several bacterial toxins modulate intracellular signaling events and thus have been widely utilized to study signaling mechanisms involved in eukaryotic responses to environmental factors. For example, cholera and pertussis toxins, produced by Vibrio cholera and Bordetella pertussis, respectively, activate adenylate cyclase/cyclic-AMP (cAMP) signaling via ADP-ribosylation of specific G-protein subunits and, consequently, have been used to elucidate these signaling pathways (29, 55, 91). This approach has yielded significant results that contribute to our understanding of many cellular processes such as the actin network rearrangements, vesicular trafficking, and signaling pathways that regulate gene expression. However, it is becoming increasingly evident that the use of pathogens is not just beneficial to our understanding of cellular physiology but also to our understanding of how microbes cause disease, which has possible therapeutic implications.
Epithelial tissues line the surfaces of many structures throughout the human body and play a critical role in protecting the human body and underlying tissues from infection and damage by serving as a barrier and the first line of defense that microbes encounter. The epithelium is also responsible for many other cellular processes including selective absorption of nutrients, detection of the extracellular environment, secretion of signaling molecules and other products that mediate cell-to-cell communication, and regulation of immune responses that fight infections, which have overcome the physical barrier established by epithelial cells. Multiple model cell lines that maintain epithelial cell morphology have been employed to study these functions, including Madin-Darby canine kidney (MDCK), Caco-2, and HeLA cells. For example, MDCK cells have been used to show that a FMDE motif is responsible for trafficking of syntaxin 3 to the apical membrane of epithelial cells (71). MDCK cells have also been utilized to study aspects of host-pathogen interactions, including the observation that Pseudomonas aeruginosa inhibits cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel recycling, in addition to their use by cell physiologists to elucidate mechanisms of cellular physiology (81). Although the epithelial barrier efficiently performs these tasks and clears noncommensal microorganisms most of the time, pathogens have developed ways of subverting signaling pathways and trafficking events at the host-pathogen interface to successfully infect and/or colonize host epithelial tissues and cause disease.
The purpose of this review is to highlight recent advances within the field of cellular microbiology and raise awareness for how pathogens manipulate cellular processes at epithelial barriers and how this has expanded our knowledge of cellular physiology.
Endocytosis
Endocytosis is an energy-dependent process that cells use to take up substances by engulfing them with the plasma membrane. This process is required for large polar molecules that cannot penetrate the hydrophobic plasma membrane of the cell. All cells participate in endocytosis, which can be categorized into four categories based on morphology: clathrin mediated, caveolae mediated, macropinocytosis, and phagocytosis. Clathrin-mediated endocytosis involves ∼100-nm vesicles containing a complex of proteins associated with the cytosolic protein clathrin and often serves to internalize receptor-ligand complexes from the cell surface. Caveolae-mediated endocytosis utilizes a different type of membrane budding, consisting of smaller 50- to 100-nm vesicles rich in cholesterol and glycolipids and containing the cholesterol-binding protein caveolin. Both macropinocytosis and phagocytosis internalize larger volumes using larger vesicles, ranging from 0.5 to 5 μm in size, but differ in their specificity for cargo. Macropinocytosis invaginations nonspecifically engulf extracellular components, while phagocytosis requires binding of cargo to the cell surface to initiate the phagocytic event. Entry into the host cell is an essential step for many pathogens and thus endocytosis is a pathway commonly exploited during infection or intoxication. We have focused below on several functional categorizations for microbial manipulation of the endocytic pathway including microbial entry into the epithelium, toxin delivery, and immune evasion (Fig. 1).
Fig. 1.
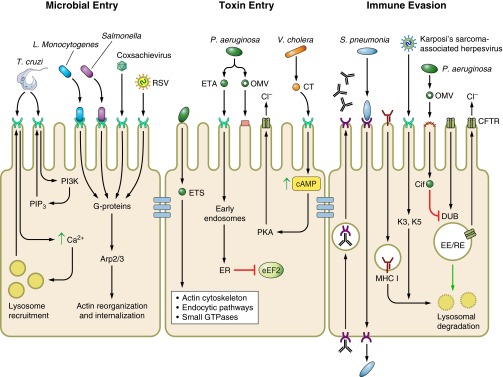
Microbial regulation of endocytosis. Microbial targeting of host endocytic pathways facilitates their invasion of host epithelial cells. Intracellular bacteria invade nonphagocytic host epithelial cells via one of two discrete mechanisms, both of which involve actin rearrangements due to activation of G proteins: zipper and trigger. Viruses exploit similar signaling pathways to facilitate actin rearrangements and invasion of host epithelial cells through three mechanisms: clathrin mediated, caveolar mediated, or macropinocytosis. Invasion of host epithelial cells by protozoa results following multiple complex interactions between the host and pathogen. As a result, protozoa invade host epithelial cells through multiple mechanisms. Trypanosoma cruzi invasion occurs as a result of either recruitment of lysosomes or phosphoinositide-3,4,5-triphosphate (PIP3) to the site of invasion. Many extracellular bacteria produce protein toxins that directly modulate host cell signaling and endocytosis to actively remodel cellular physiology. Bacterial secretion systems directly inject bacterial toxins from the bacteria into the cytosol of host cells. Bacterial toxins secreted extracellularly contain domains that facilitate binding of host receptors and translocation across cellular membranes following endocytosis, or are delivered by outer membrane vesicles (OMVs). Many pathogens successfully evade host immune responses as a result of disrupting endocytic pathways. ER, endoplasmic reticulum; PI3K, phosphatidylinositol 3-kinase; CT, cholera toxin; CFTR, cystic fibrosis transmembrane conductance regulator; MHC I, major histocompatibility complex class I; eEF2, eukaryotic elongation factor-2; RSV, respiratory syncytial virus; ETA, exotoxin A; ETS, exoenzyme S; EE/RE, early endosome/recycling endosome; PKA, protein kinase A.
Microbial Entry into Epithelial Cells
Bacterial subversion of endocytic pathways is a necessity of intracellular bacteria that do not invade phagocytic cells. As a consequence, these bacteria have developed two discrete ways of manipulating signaling molecules that facilitate their uptake by promoting plasma membrane rearrangements due to manipulation of the actin network: the zipper mechanism and the trigger mechanism.
Zipper mechanisms are initiated when bacterial surface proteins bind host receptors, which signal across the plasma membrane to promote formation of internalization vacuoles at the site of bacterial attachment. Zipper mechanism-mediated entry of intracellular bacteria has been best studied for the Gram-positive bacterium Listeria monocytogenes. L. monocytogenes has two main invasion proteins, InlA and InlB (21, 68). InlA engages E-cadherin, which results in posttranslational modification of the cytoplasmic tail of E-cadherin (8). Similarly, InlB-mediated invasion of host cells requires similar posttranslational modifications of the InlB receptor, Met (92). Interestingly, regardless of which host receptor L. monocytogenes engages, clathrin is recruited to the site of bacterial entry and presumably is required for internalization based on experiments showing that small interfering (si)RNA-mediated knockdown of clathrin abrogates L. monocytogenes entry (92). The significance of these findings cannot be understated as it was the first time that it had been shown bacterial internalization utilizes clathrin, which was previously believed to be important only for internalization of small molecules, providing a new way to study clathrin-mediated endocytosis. It has been shown that for InlA-mediated entry, caveolin also mediates internalization of L. monocytogenes via a clathrin-independent pathway (8) and highlights a key point: microorganisms have developed multiple mechanisms that facilitate invasion. In addition, InlA- and InlB-mediated L. monocytogenes entry involves actin cytoskeletal rearrangements. Upon engagement of InlA or InlB with their respective receptors, various signaling molecules are recruited to the site of attachment. Although the signaling molecules utilized appear to vary based on the receptor engaged and cell type infected, Arp2/3 is ultimately activated and promotes actin polymerization, which facilitates L. monocytogenes internalization (reviewed extensively in Ref. 68). It is clear that L. monocytogenes invasion of epithelial cells requires multiple interactions between clathrin-dependent and -independent endocytic pathways and the actin cytoskeleton, resulting in a “zippering” of the plasma membrane around the bacterium. This mechanism has also been demonstrated for other intracellular pathogens, including Yersinia species.
Conversely, trigger mechanisms result when bacterial effector proteins are injected into host cells by the type 3-secretion system (T3SS). The effector proteins act on various signaling cascades that induce cytoskeletal rearrangements. Ultimately, the trigger mechanism culminates in the formation of membrane ruffles that internalize the bacterium. Trigger mechanism-mediated entry has been well established for the Gram-negative bacterium Salmonella enterica serovar Typhimurium, which has the capacity to invade intestinal epithelial cells. Salmonella invasion is regulated by at least six effector proteins injected into host cells by the T3SS, which each regulate certain aspects of actin reorganization during this process. For example, SopE and SopE2 act as guanine exchange factors and activate the Rho GTPases Rac1 and Cdc42, which leads to recruitment of the Arp2/3 complex and induction of actin rearrangements (43, 79). SopB, an inositol polyphosphatase, also activates GTPases in host cells, but indirectly through activation of another factor identified as SH3-containing guanine nucleotide exchange factor (SGEF; Ref. 65). SopB-induced actin reorganization is dependent on SopB phosphatase activity (102), suggesting that phosphoinositide production is important for SGEF activation. SipA and SipC directly interact with actin to induce membrane ruffling at the site of bacterial invasion (1). Following internalization of Salmonella, SptP, functioning as a GTPase-activating protein (GAP), regulates Cdc42 and Rac1 to restore host cell membranes to their normal morphological state (32). Collectively, these effector proteins act to induce actin cytoskeleton reorganization to induce Salmonella invasion. Furthermore, the Salmonella effector proteins discussed here demonstrate the precision to which bacterial effector proteins are delivered into host cells and modulate specific cellular processes.
Most viruses also rely on endocytic mechanisms to facilitate their infection of host epithelial cells, although examples do exist where viruses can fuse directly with the plasma membrane to gain entry into cells. Before endocytosis, however, viruses must engage the host plasma membrane by binding to specific receptors on the cell surface. Interestingly, interactions between viral proteins and host cellular proteins stimulate the physical movement of viruses along the cell surface to entry sites, where endocytosis occurs (53). This phenomenon has been termed viron surfing and is both actin and myosin II dependent, as addition of cytochalasin D and blebbistatin disrupted murine leukemia virus and avian leukosis virus surfing and infection (53). Other examples of viruses manipulating the host actin network include work on coxsackievirus by Coyne and Bergelson (22) This work has shown that upon binding decay-accelerating factor on the apical cell surface, coxsackievirus induces activation of Abl kinase, which initiates Rac-dependent actin rearrangements that allow virus movement to tight junctions where the virus engages its receptor, coxsackievirus and adenovirus receptor. Following receptor engagement, viruses penetrate the plasma membrane and enter host cells. Virus invasion of host cells can generally be categorized into three endocytic mechanisms: 1) clathrin-mediated endocytosis, 2) caveolar-mediated endocytosis, and 3) macropinocytosis (63).
As an example, we discuss recent findings that elucidate the mechanism by which respiratory syncytial virus (RSV) invades host cells by macropinocytosis (49). Studies have shown that RSV invasion does not require clathrin-mediated endocytosis, as inhibitors of clathrin-mediated endocytosis had no effect on RSV entry into cells. Disruption of the actin network, as well as Cdc42, PAK1, and N-Wasp inhibitors, severely diminished RSV internalization and infection but disruption of microtubules had no effect. In addition to increases in dextran bead uptake, these results strongly suggested that RSV enters cells by macropinocytosis. Not surprisingly, this conclusion was confirmed by inhibition of receptor tyrosine kinases (RTKs), which generally initiate activation of macropinocytosis events. Furthermore, the results showed that the only RTK responsible for RSV entry was epidermial growth factor receptor (EGFR) and inhibition of myosin II greatly decreases RSV internalization. Together these results show that macropinocytosis of RSV is dependent on EFGR activation and downstream effectors such as Cdc42 and myosin II. Following entry into host cells, viruses exploit host cell transport machinery to traffic to their replication sites, as reviewed elsewhere (38, 59, 63, 76). This aspect of RSV internalization is still under investigation, but initial studies indicate that intracellular trafficking of RSV is Rab5 dependent (49).
Protozoa are a third class of intracellular pathogens that invade host cells. Although protozoa are often overlooked as tools for studying host cellular physiology, it is becoming clear they are able to subvert host cellular signaling pathways during the course of infection to enter host epithelial cells, much like intracellular bacteria and viruses do. As a brief example, we consider the case of Trypanosoma cruzi trypomastigotes but acknowledge that much is known about how other parasites utilize host pathways to facilitate their entry into cells, such as Toxoplasma gondii, which is reviewed elsewhere (51, 72, 73).
Initial studies revealed that T. cruzi exploit a unique strategy to gain access to host cells that involves recruitment of lysosomes to the site of parasite internalization (84). Moreover, following exposure to T. cruzi, intracellular Ca2+ levels in host cells increases, and depleting these levels inhibits T. cruzi invasion (70, 83). These early studies revealed a novel mechanism for parasite invasion that required activating Ca2+-signaling pathways to induce lysosome exocytosis. This paradigm has led to the description, albeit requiring further investigation, of lysosome-dependent and lysosome-independent invasion pathways to explain T. cruzi invasion (10). In the lysosome-dependent pathway, T. cruzi engages host cells and phospholipase C is subsequently activated, which promotes production of diacylglycerol and inositol 1,4,5-tripohsphate. The initial interactions between T. cruzi and host cells seem to be a dynamic process that involves multiple T. cruzi surface molecules and/or secreted factors, depending on the lineage and system studied (11, 57, 101). Inositol 1,4,5-tripohsphate promotes release of Ca2+ from the endoplasmic reticulum, and the increase in intracellular Ca2+ promotes lysosome recruitment to the site of parasite internalization, as reviewed extensively elsewhere (57). Alternatively, parasites enter directly through the plasma membrane as a result of host phosphatidylinositol 3-kinase activation, which leads to accumulation of phosphoinositide-3,4,5-triphosphate at the site of invasion in the lysosome-independent pathway in nonphagocytic cells (10, 95, 97). The role of actin reorganization in T. cruzi invasion has been implicated to different degrees depending on various factors, including strain of T. cruzi and host cell type, as reviewed elsewhere (23, 30, 57). For example, invasion of macrophages seems to depend on actin polymerization, whereas invasion of nonphagocytic cells occurs as a result of Ca2+-dependent actin disruption. In addition, amastigote invasion is actin dependent, whereas trypomastigote invasion is not. It is clear that T. cruzi invasion of host epithelial cells is a complex process at the molecular level, and further study is needed to connect the various signaling events implicated in this process.
Overall, intracellular bacterial invasion requires subversion of signaling cascades that culminate in cytoskeletal rearrangements, which is not different from the strategies employed by viruses and protozoa to enter host epithelial cells. However, many extracellular bacteria also modify host signaling and trafficking pathways at the host-pathogen interface, without entering host epithelial cells. The mechanisms by which these bacteria modify host cellular physiology is primarily dependent on secreted toxins, which we will discuss in the next section.
Toxin Entry
During infection, many pathogenic bacteria, particularly Gram-negative bacteria, produce protein toxins that mediate the interaction of these pathogens with the host by actively remodeling host cells to establish a niche for bacterial survival and replication. In many cases, these bacterial toxins act in the intracellular compartment of host cells, which is not surprising considering the number of signaling pathways that can be targeted to induce changes in cell physiology.
Bacteria have evolved complex protein secretion systems, including the type III secretion system (T3SS) and type VI secretion system (T6SS), to deliver toxins from the cytoplasm of infecting bacteria directly to the host cytoplasm utilizing macromolecular syringe-like transport machines (reviewed in Refs. 18, 34, 74). For example, the T3SS is a complex composed of proteins responsible for transporting effector proteins across the multimembrane envelope of Gram-negative bacteria and translocating them into host cells. P. aeruginosa is an opportunistic pathogen that uses a T3SS to inject four effector proteins, including exoenzyme S (ExoS), into eukaryotic host cells to manipulate basic cellular processes. ExoS is an example of a bifunctional toxin that has both ADP ribosyl transferease (ADPRT) and GAP activity. The ADPRT activity of ExoS has several effects on host cells including targeting the actin cytoskeleton, RAS signal transduction, and endocytic pathways (2–4, 35). Similarly, the ExoS GAP activity disrupts the host cell actin cytoskeleton but targets the small GTPases Rho, Rac and Cdc42 (36, 66). The use of a secretion system is an efficient and regulated mechanism that ensures that the toxin can access the intracellular environment of host cells. However, this mechanism only allows for localized intoxication of host cells, which potentially limits the effects bacteria have on host epithelial cells and requires bacterial contact with host epithelial cells, unlike toxins secreted into the extracellular environment, like AB toxins, and toxins delivered by bacterial-derived outer membrane vesicles (OMV).
AB toxins are two-component bacterial toxins that are composed of an A subunit, which contains enzymatic activity, and a B subunit that is responsible for mediating toxin attachment to host cells by binding receptors on the plasma membrane. The B subunit is of paramount importance to these toxins because AB toxins are not injected into host cells but instead are secreted into the extracellular environment by bacteria. Thus these toxins must facilitate their own entry into host cells. Following attachment, AB toxins are internalized via endocytic mechanisms, and the B subunit facilitates translocation of the A subunit into the cytosol of host cells following proteolytic cleavage between the A and B subunits. Once in the cytosol, A subunits are free to target host proteins by one of several enzymatic mechanisms, including ADP-ribosylation. For example, we again consider P. aeruginosa, which produce exotoxin A (ETA). Full-length ETA is composed of three major domains: a cell binding domain (domain I), a translocation domain (domain II), and an enzymatic domain that contains ADP-ribosyltransferase activity (domain III) (reviewed elsewhere in Ref. 96). Briefly, ETA binds to the CD91 receptor following cleavage of a lysine at the COOH-terminal domain of ETA and is internalized into early endosomes via clathrin-coated pits. ETA undergoes a number of conformational rearrangements and cleavage events as it is transported from early endosomes to the endoplasmic reticulum, where domain II facilitates translocation of domain III into the cytosol of host cells. Once in the cytosol, the enzymatic subunit of ETA catalyzes ADP ribosylation of eukaryotic elongation factor-2 and inhibits protein synthesis in host cells, inducing cell death. Other examples of AB toxins include diphtheria toxin (DT), produced by Corynebacterium diphtheria, which also results in inhibition of protein synthesis (99), and pertussis toxin from Bordetella pertussis, which ADP ribosylates and inactivates Gαi, an inhibitor of adenylyl cyclase in host cells, to modulate cAMP signaling (48, 55), among other toxins reviewed elsewhere (28).
Of particular interest is the example of cholera toxin (CT) produced by Vibrio cholera. CT is another AB toxin that stimulates adenylyl cyclase activity. Unlike pertussis toxin, CT catalyzes ADP-ribosylation of Gαs, which inhibits the GTPase activity of Gαs, and thereby activates adenylyl cyclase and increases cAMP levels in host cells (17). Ultimately, overstimulation of adenylyl cyclase results in intestinal fluid and electrolyte secretion and diarrhea. Secretory diarrheas induced by Vibrio cholera have been attributed to CFTR-mediated Cl− secretion as experiments have shown that fluid accumulation in CFTR−/− mice administered CT is significantly decreased compared with CFTR competent animals (33, 86). As reviewed elsewhere, CFTR activity is regulated by numerous interactions among multiple proteins that form a microdomain wherein CFTR dimerizes and is activated (39). Protein kinase A (PKA)-mediated phosphorylation of the CFTR R domain appears to be central event that leads to CFTR-mediated Cl− secretion (39), although studies suggest that PKC-mediated phosphorylation of CFTR (14, 15, 69) and TC10-mediated trafficking of CFTR (16) are also important regulators of CFTR activation. Nevertheless, CT presumably promotes activation of PKA by increasing cAMP levels and induces phosphorylation of CFTR and Cl− secretion. Similarily, Escherichia coli heat-stable toxin activates CFTR-mediated Cl− secretion via PKA (13). Not surprisingly, lysophosphatidic acid, which signals through the LPA2 receptor to activate Gαi, reduces cAMP levels and inhibits CT-induced fluid secretion (54).
OMV are proteoliposomes that pathogenic and nonpathogenic species of Gram-negative bacteria ubiquitously secrete (50). Unlike the previously described mechanisms of toxin secretion, OMV are vehicles of intercellular transport that allow for secretion of large groups of proteins and lipids from bacteria, including transport of toxins into host cells (50). Many active toxins appear to be associated with OMVs including enterotoxigenic E. coli heat labile enterotoxin (LT) and Helicobacter pylori vacuolating toxin (VacA) (31, 45). Interestingly, P. aeruginosa also secretes virulence factors via OMVs, including alkaline phosphatase, hemolytic phospholipase C, and β-lactamase (6, 47). Recent findings have shown that P. aeruginosa secreted OMVs deliver these toxins from the bacteria directly into host cytoplasm in a mechanism that is dependent on fusion of vesicles with lipid rafts in the host plasma membrane (6).
Overall, there are multiple diverse pathways by which bacterial toxins can enter epithelial cells. Interestingly, bacteria utilize multiple secretion mechanisms to alter host cell physiology and are not necessarily limited to how they deliver protein toxins to host cells, as the example of P. aeruginosa demonstrates. In addition to perturbing host signaling and trafficking pathways to facilitate entry and changes in cellular physiology that benefit colonization, pathogens also alter these events at the epithelial barrier to evade immune responses that would otherwise recognize and clear infections.
Immune Evasion
Pathogens have developed a myriad of mechanisms to evade the host immune response to enable successful infection. Several microbes target the endocytic machinery to disrupt the ability of the host to either detect and/or respond to an infecting pathogen. Some examples of this are described below.
For the Gram-positive bacteria, Streptococcus pneuomoniae, anti-capsular antibodies play a critical role in immune control of this pathogen. Pneumococcus hijacks the secretory antibody pathway in the upper respiratory tract by using host endocytosis machinery to interfere with IgM and IgA secretion. The polymeric immunoglobulin receptor (PigR), expressed on the basolateral membrane of epithelial cells, binds to the J chain of IgM and dimeric IgA that have been secreted by B lymphocytes in the submucosa. The bound antibodies are then transcytosed in a vesicle through the cell and released at the apical membrane, where protease cleavage of the receptor at the membrane surface releases the antibody and keeps the secretory component of PigR attached. S. pneumoniae, on the apical side of the respiratory epithelium, expresses a cell surface protein termed PspC, which binds to PigR at the apical membrane. This binding of PspC to PigR facilitates reverse transcytosis of the pneumococcus bacteria from the apical lumen of the airways to the submucosa, where the bacteria can now disseminate. S. pneumoniae is adept at manipulating the host endocytic machinery in the airway epithelium to not only initiate its own transcytosis across the epithelial barrier but also by competitively binding the PigR with PspC to prevent antibody secretion into the apical compartment. This serves to prevent an important component of the host detection and clearance of the pathogen.
Another respiratory pathogen that targets the endocytic pathway to disrupt host defenses is the opportunistic pathogen, Pseudomonas aeruginosa. An essential component of the host innate immune response to respiratory pathogens is mucociliary clearance, where cilia on the respiratory epithelium beat in a coordinated manner to transport microbes and debris caught in a mucus layer lining the respiratory epithelium toward the pharynx to be swallowed and killed by the acidity of the stomach or expelled by coughing. In a recent study, we demonstrated that P. aeruginosa targets mucociliary clearance in the airway epithelium by secreting a virulence factor, termed Cif, which prevents the deubiquitination of the CFTR chloride channel, and thus promotes the lysosomal degradation of CFTR (7). Chloride secretion by CFTR is crucial in maintaining the airway surface liquid layer (ASL) and effective mucociliary clearance in the airways, so excessive degradation of CFTR will cause dehydration of the ASL and loss of mucociliary clearance. Butterworth et al. (12) reported another mechanism P. aeruginosa uses to target mucociliary clearance in the airway by targeting the activation status of the epithelial sodium channel (ENaC). These authors demonstrated that the P. aeruginosa secreted protease AprA cleaves and activates ENaC for sodium conductance into airway epithelial cells. Protease cleavage of ENaC increases its apical plasma membrane half-life, thereby providing a second mechanism by which P. aeruginosa can dehydrate the ASL and prevent effective mucociliary clearance.
Viral pathogens also commandeer the endocytic pathway to prevent their detection and immune clearance. Since viral detection and clearance are regulated in large part by major histocompatibility complex (MHC) antigen presentation and signaling pathways, its not surprising that numerous viruses target the endocytosis of MHC class I receptor in the plasma membrane to reduce the host immune response to virus infection. A detailed review describing the myriad viral proteins that target the MHC class I complex is reviewed comprehensively by Petersen et al. (67). To highlight a virus that encodes proteins that target the endocytic trafficking of MHC class I molecules, we will focus on the Kaposi's sarcoma-associated herpesvirus. This virus encodes two gene products, K3 and K5, which facilitate the endocytosis of MHC class I molecules and by altering interactions of MHC class I with TSG-101, promoting downregulation from the cell surface and routing class I molecules for lysosomal degradation (19, 80). This is accomplished by increasing the ubiquitination status of MHC class I via the ubiquitin ligase activity of K3 and K5 (56, 62).
While this is not an exhaustive list, we hope to emphasize the variety in mechanisms microbes employ to target the endocytic pathway and thus disrupt the host immune response to infection.
Exocytosis
Exocytosis is a cellular process that utilizes energy to secrete molecules from the intracellular environment into the extracellular milleu in a process that involves vesicle fusion with the plasma membrane. Exocytosis is a ubiquitious process in cells but can be either calcium dependent or independent and occur constitutively or in a regulated manner. In this review, we will focus on several functional outcomes of microbial regulation of exocytic events in an epithelial cell, namely microbial regulation of exocytosis to escape infected cells (Fig. 2) and for the acquisition of nutrients by extracellular microbes.
Fig. 2.

Microbial regulation of exocytosis. Microbial escape from host epithelial cells utilizes host exocytic pathways. Acidification of the phagocytic vacuole activates listeriolysin O (LLO) and facilitates Listeria monocytogenes entry into host cytosol. Following replication, L. monocytogenes escapes host cells by actin-based motility and spreads to neighboring cells in a nonlytic manner. Uropathogenic Escherichia coli (UPEC) invade host epithelial cells by zipper mechanism, and then reside in CD63/Rab27b/caveolin-1 positive fusiform vesicles. Activation of Toll-like receptor-4 (TLR4) by UPEC-derived LPS increases intracellular cAMP, and results in vesicle exocytosis and UPEC escape. Viral escape from host epithelial cells is dependent on the site of replication. Viruses that replicate in the nucleus are transported through the ER to the trans-Golgi network (TGN), where sorting and targeting to the apical or basolateral membranes occur.
Microbial Exit
In the previous section, we discussed L. monocytogenes as a model for zipper mechanism endocytosis. Following endocytosis, the phagocytic vesicle carrying L. monocytogenes travels down the endocytic pathway and acidifies as it does so. Acidification of the vacuole activates listeriolysin O (LLO), a pore-forming toxin produced by L. monocytogens that is optimally active at low pH and mediates escape of L. monocytogenes from the phagocytic vacuole into the cytosol (25). At this point, internalized bacteria either exit the vacuole or are killed upon fusion of the vacuole with lysosomes. Once in the cytosol, L. monocytogenes establishes a replication niche.
In order for infection to propagate to other cells, L. monocytogenes escapes from host cells utilizing actin-based motility, an organized, nonlytic mechanism that has been well characterized in this system as well as others including Shigella (37, 40). In L. monocytogenes, the bacterial surface protein ActA drives this process by promoting formation of actin comet tails via the recruitment and activation of the Arp2/3 complex (37, 94). Actin comet tails facilitate movement of bacteria and eventually facilitate exit and cell-to-cell spread of L. monocytogenes from host cells into adjacent cells. This escape process is initiated when L. monocytogenes reaches the plasma membrane and induces protrusions that extend into neighboring cells (88), which results in formation of two-membrane vacuoles in neighboring cells. The two-membrane vacuole is then lysed by the action of LLO and phospholipase C (PLC), which allows a new cycle of infection to begin (reviewed in Ref. 41).
As another example, we consider a well-studied escape mechanism described for uropathogenic E. coli (UPEC). UPEC is a Gram-negative bacterium that is responsible for the vast majority of urinary tract infections. These infections represent significant economic and medical burdens to the healthcare system. Classically, UPEC has been considered an extracellular pathogen. However, many studies over the last decade have challenged this notion, showing that UPEC can invade bladder epithelial cells via a zipper mechanism (61). FimH, the UPEC type I pilus adhesion, binds α3- and β1-integrins on epithelial cells (27). Focal adhesion kinase, phosphatidylinositol 3-kinase, and Rho GTPases Cdc42 and Rac1 then mediate actin rearrangements that culminate UPEC invasion (27, 60, 61, 100). FimH also binds UPIa, one of four uroplakins that form receptor-plaque complexes on the luminal membrane of umbrella cells (103). While studies are still underway to characterize the events leading to bacterial invasion via this pathway, initial results show engagement of UPIa is followed by phosphorylation of the cytoplasmic tail of UPIIIa and increased intracellular calcium levels (87, 93). Furthermore, caveolin-1 and lipid rafts appear necessary for UPEC invasion (26). Following invasion, UPEC reside in CD63/Rab27b/caveolin-1-positive secretory vesicles called fusiform vesicles, as shown by immunofluorescence studies (5). Interestingly, increasing intracellular cAMP concentrations within bladder epithelial cells, with the drug forskolin, induces exocytosis of these vesicles, and this result was confirmed with siRNA-mediated knockdown of adenylyl cyclase (5, 77). These results strongly suggest that UPEC expulsion from bladder epithelial cells is dependent on a cAMP signaling pathways, which were previously shown to be strongly induced upon recognition of UPEC-derived LPS by Toll-like receptor-4 (TLR4; Ref. 78). Subsequently, TLR4 recognition of LPS was shown to be necessary for exocytosis of UPEC-containing fusiform vesicles as UPEC strains with mutant LPS or bladder epithelial cells with knocked down TLR4 failed to clear UPEC infection as efficiently as when LPS was not mutated and TLR4 was fully functional (77). Furthermore, exocytosis of UPEC fusiform vesicles is dependent on Rab27b and caveolin-1, as siRNA-mediated knockdown of either significantly reduces bacterial expulsion (77). Taken together, these results represent an example of a tightly linked endocytic/exocytic mechanism that a bacterium is able to manipulate, which provides UPEC with a protective niche that contributes to pathogenesis (reviewed extensively in Refs. 24, 42).
Much like intracellular bacteria, viral infection and propagation require that fully assembled viruses exit host cells. This process is much less defined than the mechanisms described for viral attachment and entry into epithelial cells. However, an emerging body of literature is beginning to explain how viruses utilize host secretory pathways to facilitate their egress from host epithelial cells. Generally, nonenveloped viruses induce host cell lysis, which allows viral egress. However, this represents an overgeneralization to describe nonenveloped viral egress as some studies have provided some experimental support that poliovirus, a nonenveloped enterovirus, exits host cells from the apical membrane in a nonlytic manner (85, 89). The mechanism by which enveloped viruses exit host cells is dependent on the location of viral assembly. For enveloped viruses that assemble at the plasma membrane, host secretory pathways are only needed to transport necessary viral proteins, such as structural proteins, and viral genomes to the plasma membrane. Conversely, enveloped viruses that assemble at internal membranes, as opposed to the plasma membrane, rely on exocytic machinery to facilitate viral egress to the plasma membrane. For example, herpesviruses infect mucociliary epithelial cells, such as corneal cells, and assemble in the nucleus. Following assembly the virons undergo transport through the endoplasmic reticulum to the trans-Golgi network, where the viral envelope is acquired, and are ultimately sorted to cell-cell junctions or the basolateral membrane of cells, as reviewed elsewhere (46). In another example, the mechanism of RSV budding from polarized epithelial cells directs RSV to the apical membrane of cells in a Vps4-independent, Rab11-apical recycling endosome-dependent process (9, 90).
In comparison to viral entry, relatively few studies have addressed the mechanisms by which viruses bud from epithelial host cells or examined the role host cellular proteins play in this process. More examination is needed to characterize which host exocytic pathways are hijacked by viruses following assembly that lead to budding at epithelial cell surfaces, as this understanding this aspect of the viral life cycle will help design therapies that prevent cell-to-cell spread of viruses. Furthermore, very little is known about the dynamic interplay between viral proteins and host cellular proteins that directs viral egress. This will clearly be an intriguing field of study moving forward as many questions still remain unanswered.
Nutritional Acquisition
Bacterial pathogens must have mechanisms to facilitate their acquisition of nutrients, including transition metals, from the environments they occupy to successfully survive during the course of infection. For example, iron (Fe) has been shown to be important for colonization of many pathogens including P. aeruginosa (64, 75), Vibrio cholerae (98), and Staphylococcus aureus (58). However, host cells strongly oppose this process, restricting how accessible metals are to pathogens by utilizing a number of Fe-binding proteins to prevent infection, a process recently termed nutritional immunity. For example, at physiological pH, Fe is oxidized from Fe2+ to Fe3+, which binds transferrin with high affinity in the extracellular environment. As a consequence of this evolutionary pressure, bacteria have developed Fe acquisition mechanisms, which can be categorized as siderophore, haem, and free Fe2+ iron acquisition systems, to circumvent the attempt of the host to sequester iron (reviewed by Ref. 44). Briefly, bacterial uptake of siderophores and haem is dependent on ATP-binding cassette transporters and, in the case of Gram-negative bacteria, TonB-dependent receptors that shuttle siderophores into the periplasm. In addition, some Gram-negative bacteria express TonB-dependent receptors that directly bind host Fe3+-binding proteins and extract Fe3+ before transport across the outer membrane, or transport free Fe2+ across the cytoplasmic membrane. For more extensive details about the topic of nutritional immunity we refer readers to for a more detailed discussion of this topic (reviewed by Ref. 44), which also discusses bacterial acquisition of manganese, zinc and copper.
At the host-pathogen interface, it is vital that bacteria acquire nutrients from the surrounding environment to survive. Recent years have seen a significant increase in the body of literature describing the mechanisms of iron import, as outline above, and other nutrients. However, the field is still lacking in understanding of how microbes alter nutrient secretion from host cells. As an example of this expanding field of study, we will discuss a recent study that begins to describe how Helicobacter pylori, which colonizes at the apical membrane of gastrointestinal epithelial cells, perturbs iron trafficking of host epithelial cells (82). In this study, the authors used in vitro polarized epithelial cells to show that CagA and VacA deficient H. pylori are unable to colonize epithelial cells as efficiently as wild-type H. pylori but supplementing the apical compartment with Fe3+ rescues this phenotype; supplementing with halotransferrin was found to also rescue CagA-deficient H. pylori mutant (ΔCagA). During infection, transferrin receptor internalization at the basolateral membrane is dependent on CagA, as ΔCagA H. pylori are unable to induce internalization based on confocal microscopy studies. Furthermore, transferrin receptor and other basolateral proteins are mislocalized to the apical membrane at the site of H. pylori microcolonies. Together, these results strongly suggest that both CagA and VacA activity are essential for colonization of host polarized epithelial cells, and more interestingly, work in concert to induce changes in transferrin recycling that targets this source of iron to sites of bacterial infection, where it presumably promotes colonization. This conclusion was confirmed by experiments that showed biotinylated transferrin is transcytosed from the basolateral medium to the apical chamber of the author's epithelial cell model during H. pylori infection, but this effect is lost when infection occurs with ΔCagA H. pylori. Overall, this study demonstrates that iron acquisition and colonization of polarized epithelial cells by H. pylori is a dynamic process that involves both CagA and VacA toxins and, more universally, the ability of microbes to alter nutrient homeostasis pathways during infection.
Summary and Conclusions
Pathogens disrupt signaling and trafficking events at the epithelial barrier to change cellular physiology in a way that benefits colonization of and survival of the microbe. A key theme that has emerged in studies to date is that microorganisms have developed multiple mechanisms to disrupt normal cellular physiology. Although a simple concept in principle, understanding that pathogens have various strategies for disrupting normal cellular processes at epithelial barriers is beneficial on two fronts. First, this increases the number of tools available for studying questions about cell biology of epithelial cells. Second, this awareness will be crucial as studies continue to examine the host-pathogen interface with the intention of understanding how microbes cause disease and designing therapies to improve disease outcome. There is still much to be learned about how microbes perturb cellular trafficking events at the host-pathogen interface, but as this review suggests, microbes exert their own control at this interface.
GRANTS
This work was supported by National Institutes of Health Grants R00-HL-098342 and P30-DK-072506 (to J. M. Bomberger).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.R.H. and J.M.B. prepared figures; M.R.H. and J.M.B. drafted manuscript; M.R.H. and J.M.B. edited and revised manuscript; M.R.H. and J.M.B. approved final version of manuscript.
REFERENCES
- 1.Agbor TA, McCormick BA. Salmonella effectors: important players modulating host cell function during infection. Cell Microbiol 13: 1858–1869, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angus AA, Evans DJ, Barbieri JT, Fleiszig SM. The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun 78: 4500–4510, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbieri AM, Sha Q, Bette-Bobillo P, Stahl PD, Vidal M. ADP-ribosylation of Rab5 by ExoS of Pseudomonas aeruginosa affects endocytosis. Infect Immun 69: 5329–5334, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbieri JT, Sun J. Pseudomonas aeruginosa ExoS and ExoT. Rev Physiol Biochem Pharmacol 152: 79–92, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Bishop BL, Duncan MJ, Song J, Li G, Zaas D, Abraham SN. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med 13: 625–630, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton BA. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog 5: e1000382, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bomberger JM, Ye S, Maceachran DP, Koeppen K, Barnaby RL, O'Toole GA, Stanton BA. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog 7: e1001325, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonazzi M, Veiga E, Pizarro-Cerda J, Cossart P. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol 10: 2208–2222, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Brock SC, Goldenring JR, Crowe JE., Jr Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc Natl Acad Sci USA 100: 15143–15148, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burleigh BA. Host cell signaling and Trypanosoma cruzi invasion: do all roads lead to lysosomes? Sci STKE 2005: pe36, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Burleigh BA, Woolsey AM. Cell signalling and Trypanosoma cruzi invasion. Cell Microbiol 4: 701–711, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Butterworth MB, Zhang L, Heidrich EM, Myerburg MM, Thibodeau PH. Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J Biol Chem 287: 32556–32565, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao AC, de Sauvage FJ, Dong YJ, Wagner JA, Goeddel DV, Gardner P. Activation of intestinal CFTR Cl− channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J 13: 1065–1072, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chappe V, Hinkson DA, Howell LD, Evagelidis A, Liao J, Chang XB, Riordan JR, Hanrahan JW. Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 101: 390–395, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chappe V, Hinkson DA, Zhu T, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol 548: 39–52, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng J, Wang H, Guggino WB. Regulation of cystic fibrosis transmembrane regulator trafficking and protein expression by a Rho family small GTPase TC10. J Biol Chem 280: 3731–3739, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett 266: 129–137, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coburn B, Sekirov I, Finlay BB. Type III secretion systems and disease. Clin Microbiol Rev 20: 535–549, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coscoy L, Ganem D. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA 97: 8051–8056, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cossart P, Boquet P, Normark S, Rappuoli R. Cellular microbiology emerging. Science 271: 315–316, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Cossart P, Roy CR. Manipulation of host membrane machinery by bacterial pathogens. Curr Opin Cell Biol 22: 547–554, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124: 119–131, 2006 [DOI] [PubMed] [Google Scholar]
- 23.de Souza W, de Carvalho TM, Barrias ES. Review on Trypanosoma cruzi: host cell interaction. Int J Cell Biol 2010: pii: 295394, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhakal BK, Kulesus RR, Mulvey MA. Mechanisms and consequences of bladder cell invasion by uropathogenic Escherichia coli. Eur J Clin Invest 38, Suppl 2: 2–11, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Dramsi S, Cossart P. Listeriolysin O: a genuine cytolysin optimized for an intracellular parasite. J Cell Biol 156: 943–946, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duncan MJ, Li G, Shin JS, Carson JL, Abraham SN. Bacterial penetration of bladder epithelium through lipid rafts. J Biol Chem 279: 18944–18951, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Eto DS, Jones TA, Sundsbak JL, Mulvey MA. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog 3: e100, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falnes PO, Sandvig K. Penetration of protein toxins into cells. Curr Opin Cell Biol 12: 407–413, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Fasano A. Cellular microbiology: can we learn cell physiology from microorganisms? Am J Physiol Cell Physiol 276: C765–C776, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Fernandes MC, Andrews NW. Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol Rev 36: 734–747, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiocca R, Necchi V, Sommi P, Ricci V, Telford J, Cover TL, Solcia E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J Pathol 188: 220–226, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Fu Y, Galan JE. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401: 293–297, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266: 107–109, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type. III. Secretion machines. Nature 444: 567–573, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Ganesan AK, Vincent TS, Olson JC, Barbieri JT. Pseudomonas aeruginosa exoenzyme S disrupts Ras-mediated signal transduction by inhibiting guanine nucleotide exchange factor-catalyzed nucleotide exchange. J Biol Chem 274: 21823–21829, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Goehring UM, Schmidt G, Pederson KJ, Aktories K, Barbieri JT. The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J Biol Chem 274: 36369–36372, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Gouin E, Welch MD, Cossart P. Actin-based motility of intracellular pathogens. Curr Opin Microbiol 8: 35–45, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Gruenberg J. Viruses and endosome membrane dynamics. Curr Opin Cell Biol 21: 582–588, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7: 426–436, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Haglund CM, Welch MD. Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol 195: 7–17, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamon MA, Ribet D, Stavru F, Cossart P. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol 20: 360–368, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Hannan TJ, Totsika M, Mansfield KJ, Moore KH, Schembri MA, Hultgren SJ. Host-pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev 36: 616–648, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galan JES. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93: 815–826, 1998 [DOI] [PubMed] [Google Scholar]
- 44.Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10: 525–537, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horstman AL, Kuehn MJ. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J Biol Chem 275: 12489–12496, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9: 382–394, 2011 [DOI] [PubMed] [Google Scholar]
- 47.Kadurugamuwa JL, Beveridge TJ. Virulence factors are released from Pseudomonas aeruginosa in association with membrane vesicles during normal growth and exposure to gentamicin: a novel mechanism of enzyme secretion. J Bacteriol 177: 3998–4008, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaur T, Ganguly NK. Modulation of gut physiology through enteric toxins. Mol Cell Biochem 253: 15–19, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9: e1003309, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev 19: 2645–2655, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Laliberte J, Carruthers VB. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell Mol Life Sci 65: 1900–1915, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lalli G, Bohnert S, Deinhardt K, Verastegui C, Schiavo G. The journey of tetanus and botulinum neurotoxins in neurons. Trends Microbiol 11: 431–437, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170: 317–325, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, Naren AP. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med 202: 975–986, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Locht C, Coutte L, Mielcarek N. The ins and outs of pertussis toxin. FEBS J 278: 4668–4682, 2011 [DOI] [PubMed] [Google Scholar]
- 56.Lorenzo ME, Jung JU, Ploegh HL. Kaposi's sarcoma-associated herpesvirus K3 utilizes the ubiquitin-proteasome system in routing class major histocompatibility complexes to late endocytic compartments. J Virol 76: 5522–5531, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maeda FY, Cortez C, Yoshida N. Cell signaling during Trypanosoma cruzi invasion. Front Immunol 3: 361, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maresso AW, Schneewind O. Iron acquisition and transport in Staphylococcus aureus. Biometals 19: 193–203, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Marsh M, Helenius A. Virus entry: open sesame. Cell 124: 729–740, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez JJ, Hultgren SJ. Requirement of Rho-family GTPases in the invasion of Type 1-piliated uropathogenic Escherichia coli. Cell Microbiol 4: 19–28, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J 19: 2803–2812, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Means RE, Ishido S, Alvarez X, Jung JU. Multiple endocytic trafficking pathways of MHC class I molecules induced by a herpesvirus protein. EMBO J 21: 1638–1649, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem 79: 803–833, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Moreau-Marquis S, Bomberger JM, Anderson GG, Swiatecka-Urban A, Ye S, O'Toole GA, Stanton BA. The ΔF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. Am J Physiol Lung Cell Mol Physiol 295: L25–L37, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patel JC, Galan JE. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol 175: 453–463, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pederson KJ, Vallis AJ, Aktories K, Frank DW, Barbieri JT. The amino-terminal domain of Pseudomonas aeruginosa ExoS disrupts actin filaments via small-molecular-weight GTP-binding proteins. Mol Microbiol 32: 393–401, 1999 [DOI] [PubMed] [Google Scholar]
- 67.Petersen JL, Morris CR, Solheim JC. Virus evasion of MHC class I molecule presentation. J Immunol 171: 4473–4478, 2003 [DOI] [PubMed] [Google Scholar]
- 68.Pizarro-Cerda J, Kuhbacher A, Cossart P. Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb Perspect Med 2: 11, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raghuram V, Hormuth H, Foskett JK. A kinase-regulated mechanism controls CFTR channel gating by disrupting bivalent PDZ domain interactions. Proc Natl Acad Sci USA 100: 9620–9625, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodriguez A, Rioult MG, Ora A, Andrews NW. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J Cell Biol 129: 1263–1273, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma N, Low SH, Misra S, Pallavi B, Weimbs T. Apical targeting of syntaxin 3 is essential for epithelial cell polarity. J Cell Biol 173: 937–948, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sibley LD. Intracellular parasite invasion strategies. Science 304: 248–253, 2004 [DOI] [PubMed] [Google Scholar]
- 73.Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunol Rev 240: 72–91, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol 66: 453–472, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh PK, Parsek MR, Greenberg EP, Welsh MJ. A component of innate immunity prevents bacterial biofilm development. Nature 417: 552–555, 2002 [DOI] [PubMed] [Google Scholar]
- 76.Sodeik B. Mechanisms of viral transport in the cytoplasm. Trends Microbiol 8: 465–472, 2000 [DOI] [PubMed] [Google Scholar]
- 77.Song J, Bishop BL, Li G, Grady R, Stapleton A, Abraham SN. TLR4-mediated expulsion of bacteria from infected bladder epithelial cells. Proc Natl Acad Sci USA 106: 14966–14971, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song J, Duncan MJ, Li G, Chan C, Grady R, Stapleton A, Abraham SN. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Pathog 3: e60, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol 36: 1206–1221, 2000 [DOI] [PubMed] [Google Scholar]
- 80.Stevenson PG, Efstathiou S, Doherty PC, Lehner PJ. Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc Natl Acad Sci USA 97: 8455–8460, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Swiatecka-Urban A, Moreau-Marquis S, Maceachran DP, Connolly JP, Stanton CR, Su JR, Barnaby R, O'Toole GA, Stanton BA. Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. Am J Physiol Cell Physiol 290: C862–C872, 2006 [DOI] [PubMed] [Google Scholar]
- 82.Tan S, Noto JM, Romero-Gallo J, Peek RM, Jr, Amieva MR. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog 7: e1002050, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tardieux I, Nathanson MH, Andrews NW. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+ transients. J Exp Med 179: 1017–1022, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, Andrews NW. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71: 1117–1130, 1992 [DOI] [PubMed] [Google Scholar]
- 85.Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular release of poliovirus. J Virol 83: 6599–6609, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thiagarajah JR, Verkman AS. CFTR pharmacology and its role in intestinal fluid secretion. Curr Opin Pharmacol 3: 594–599, 2003 [DOI] [PubMed] [Google Scholar]
- 87.Thumbikat P, Berry RE, Zhou G, Billips BK, Yaggie RE, Zaichuk T, Sun TT, Schaeffer AJ, Klumpp DJ. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathog 5: e1000415, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109: 1597–1608, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tucker SP, Thornton CL, Wimmer E, Compans RW. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J Virol 67: 4274–4282, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE., Jr Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci USA 105: 10209–10214, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vanden Broeck D, Horvath C, De Wolf MJ. Vibrio cholerae: cholera toxin. Int J Biochem Cell Biol 39: 1771–1775, 2007 [DOI] [PubMed] [Google Scholar]
- 92.Veiga E, Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol 7: 894–900, 2005 [DOI] [PubMed] [Google Scholar]
- 93.Wang H, Min G, Glockshuber R, Sun TT, Kong Uropathogenic EXP. coli adhesin-induced host cell receptor conformational changes: implications in transmembrane signaling transduction. J Mol Biol 392: 352–361, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385: 265–269, 1997 [DOI] [PubMed] [Google Scholar]
- 95.Wilkowsky SE, Barbieri MA, Stahl P, Isola EL. Trypanosoma cruzi: phosphatidylinositol 3-kinase and protein kinase B activation is associated with parasite invasion. Exp Cell Res 264: 211–218, 2001 [DOI] [PubMed] [Google Scholar]
- 96.Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol 299: 161–176, 2009 [DOI] [PubMed] [Google Scholar]
- 97.Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci 116: 3611–3622, 2003 [DOI] [PubMed] [Google Scholar]
- 98.Wyckoff EE, Mey AR, Payne SM. Iron acquisition in Vibrio cholerae. Biometals 20: 405–416, 2007 [DOI] [PubMed] [Google Scholar]
- 99.Yates SP, Jorgensen R, Andersen GR, Merrill AR. Stealth and mimicry by deadly bacterial toxins. Trends Biochem Sci 31: 123–133, 2006 [DOI] [PubMed] [Google Scholar]
- 100.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol 65: 761–789, 2003 [DOI] [PubMed] [Google Scholar]
- 101.Yoshida N, Cortez M. Trypanosoma cruzi: parasite and host cell signaling during the invasion process. Subcell Biochem 47: 82–91, 2008 [DOI] [PubMed] [Google Scholar]
- 102.Zhou D, Chen LM, Hernandez L, Shears SB, Galan JE. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol 39: 248–259, 2001 [DOI] [PubMed] [Google Scholar]
- 103.Zhou G, Mo WJ, Sebbel P, Min G, Neubert TA, Glockshuber R, Wu XR, Sun TT, Kong XP. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J Cell Sci 114: 4095–4103, 2001 [DOI] [PubMed] [Google Scholar]