

Abstract
The mechanisms underlying the establishment, development, and maintenance of the renal vasculature are poorly understood. Here, we propose that the transcription factor “recombination signal binding protein for immunoglobulin kappa J region” (RBP-J) plays a key role in the differentiation of the mural cells of the kidney arteries and arterioles, as well as the mesangial cells of the glomerulus. Deletion of RBP-J in renal stromal cells of the forkhead box D1 (FOXD1) lineage, which differentiate into all the mural cells of the kidney arterioles along with mesangial cells and pericytes, resulted in significant kidney abnormalities and mortality by day 30 postpartum (P30). In newborn mutant animals, we observed a decrease in the total number of arteries and arterioles, along with thinner vessel walls, and depletion of renin cells. Glomeruli displayed striking abnormalities, including a failure of FOXD1-descendent cells to populate the glomerulus, an absence of mesangial cells, and in some cases complete loss of glomerular interior structure and the development of aneurysms. By P30, the kidney malformations were accentuated by extensive secondary fibrosis and glomerulosclerosis. We conclude that RBP-J is essential for proper formation and maintenance of the kidney vasculature and glomeruli.
Keywords: renal arterioles, mesangial cell, fibrosis, renin, vasculature, development
the kidney is a highly vascularized organ, receiving over 20% of the cardiac output in the adult human. However, the developmental cues and mechanisms necessary for proper formation of the complex renal arterial tree are poorly understood. Our lab and others previously showed that the progenitors for the kidney vasculature are present in the early embryonic kidney, well before vessels can be discerned (14, 28). Prior work using cell-specific lineage tracing demonstrated that within the embryonic kidney there is a population of stromal mesenchyme cells marked by expression of the transcription factor forkhead box D1 (FOXD1) (10, 13). Cells derived from the Foxd1-lineage differentiate to form all the cell types that comprise the mural cell layer of the kidney arteries and arterioles [vascular smooth muscle cells (VSMCs), renin cells, and perivascular fibroblasts], along with glomerular mesangial cells, and various cell types of the kidney interstitium including pericytes (27).
In many organisms, the Notch signaling pathway is involved in key cell fate and developmental decisions (1, 12). Interestingly, all elements of the Notch pathway share a common downstream transcription factor, “recombination signal binding protein for immunoglobulin kappa J region” (RBP-J) (15). Prior work in our lab determined that RBP-J, and likely Notch signaling, is essential for establishing and maintaining the identity of renin-expressing cells in adulthood (3). However, it is not currently known whether RBP-J plays a role in the differentiation of FOXD1+ progenitors into the downstream derivative cell types, which include renin cells along with the previously mentioned cell types. Using the cre-lox system, we examined the effects of conditional deletion of RBP-J in the FOXD1+ precursor cell population and found severe vascular defects in the mural cell layer of arteries and arterioles accompanied by a depletion of renin-expressing cells, and serious glomerular defects characterized by a lack of mesangial cells and development of glomerular aneurysms. Later in life, these developmental alterations were aggravated by striking reactive alterations including glomerulosclerosis, tubular dilations, and fibrosis, suggesting that developmental defects are further complicated by secondary injury and/or repair mechanisms.
MATERIALS AND METHODS
Mice.
To study the role of RBP-J in FOXD1-lineage cells, FOXD1cre/+; RBP-Jfl/+ animals were crossed to RBP-Jfl/fl animals to produce FOXD1cre/+; RBP-Jfl/fl animals (termed FOXD1RBPJ−/−), at an expected Mendelian ratio of 25%. Control animals either lacked cre-recombinase (FOXD1+/+; RBP-Jfl/fl) or had conditional deletion of only one RBP-J allele (FOXD1cre/+; RBP-J+/fl). For lineage study, we crossed in the Rosa26 (R26R) allele by breeding FOXD1cre/+; RBP-Jfl/+ animals with RBP-Jfl/fl; R26R+/+ to produce FOXD1cre/+; RBP-Jfl/fl; R26R+/− animals (termed FOXD1RBP-J−/−;R26R). RBP-Jfl mice were a kind gift of Dr. Tasuku Honjo, and FOXD1cre animals were generated by the McMahon laboratory (10). R26R animals were obtained from Jackson Laboratories. All procedures were performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Virginia Animal Care and Use Committee.
Deletion of RBP-J in FOXD1RBPJ−/− animals was confirmed using RT-PCR analysis of mRNA extracted from whole mouse kidney. Using primer pair GGCTAAAATGTTTGCCACCAGA (forward) and GACCAGTGGCTCTCAACTCC (reverse) generates a 329-bp product in animals with deletion of RBP-J which is absent in controls.
Histological analysis and immunostaining.
Bouins-fixed and paraffin-embedded kidneys were stained with hematoxylin and eosin, Sirius red, β-galactosidase (X-Gal) reaction, or immunostained with antibodies against renin (1:500 rabbit polyclonal) as previously described (2, 25, 26), α-smooth muscle actin (ACTA2; 1:10,000 mouse monoclonal, Sigma, St. Louis, MO), smooth muscle-myosin heavy chain (SM-MHC; 1:1,000 rabbit polyclonal, Sigma), podocalyxin (1:200 goat polyclonal, R&D Systems), Slit3 (1:100 rabbit polyclonal, Abcam), or phospho-histone H3 (Cell Signaling Technology, Danvers, MA), as described previously (26). Kidneys were viewed using a DM 5500 B microscope (Leica Camera, Solms, Germany) and pictures were taken using a DFC310 FX camera (Leica). Paired images are of equal magnification unless otherwise indicated.
Arteriole and glomerular quantitation.
Fields (1,400 × 1,200 μm) of comparable kidney sections were examined, and all blood vessels and glomeruli within the area were counted. Vessel thickness was calculated as the difference between inner and outer vessel diameter as marked by staining for appropriate VSMC markers. Glomerular diameter was calculated as the average of the glomerulus at its widest point and on an axis perpendicular to that point. For newborn kidneys all fields within the kidney were counted, whereas for adults at least five fields per kidney were randomly selected and examined. Glomeruli were scored as “normal” or “abnormal” based on general morphology and staining/lack of staining for appropriate markers, while glomeruli filled with at least 50% red blood cells were classified as “RC-filled.”
Statistical analysis.
Values are expressed as means ± SE. Statistical significance was calculated using Student's t-test and ANOVA. A P < 0.05 was considered significant.
RESULTS
To examine the effects of RBP-J deletion on FOXD1-lineage stromal cells and their descendents, we generated animals with conditional deletion of RBP-J in FOXD1-cells (FOXD1cre/+; RBP-Jfl/fl, hereby termed FOXD1RBPJ−/−). Confirmation of RBP-J deletion was conducted using RT-PCR on whole kidney isolated from mutant or control animals (not shown). FOXD1RBPJ−/− mice were born at expected Mendelian ratios (7 out of 24, 25% expected), and on the first day of life [postnatal day 0 (P0)] they were indistinguishable at a macroscopic level from control animals with the exception of a shorter tail length (Fig. 1A), a feature that was retained in older animals at 30 days of postnatal life (see Fig. 4A). Examination of kidneys harvested from FOXD1RBPJ−/− animals at day of birth however, revealed striking renal defects. Newborn FOXD1RBPJ−/− mice had significantly fewer SMC-coated renal arteries than controls, as shown by immunostaining for ACTA2 (Fig. 1B, arrows), a marker for SMCs. Arterioles appeared to have significantly thinner vessel walls, and the cells comprising the mural cell layer were thinner in appearance. Endothelial cells however, as marked by staining with platelet and endothelial cell adhesion molecule, showed no morphological alterations, and we did not find evidence for uncoated endothelial vessels lacking a SMC coating (not shown). Quantification of vessel numbers showed a nearly sixfold decrease in smooth muscle-coated arteries per area surveyed and a significant reduction in vessel wall thickness, with control animals having an average of 6.1 ± 0.4 (n = 5) vessels per area surveyed, while FOXD1RBPJ−/− kidneys had on average 1.1 ± 0.2 (n = 5) vessels per area surveyed (Fig. 1C; P < 0.05). In addition, the smooth muscle layer of mutant vessels was thinner, 7.4 ± 0.3 μm (n = 5) in mutants vs. 9.5 ± 0.5 μm (n = 5) in controls (P < 0.005; Fig. 1D). A distribution of the measurements of vessel diameter showed that mutant kidneys had comparable numbers of large-diameter vessels but significantly fewer small-diameter vessels (Fig. 1E), indicating that smaller-diameter vessels, mostly afferent arterioles, were the most affected by the mutation.
Fig. 1.

Kidneys from newborn FOXD1RBPJ−/− animals show pronounced vascular and glomerular defects. A: newborn [postnatal day 0 (P0)] FOXD1RBPJ−/− and control pups are indistinguishable other than by tail length (yellow boxes). WT, wild-type. B: staining for α-smooth muscle actin (ACTA2; brown) in kidney sections. Mutant animals have significantly fewer arteries (arrows), and comparable vessels are thinner in FOXD1RBPJ−/− animals than in controls. Mutant glomeruli (circles) are dilated and have distorted morphology, while some are aneurysmatic and lacking structural cells (solid circles, also in Fig. 1F). We also note that the nephrogenic zone (brackets) is wider in mutant animals. C: quantification of arteries shows significant decreases in vessel number per area surveyed in FOXD1RBPJ−/− animals (*P < 0.05). D: quantification of average vessel thickness shows that FOXD1RBPJ−/− animals have thinner vessels (**P < 0.005). E: in regards to the distribution of arterial diameters, newborn mutant animals show a deficit in the percentage of smaller-diameter vessels, but a higher proportion of larger-diameter vessels. F: immunostaining for renin (brown, arrowheads) in control animals can be found extending along afferent arterioles and adjacent to glomeruli. Meanwhile, FOXD1RBPJ−/− animals show a significant depletion of renin cells, with almost no renin-positive cells throughout the kidney. The solid circle denotes an aneurysmatic glomeruli(see Fig. 1B).
Fig. 4.

FOXD1RBPJ−/− adult animals have growth defects. A: by time of death, around day 30 postpartum (P30), FOXD1RBPJ−/− mice (n = 8) are half the size of their control littermates (n = 6; *P < 0.05). B: FOXD1RBPJ−/− kidneys are significantly smaller than controls (*P < 0.05).
Because our previous work indicated that RBP-J plays a key role in establishing and maintaining the identity of renin cells, and because renin cells are derived from FOXD1-expressing cells, we investigated the expression of renin in FOXD1RBPJ−/− animals by immunostaining. In wild-type animals at day of birth, renin cells can typically be found along afferent arterioles, in addition to larger vessels. However, in FOXD1RBPJ−/− animals, we found an almost complete absence of immunoreactive renin (Fig. 1F), with very few cells positively stained, and renin expression limited to a few juxtaglomerular regions rather than extending along the arterioles as is commonly observed in newborn mammals. Furthermore, we occasionally found that some kidney areas showed tubular dilations and disruption of tubular structure in mutant animals (not shown).
Further examination of newborn FOXD1RBPJ−/− kidneys also revealed that in mutant animals glomeruli displayed striking morphological abnormalities. Hematoxylin and eosin staining (Fig. 2A) showed that glomeruli from FOXD1RBPJ−/− animals (circles) contained an abnormally elevated number of red blood cells and that in some cases entire glomeruli were replaced by glomerular aneurysms filled with blood (Fig. 2A, also Fig. 1, B and F, solid circles). Quantification of glomerular alterations showed that more than half of the glomeruli in mutant kidneys appeared abnormal, with nearly a quarter having aneurysms (Fig. 2B). The likely cause of the aforementioned glomerular alterations was suggested by the unusual ACTA2 staining, which during development stains mesangial cells, but was diffuse or absent in the glomeruli of mutant kidneys (Fig. 1B). To confirm an alteration in the endowment of glomerular mesangium, we stained for platelet-derived growth factor receptor β (PDGFR-β), a marker for mesangial cells and a key protein in mesangial differentiation (30). In control kidneys, PDGFR-β-positive mesangial cells populate the interior of the glomerulus. FOXD1RBPJ−/− kidneys, however, displayed a significant reduction or complete absence of PDGFR-β staining within the glomerulus (Fig. 2C), indicating a marked decrease in mesangial cells. As expected and in contrast to the mesangial alterations, PDGFR-β-positive interstitial cells in between the tubules were unaffected and stained positive in control and mutant animals. Alterations to the glomerular mesangium resulted in changes to other cell types that populate the glomerulus. Staining for podocytes using a podocalyxin antibody (PODXL; Fig. 2D) demonstrated that podocytes remained in glomeruli (circles), although their localization and structure were severely affected, with the PODXL-positive cells often confined to a single thick, peripheral ring instead of being spread more uniformly throughout the glomerulus. Remarkably, many podocytes persisted even in cases of severe glomerular aneurysm (solid circle). Glomerular capillaries also showed significant alterations. Staining for Slit3, a marker for glomerular capillaries, shows that the endowment of glomerular capillaries is unaffected, as they can be found even in aneurysmatic glomeruli (Fig. 2E). However, in FOXD1RBPJ−/− animals, the typical arrangement of multiple capillary loops seen in wild-type animals is lost, with the glomerular capillaries collapsing into fewer loops or a single, giant loop.
Fig. 2.

Glomeruli of FOXD1RBPJ−/− kidneys lack mesangial cells and are aneurysmatic. A: high-power view of glomeruli (dashed circles) with hematoxylin and eosin staining in control animals shows well-formed glomeruli with few interior red blood cells (RBCs), while a large number of RBCs are found in the interior of glomeruli from FOXD1RBPJ−/− animals. Mutant glomeruli are at different stages of aneurysm formation and glomerular hemorrhage. A glomerulus completely devoid of mesangial cells and filled with RBCs is indicated by the solid circle. B: analysis of the prevalence of glomerular abnormalities shows that newborn FOXD1RBPJ−/− animals display alterations in almost two-thirds of their glomeruli and that an even greater proportion (75%) shows abnormalities by day 30 of life. We also examined the proportion of glomeruli that were devoid of structure and filled with RBCs. This subtype was completely absent in wild-type kidneys, but comprised 31% of newborn FOXD1RBPJ−/− glomeruli, decreasing slightly to 26% in adults. Right: example of a FOXD1RBPJ−/− kidney at P30 stained for ACTA2 with normal (dark blue), abnormal (light blue), and RBC-filled (red) glomeruli. C: staining for platelet-derived growth factor receptor β (PDGFR-β), a marker for glomerular mesangium (along with vascular smooth muscle cells and pericytes), is absent in the glomeruli in mutants, suggesting a lack of mesangial cells in FOXD1RBPJ−/− animals. D: staining for podocalyxin (PODXL) shows that podocytes persist in FOXD1RBPJ−/− glomeruli, even in aneurysmatic glomeruli, although their location is altered. E: staining for Slit3 labels glomerular endothelial cells. The glomerular capillaries in FOXD1RBPJ−/− kidneys have collapsed into a single capillary loop, in contrast to the multiple capillary loops seen in wild-type animals.
To examine the fate of FOXD1+ cells upon deletion of RBP-J and assess their contribution to the resultant disease processes seen in the vasculature and glomeruli, we traced the lineage of FOXD1+ cells by incorporating the R26R reporter system. We bred our FOXD1RBPJ animals to mice homozygous for the R26R allele, which expresses β-Gal upon cre-mediated recombination. In these animals, termed FOXD1RBPJ−/−;R26R, FOXD1-lineage cells can be visualized blue upon reaction with X-Gal. As previously reported, in newborn wild-type animals, FOXD1-lineage cells comprised the entirety of the mural cell layer of arteries and arterioles, along with undifferentiated kidney stroma, interstitial cells, and mesangial cells (Fig. 3). Whereas in control animals afferent and efferent FOXD1-derived arterioles can be clearly seen branching off larger vessels and entering glomeruli (Fig. 3, arrows), in mutant animals the arterioles do not branch adequately, are poorly formed, and are markedly more disorganized, making thin connections with nearby glomeruli. FOXD1RBPJ−/−;R26R mice at day of birth also showed an absence of blue FOXD1-lineage cells from the glomerulus (Fig. 3, circles). At this age, FOXD1-lineage mesangial cells can be typically found in the center of the glomerulus, and their absence indicates a failure of those cells to populate the glomerulus, likely leading to the glomerular aneurysms described above.
Fig. 3.

Lineage study of FOXD1 cells in newborn animals upon RBP-J deletion. Newborn kidneys were examined for expression of the β-galactosidase (β-Gal) reporter after reaction with X-Gal. FOXD1-lineage cells are labeled blue and can be found in control mice in undifferentiated stroma, interstitium, the mural cell layer of arteries (arrows), and mesangial cells. Labeled cells in mutants meanwhile show poor vascular wall integrity and branching. FOXD1RBPJ−/− animals also lose expression of the reporter within the interior of the glomerulus (circles), likely corresponding to a loss of mesangial cells.
Given the severe developmental alterations seen in the newborn kidney, we wished to examine the progression of kidney alterations in the maturing animal. We observed that a portion of mutant animals died some time after birth, as genotyping of weaned animals [21 days postpartum (P21)] showed that, in contrast to newborn animals, P21 animals no longer conformed to expected Mendelian ratios (10 out of 58, 25% expected, x2 = 2.68, P < 0.05). Subsequently, surviving FOXD1RBPJ−/− mice all died by 30 days postpartum (P30). By this age, FOXD1RBPJ−/− animals showed significant secondary defects, as they were on average approximately half the weight of control animals (Fig. 4A; P < 0.05). The kidneys of mutant animals were also smaller than those of controls, being on average half the weight of controls, and were paler in color (Fig. 4B; P < 0.05). However, the ratio of kidney weight to body weight did not differ between control and mutant animals, indicating that RBP-J deletion in the FOXD1-lineage had additional somatic effects (not shown).
Given the developmental defects seen and subsequent early death of adult FOXD1RBPJ−/− animals, we examined FOXD1RBPJ−/− kidneys perimortem at around P30, where we found an even more pronounced and complex phenotype compared with newborn animals. In mutant animals staining for ACTA2 (Fig. 5A, top) was no longer limited to vessels (arrows), but instead showed extensive expression across multiple regions of the kidney, particularly in interstitial areas surrounding vessels, and within and around glomeruli. Interestingly, there was expansion of ACTA2 staining in structures outside known FOXD1 descendents, including tubules and capsular cells, indicating a secondary phenotype in response to vascular damage. As ACTA2 is expressed not only in SMCs but also in cells undergoing injury and repair, we conducted staining for another marker of vascular smooth muscle, myosin heavy chain (SM-MHC, Fig. 5A, bottom), which showed prominent alterations in the perivascular compartment. As in the newborn FOXD1RBPJ−/− animals, at P30 arteries and arterioles were significantly reduced in number (Fig. 5B; P < 0.005). In contrast to newborn animals, however, at P30 there was significant thickening of vessel walls (Fig. 5A, arrows), in some occasions to the point of almost occluding the vessel (white arrow). Quantitation of vessel thickness in FOXD1RBPJ−/− animals again showed a deficiency of thin-diameter vessels but an enrichment of the thickest vessels (data not shown). Staining for renin also demonstrated that renin cells were nearly completely absent (data not shown).
Fig. 5.

Adult (P30) kidneys of FOXD1RBPJ−/− animals display active vascular remodeling and fibrosis. A: immunostaining for ACTA2 (top) and smooth muscle-myosin heavy chain (SM-MHC; bottom) in kidney sections, with blood vessels marked with arrows. In wild-type animals, ACTA2 and SM-MHC expression is limited to the vascular smooth muscle cells. Expression of ACTA2 expands significantly in mutant animals and can be seen in peritubular and perivascular areas, within tubules, and also in the interior and around glomeruli. SM-MHC in mutants meanwhile stains more specifically for vascular smooth muscle cells, but also shows expansion in perivascular areas and within glomeruli, occasionally to the point of almost occluding a vessel (white arrow). B: quantification of arterioles as assessed by SM-MHC staining shows significantly fewer vessels in FOXD1RBPJ−/− animals (**P < 0.005). C: Sirius red staining for collagen stains collagen fibers red under visible light (top), while when viewing under polarized light birefringence is an indication of thicker collagen fibers (bottom). In wild-type animals, only the areas comprising the vascular smooth muscle cell layer (arrows) stain red and are also birefringent (arrows). FOXD1RBPJ−/− animals show significant expansion of collagen in perivascular areas, with large regions of birefringence around the vessel. D: staining for phospho-histone H3, a marker for actively dividing cells, in FOXD1RBPJ−/− kidneys shows multiple proliferating cells (arrowheads) around vessel areas (arrows), while areas surrounding wild-type vessels are devoid of proliferating cells.
The expansion of SMC markers suggested an ongoing fibrotic/reparative process in the adult animals. To investigate this possibility, we performed Sirius red staining for collagen (Fig. 5C). In FOXD1RBPJ−/− kidneys, the areas surrounding vessels (arrow) showed staining for multiple layers of dense collagen fibers that were also birefringent (indicative of thicker collagen fibers), contrasting with control animals where staining is limited to the mural cell layer proper, with collagen fibers less frequently birefringent. Also, in support of an active fibrotic process, we found that areas surrounding the vessels showed marked increases in fibroblasts (data not shown) and ongoing cellular proliferation, with multiple cells in the perivascular area stained positive for phospho-histone H3 (Fig. 5D, arrows). As an indicator of the extent of kidney damage, we also noted signs of tubular atrophy as indicated by tubular dilation and protein casts (data not shown).
Lineage tracing of FOXD1 cells again using the FOXD1RBPJ−/−;R26R reporter mouse showed significant expansion of FOXD1-lineage cells in perivascular areas, especially around larger vessels, compared with controls (Fig. 6). These cells stain positive for ACTA2 (data not shown) and are likely indicative of vascular damage and fibrosis. We also found an expansion in the number of FOXD1-lineage interstitial cells in the areas between tubules, suggesting an expansion of the pericyte compartment (Fig. 6, asterisks), which was confirmed using quantification of cellular proliferation markers (not shown).
Fig. 6.
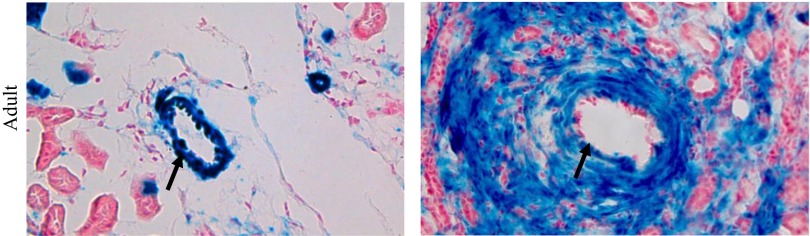
Lineage study of FOXD1 cells in adult animals upon rbp-j deletion. Examination of FOXD1-lineage cells (blue) in adult animals in perivascular areas indicates significant proliferation of FOXD1-lineage cells in areas surrounding blood vessels (arrows), in addition to an increase in interstitial cells.
Also at P30, glomeruli showed thickening characteristic of glomerulosclerosis, and stained positive for smooth muscle markers that are typically absent by this age in control animals (Fig. 5A). Interestingly, mutant animals displayed a decrease in glomerular number (Fig. 7A; P < 0.005), accompanied by significant increases of glomerular size in mutant animals compared with controls (P < 0.05); no such differences were found at newborn age (not shown). Glomerular morphology in FOXD1RBPJ−/− adult kidneys was distorted and sometimes aneurysmatic (Fig. 7B, also Fig. 5A), but at a higher occurency compared with newborn animals (Fig. 2B), all indicative of a continuous injurious process. As in the newborn animals, glomeruli in mutant kidneys at P30 showed a significant decrease in PDGFR-β staining, indicating a lack of mesangial cells (Fig. 7B).
Fig. 7.

FOXD1RBPJ−/− adult mice have larger and fewer glomeruli, and they lack mesangial cells. A: quantification of glomerular size and number indicates on average fewer (**P < 0.005) but larger (*P < 0.05) glomeruli per region in FOXD1RBPJ−/− adult animals, indicating an incomplete glomerular complement accompanied by glomerular hypertrophy. B: decrease in immunostaining for PDGFR-β in mutant glomeruli demonstrates that mesangial cells are absent in the glomeruli of FOXD1RBPJ−/− adults, as in newborn mutant animals.
DISCUSSION
This study demonstrates that RBP-J, the final transcriptional effector of Notch signaling, is crucial for kidney vascular development, and indispensable for establishing the identity of the cells of the renal arterial mural cell layer and the glomerular mesangium. Lack of RBP-J in the FOXD1-positive precursor cell population results in kidneys with a significant decrease in the number of vessels. Remaining vessels show striking abnormalities, including a thinner SMC layer, loss of renin cells, and eventually in the adult animal perivascular fibrosis and associated perivascular hypertrophy. In addition, we found that differentiation of mesangial cells within the glomerulus is also dependent on RBP-J. In summary, our results indicate that differentiation of stromal FOXD1+ progenitors is severely disrupted by RBP-J deletion, resulting in abnormal development of the kidney vasculature and glomeruli.
While the role of Notch signaling and RBP-J with regards to endothelial cell differentiation and proliferation has been suggested by several investigators (6, 8, 17), less understood is the role that canonical Notch signaling, and thus RBP-J, plays in the recruitment of the SMC coating as the endothelial layer is being established. Although it is known that downstream effectors of the Notch pathway directly bind to and regulate specific smooth muscle genes (21, 22), reports on the effects of Notch signaling on vascular smooth muscle have been contradictory. Studies variously suggest that Notch signaling can repress (21, 24, 33) or activate (9, 19), the SMC program. The discrepancies in the data are likely due to a context-dependent effect of Notch signaling and interaction with other signaling pathways such as SRF/Myocardin (7) or TGF-β (16). Our data strongly suggest a requirement for RBP-J for the proper development of the mural cell layer of the renal arteries. At day of birth, mice that lacked RBP-J in FOXD1-lineage cells displayed vessels that had a significantly thinner SMC layer, indicating that RBP-J is necessary for proper recruitment and/or differentiation of the mural cell layer during intrauterine life. It would be interesting to define how early in the embryo RBP-J effect(s) in the renal vasculature become established. Similarly, whether this effect proceeds through canonical Notch signaling, and if so by which receptors and ligands, remains to be determined.
Interestingly, although the role of Notch signaling in endothelial cells regarding vascular branching is well-established, we found that disruption of the Notch pathway in VSMCs through deletion of RBP-J also affects vascular patterning. Mutant mice lacking RBP-J in the vascular smooth muscle precursors exhibit a significant decrease in total vessel number (without a concomitant increase in uncoated vessels), as well as poor arteriolar branching. Also, in support of a possible role of RBP-J in SMCs in vascular patterning, we note that FOXD1RBPJ−/− animals displayed a reduced complement of smaller-diameter vessels, corresponding to arterioles. Although there is evidence that suggests the identity of endothelial cells and VSMCs is established independently, as seen in the persistence of normal endothelial cell marker expression in instances of VSMC perturbation (5) or the endothelial cell-autonomous role of S1P1 signaling (2), there is increasing evidence for several possible mechanisms for VSMCs to affect vascular patterning. For example, direct signaling from VSMCs through nitric oxide production may play a vasoprotective role for endothelial cells and promote their survival (4). In addition, SMCs have proven to be key mediators of vascular remodeling after injury, as the perturbation of key VSMC genes can alter the response of the vasculature to injury (25). Alternatively, the interaction of VSMCs with other cell types, such as monocytes (11), may induce the expression of key vascular growth factors. Our data support a model in which signaling between the two cell types and their respective vascular layers may be necessary for proper vessel patterning.
In addition to the developmental defects mentioned above, we observed significant ongoing perivascular remodeling as the animals aged. At 30 days of postnatal life, around time of death, we found a marked expansion of proliferating ACTA2-expressing cells surrounding defective arterioles, accompanied by increased interstitial collagen accumulation as indicated by Sirius red staining. The reason(s) for this additional reactive phenotype is not entirely clear. It is known that perivascular fibroblasts and pericytes descend from FOXD1-expressing progenitors. Thus, absence of RBP-J in either or both these cell types may have led to a change in cell fate and expansion of the myofibroblast population, thus explaining the subsequent ongoing fibrosis seen in our animals. Alternatively, it is possible that the perivascular fibrosis is due to renal hypoperfusion resulting from abnormal vascular architecture and a lack of renin cells, and is thus a secondary, reparative phenotype resulting from the primary defect, the deletion of RBP-J. We previously showed that mice with deletion of the renin gene or reduction in renal renin expression show a similar phenotype, with significant hypertrophy in areas surrounding renal vessels and regions of marked fibrosis, possibly due to tissue ischemia resulting from hypotension and renal hypoperfusion (23, 29, 32). It remains to be determined whether lack of RBP-J in the renal vascular and perivascular cells leads to a dysregulation of the pathways that control tissue oxygenation of the perivascular compartment.
The striking lack of renin cells in FOXD1RBPJ−/− animals also strongly supports our earlier studies demonstrating the integral role of RBP-J in the identity of the renin cells (2). However, we noted a more severe decrease in renin expression, as renin cells were nearly completely absent in FOXD1RBPJ−/− kidneys. Our data also showed an earlier depletion in renin cells, at day of birth in FOXD1RBPJ−/− animals, while deletion of RBP-J in renin cells did not cause an appreciable alteration in renin expression until adulthood (2). It is likely that deletion of RBP-J in the earlier progenitor cell, the FOXD1+ stromal cell, results in a more profound phenotype.
Deletion of RBP-J in FOXD1 cells also caused severe glomerular defects. Mutant glomeruli exhibited a range of phenotypes, either lacking mesangial cells, or lacking all of the interior cells including mesangium, podocytes, and the glomerular tuft. This phenotype mimics the mesangial defects found in mice lacking PDGF-β (18) or its receptor (31). It is unknown at this time, however, whether a lack of RBP-J causes a defect in the development of the entire glomerular tuft, or whether the primary defect is on mesangial cells, which are derived from the FOXD1-lineage stromal cells, and then the other cell types are subsequently lost due to lack of physical support and integrity. Interestingly, it has been shown that mice expressing a hypomorphic allele of Notch 2 exhibit improper differentiation and development of the glomerular capillaries and mesangial cells (20), which in combination with our data demonstrate an important role for Notch signaling in glomerular development. Later in life, glomeruli show signs of hypertrophy and glomerulosclerosis, as the kidney attempts to repair and compensate for the function lost due to the renal damage.
The cause(s) of the early mortality seen in FOXD1RBPJ−/− animals is currently unknown. The timing of the animals' death, corresponding with the maximum degree of observed kidney injury (glomerular aneurysms and sclerosis, vascular degeneration and scarring), implies that kidney failure plays a significant role. This conclusion is supported by limited urinalysis of FOXD1RBPJ−/− mice, which showed proteinuria and dilute urine (data not shown). However, Foxd1-lineage cells can be found in multiple other tissues, including the brain, retina, along with the lungs, and the SMCs lining the aorta (unpublished data from our lab). Although we do not observe abnormalities in those tissues, it is possible that more subtle alterations to those organs could over time result in early death.
In summary, RBP-J regulates the fate of stromal FOXD1+ vascular progenitors and glomerular mesangium. Figure 8 illustrates the consequences of RBP-J deletion on this early progenitor. At day of birth, kidneys from FOXD1RBPJ−/− animals have fewer and thinner vessels, lack mesangial and renin cells, and have abnormal glomeruli with aberrant structure and glomerular aneurysms. By day 30 of life, these animals display an additional, possibly secondary and reactive phenotype in the kidney including perivascular fibrosis, glomerulosclerosis, tubular degeneration, and early death.
Fig. 8.

Deletion of RBP-J in the FOXD1-positive stromal precursors results in severe vascular and glomerular abnormalities. This figure summarizes our findings on the effects on the kidney upon RBP-J deletion in FOXD1-lineage cells at newborn (P0) and adult (P30) ages. FOXD1RBPJ−/− newborns have fewer arteries and arterioles with poor branching, accompanied by a decrease in vessel wall thickness. These animals also lack renin cells and glomerular mesangial cells. In adulthood, FOXD1RBPJ−/− adults also exhibit mural cell proliferation and fibrosis, accompanied by glomerulosclerosis and tubular degeneration. WT, wild-type.
GRANTS
We acknowledge funding from National Institutes of Health Grants RO1 DK-091330 (to M. L. S. Sequeira-Lopez) and R37 HL066242 and RO1 HL096735, the Center of Excellence in Pediatric Nephrology P50 DK096373 (to R. A. Gomez), and the American Heart Association Predoctoral Fellowship 13PRE16920043 (to E. E. Lin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.E.L., M.L.S.S.-L., and R.A.G. conception and design of research; E.E.L. performed experiments; E.E.L., M.L.S.S.-L., and R.A.G. analyzed data; E.E.L., M.L.S.S.-L., and R.A.G. interpreted results of experiments; E.E.L. prepared figures; E.E.L. drafted manuscript; E.E.L., M.L.S.S.-L., and R.A.G. edited and revised manuscript; M.L.S.S.-L. and R.A.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Ellen S. Pentz for helping to revise the manuscript, PCR primer design, and for the excellent discussions and guidance. We also thank Takele Yazew, Kimberly Hilsen, and Cristina Monteagudo for excellent technical assistance.
REFERENCES
- 1.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science 268: 225–232, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Ben SA, Malkinson G, Krief S, Shwartz Y, Ely Y, Ferrara N, Yaniv K, Zelzer E. S1P1 inhibits sprouting angiogenesis during vascular development. Development 139: 3859–3869, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics 43: 1021–1028, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de FT, de Miguel LS, Garcia-Duran M, Gonzalez-Fernandez F, Rodriguez-Feo JA, Monton M, Guerra J, Farre J, Casado S, Lopez-Farre A. NO from smooth muscle cells decreases NOS expression in endothelial cells: role of TNF-α. Am J Physiol Heart Circ Physiol 277: H1317–H1325, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18: 2730–2735, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou GR, Wang YC, Hu XB, Hou LH, Wang CM, Xu JF, Wang YS, Liang YM, Yao LB, Yang AG, Han H. RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J 22: 1606–1617, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol 23: 2425–2437, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gridley T. Notch signaling in vascular development and physiology. Development 134: 2709–2718, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Grieskamp T, Rudat C, Ludtke TH, Norden J, Kispert A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ Res 108: 813–823, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev 10: 1467–1478, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Hojo Y, Ikeda U, Maeda Y, Takahashi M, Takizawa T, Okada M, Funayama H, Shimada K. Interaction between human monocytes and vascular smooth muscle cells induces vascular endothelial growth factor expression. Atherosclerosis 150: 63–70, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Honjo T. The shortest path from the surface to the nucleus: RBP-J kappa/Su(H) transcription factor. Genes Cells 1: 1–9, 1996 [DOI] [PubMed] [Google Scholar]
- 13.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyink DP, Abrahamson DR. Origin of the glomerular vasculature in the developing kidney. Semin Nephrol 15: 300–314, 1995 [PubMed] [Google Scholar]
- 15.Kato H, Sakai T, Tamura K, Minoguchi S, Shirayoshi Y, Hamada Y, Tsujimoto Y, Honjo T. Functional conservation of mouse Notch receptor family members. FEBS Lett 395: 221–224, 1996 [DOI] [PubMed] [Google Scholar]
- 16.Kennard S, Liu H, Lilly B. Transforming growth factor-β (TGF-1) downregulates Notch3 in fibroblasts to promote smooth muscle gene expression. J Biol Chem 283: 1324–1333, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14: 1343–1352, 2000 [PMC free article] [PubMed] [Google Scholar]
- 18.Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev 8: 1875–1887, 1994 [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B. Notch3 is critical for proper angiogenesis and mural cell investment. Circ Res 107: 860–870, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 128: 491–502, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Morrow D, Scheller A, Birney YA, Sweeney C, Guha S, Cummins PM, Murphy R, Walls D, Redmond EM, Cahill PA. Notch-mediated CBF-1/RBP-Jκ-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am J Physiol Cell Physiol 289: C1188–C1196, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth muscle alpha-actin is a direct target of Notch/CSL. Circ Res 98: 1468–1470, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA. Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol 286: R474–R483, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem 280: 8994–9004, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest 106: 1139–1147, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sequeira Lopez ML, Chernavvsky DR, Nomasa T, Wall L, Yanagisawa M, Gomez RA. The embryo makes red blood cell progenitors in every tissue simultaneously with blood vessel morphogenesis. Am J Physiol Regul Integr Comp Physiol 284: R1126–R1137, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Sequeira Lopez ML, Gomez RA. Development of the renal arterioles. J Am Soc Nephrol 22: 2156–2165, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol 281: F345–F356, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Sequeira-Lopez ML, Weatherford ET, Borges GR, Monteagudo MC, Pentz ES, Harfe BD, Carretero O, Sigmund CD, Gomez RA. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol 21: 460–467, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shultz PJ, DiCorleto PE, Silver BJ, Abboud HE. Mesangial cells express PDGF mRNAs and proliferate in response to PDGF. Am J Physiol Renal Fluid Electrolyte Physiol 255: F674–F684, 1988 [DOI] [PubMed] [Google Scholar]
- 31.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev 8: 1888–1896, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Takahashi N, Lopez ML, Cowhig JE, Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O. Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16: 125–132, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res 102: 661–668, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]