

Abstract
Our previous work has shown that gene knockout of the sodium-glucose cotransporter SGLT2 modestly lowered blood glucose in streptozotocin-diabetic mice (BG; from 470 to 300 mg/dl) and prevented glomerular hyperfiltration but did not attenuate albuminuria or renal growth and inflammation. Here we determined effects of the SGLT2 inhibitor empagliflozin (300 mg/kg of diet for 15 wk; corresponding to 60–80 mg·kg−1·day−1) in type 1 diabetic Akita mice that, opposite to streptozotocin-diabetes, upregulate renal SGLT2 expression. Akita diabetes, empagliflozin, and Akita + empagliflozin similarly increased renal membrane SGLT2 expression (by 38–56%) and reduced the expression of SGLT1 (by 33–37%) vs. vehicle-treated wild-type controls (WT). The diabetes-induced changes in SGLT2/SGLT1 protein expression are expected to enhance the BG-lowering potential of SGLT2 inhibition, and empagliflozin strongly lowered BG in Akita (means of 187–237 vs. 517–535 mg/dl in vehicle group; 100–140 mg/dl in WT). Empagliflozin modestly reduced GFR in WT (250 vs. 306 μl/min) and completely prevented the diabetes-induced increase in glomerular filtration rate (GFR) (255 vs. 397 μl/min). Empagliflozin attenuated increases in kidney weight and urinary albumin/creatinine ratio in Akita in proportion to hyperglycemia. Empagliflozin did not increase urinary glucose/creatinine ratios in Akita, indicating the reduction in filtered glucose balanced the inhibition of glucose reabsorption. Empagliflozin attenuated/prevented the increase in systolic blood pressure, glomerular size, and molecular markers of kidney growth, inflammation, and gluconeogenesis in Akita. We propose that SGLT2 inhibition can lower GFR independent of reducing BG (consistent with the tubular hypothesis of diabetic glomerular hyperfiltration), while attenuation of albuminuria, kidney growth, and inflammation in the early diabetic kidney may mostly be secondary to lower BG.
Keywords: diabetes, diabetic nephropathy, proximal tubule, renal growth, inflammation, gluconeogenesis, phosphoenolpyruvate carboxykinase
diabetes-associated kidney disease is the commonest cause of end-stage renal disease in most countries of the world. The pathophysiology, however, is still incompletely understood. Control of blood glucose levels is important in diabetic patients but often associated with hypoglycemia and weight gain. Pharmacological inhibition of the renal sodium-glucose cotransporter SGLT2 (SLC5A2) is a new approach that inhibits the renal reabsorption of filtered glucose, thereby lowering blood glucose levels without increasing body weight (6, 17, 40).
The concept to inhibit SGLT2 in diabetes is based on the fact that SGLT2, which is localized in the early proximal tubule, mediates the bulk of tubular glucose uptake across the apical membrane of the kidney (27, 36, 43). In comparison, SGLT1 is thought to have a lower capacity for glucose reabsorption, which, in euglycemia, “cleans up” most of the remaining luminal glucose in further distal parts of the proximal tubule (1, 11, 27, 43). Besides their role in glucose reabsorption, SGLTs contribute to the increase in Na+ and fluid reabsorption in the proximal tubule of the early diabetic kidney (37). Increased proximal reabsorption contributes to diabetic glomerular hyperfiltration by lowering the Na-Cl-K concentrations at the macula densa and increasing glomerular filtration rate (GFR) through the physiology of tubuloglomerular feedback (32, 37, 39). Direct luminal application of the SGLT-inhibitor, phlorizin, into the free-flowing early proximal tubule, just downstream of Bowman space, acutely normalized the Na-Cl-K concentrations at the macula densa as well as single-nephron GFR in streptozotocin (STZ)-diabetic rats (37). These studies demonstrated the contribution of SGLTs to the diabetic hyperreabsorption and that inhibition of SGLTs can lower glomerular hyperfiltration without altering blood glucose levels. Selective pharmacological inhibition of SGLT2 with dapagliflozin has recently been proposed to lower GFR in STZ-diabetic rats through the same macula densa mechanism (33), and gene knockout of Sglt2 prevented glomerular hyperfiltration in STZ-diabetic mice (38). Both studies provided additional evidence that this response did not require effects on blood glucose levels. Similarly, studies in patients with type 1 diabetes showed that the SGLT2 inhibitor, empagliflozin, lowers GFR independent of effects on blood glucose levels (4).
Besides its role in GFR control, renal glucose uptake via SGLT2 may contribute to kidney growth in the early diabetic kidney. The latter involves diabetes-induced growth of the proximal tubule (5, 23, 30), and diabetic kidney growth has been linked to the development of nephropathy (2, 3, 13, 26, 35, 44). Using the model of low-dose STZ-induced type 1 diabetes in mice lacking Sglt2, we recently concluded that glucose uptake via SGLT2 is not a critical stimulus for diabetic kidney growth or the development of kidney inflammation and injury (38). The study further indicated that, in steady state, urinary glucose excretion was not affected by the absence of Sglt2 in STZ-diabetic mice, i.e., the lack of early proximal glucose reabsorption via SGLT2 was balanced by the resulting lowering of glomerular glucose filtration (lower blood glucose × lower GFR). As a consequence, STZ-induced glucose delivery downstream of the SGLT2-expressing early proximal tubule was likely not different between WT and Sglt2−/− mice. If luminal glucose delivery downstream of the early proximal tubule is an important stimulus for kidney growth, inflammation, and injury in diabetes, then this may explain why these changes were independent of SGLT2. Alternatively, growth and other tubulointerstitial changes may be triggered by sensing of hyperglycemia via the basolateral tubular membrane and/or glomerular filtration of specific factors, and the absence of SGLT2, which lowered blood glucose in this model from ∼470 mg/dl in wild-type mice to ∼300 mg/dl in Sglt2−/− mice, just did not lower blood glucose enough to limit kidney growth and inflammation (38).
The previous experiments, testing for a role of SGLT2 in STZ-diabetic mice, were also confounded by a 60% decline in renal SGLT2 expression following STZ administration in wild-type mice, which may have masked some effects of knocking out the Sglt2 gene (38). This problem of reduced SGLT2 expression probably owes to a direct effect of STZ on the mouse proximal tubule, since SGLT2 expression is actually increased in both the db/db model of type 2 diabetes and the Akita model of type 1 diabetes (38). Hence, to more fully assess the potential of SGLT2 inhibition in the early diabetic kidney, we tested in diabetic Akita mice the effect of the selective SGLT2 inhibitor, empagliflozin (12), on blood glucose control, GFR, kidney growth, and markers of renal inflammation.
METHODS
Animals.
All animal experimentation was conducted in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD) and was approved by the local Institutional Animal Care and Use Committee. Akita mice (Ins2+/C96Y; Akita/+) [and littermate Ins2+/+ controls (WT)] were used as a nonobese insulin-dependent model of spontaneous type 1 diabetes (The Jackson Laboratories, Bar Harbor, ME). All studies were performed in male mice, which were housed in the same animal room with a 12:12 h light-dark cycle and free access to standard rodent chow and tap water. At 4 wk of age, mice received a diet containing empagliflozin [300 mg/kg of diet; in regular rodent diet (in %: 19 protein, 3.3 fat, 4.9 fiber, 6.4 ash, 36.5 starch, 4.7 sugar, 12.8 MJ ME/kg); synthesized at ssniff Spezialdiäten, Soest, Germany] or repelleted control diet (vehicle) for 15 wk (n = 10–13/group). Empagliflozin was provided by Boehringer Ingelheim Pharma, Biberach, Germany.
Food and fluid intake, and blood and urine collection.
Food and fluid intake was determined while the mice were maintained in their regular cages. Urine was obtained by picking up the mice to elicit reflex urination and holding them over a clean Petri dish for sample collection. For paired glucose measurements, blood was collected by tail snip immediately after urine collection in awake mice. For other blood/plasma analyses, blood was collected from the retrobulbar plexus under brief isoflurane anesthesia.
Systolic blood pressure and heart rate in awake mice.
Systolic blood pressure and heart rate were determined using an automatic tail-cuff system (Visitech-Systems, Apex, NC) after appropriate training as described (36).
Measurement of GFR in awake mice.
GFR measurements were performed in conscious mice (at 14 wk after starting treatment with empagliflozin or vehicle) using the plasma kinetics of FITC-Sinistrin (Fresenius-Kabi, Linz, Austria) (19) following a single-dose intravenous injection of this GFR marker as described (24, 38, 39). Briefly, FITC-Sinistrin (2% in 0.85% NaCl, which also served to establish the standard curve) was injected into the retro-orbital plexus (2 μl/g body wt) during brief isoflurane anesthesia. At 3, 5, 7, 10, 15, 35, 56, and 75 min after injection, blood was collected from the end of the tail into a Na+-heparinized 10 μl microcap (Hirschmann Laborgeräte, Eberstadt, Germany). After centrifugation plasma was diluted 1:10 in 0.5 mol/l HEPES (pH 7.4) and fluorescence determined using a Nanodrop ND-3300 fluorospectrometer (Nanodrop Technologies, Wilmington, DE) by pipetting 2 μl of samples onto the pedestal. GFR was calculated using a two-compartment model of two-phase exponential decay (GraphPad Prism, San Diego, CA).
Blood and urine analysis.
Blood glucose was determined using the Ascensia Elite XL glucometer (Bayer, Mishawaka, IN). Urine glucose was determined by the hexokinase/glucose-6-phosphate dehydrogenase method (Infinity, Thermo Electron, Louisville, CO). Concentrations of plasma aldosterone (Beckman Coulter, Brea, CA) and urinary albumin (Exocell, Philadelphia, PA) and creatinine (Thermo Fisher Scientific, Waltham, MA) were measured using commercial assays.
Glomerular size.
Kidneys were harvested under terminal isoflurane anesthesia and fixed in 4% paraformaldehyde (PFA), embedded in O.C.T. compound (Sakura Finetek, Torrance, CA), sliced at a thickness of 10 μm using a cryostat microtome, and placed on Superfrost Plus microscope slides (Thermo Fisher Scientific, San Diego, CA). After being air-dried, sections were blocked in 5% dry milk (in PBST), and incubated with Alexa Fluor 555 Phalloidin (Invitrogen, Carlsbad, CA) for 30 min at room temperature. Sections were washed 3 times for 5 min in PBST and mounted with Prolong (Invitrogen) and imaged on an Olympus DSU confocal microscope. The area (in pixel units) of 20 random glomeruli was measured per mouse kidney using Adobe Photoshop 7.0 software.
Western blot analysis.
Whole kidneys were harvested under terminal isoflurane anesthesia and prepared for Western blot analysis of SGLT2 and SGLT1 in the membrane fraction as previously described (36). For the former and the analysis of other proteins [cyclin-dependent kinase inhibitors p27 and p21, and heme oxygenase 1 (HO-1)], kidneys were harvested and homogenized in a buffer containing 250 mM sucrose, 10 mM triethanolamine, and protease inhibitors (Complete protease cocktail, Roche Molecular Biochemicals, Mannheim, Germany) with a tissue homogenizer (Tissumizer; Tekmar, Cincinnati, OH). Cell fractionation was achieved by differential centrifugation at 4,000 g for 10 min (nuclear pellet for p27 and p21) followed by 16,000 g for 1 h (membrane pellet for SGLT2/SGLT1 and cytosolic supernatant for HO-1). Protein concentration was determined with a DC Protein Assay (Bio-Rad). Lysates at 40 μg/lane were resolved on NuPAGE gels in MOPS buffer (Invitrogen, Carlsbad, CA). Gel proteins were transferred to nitrocellulose membranes and immunoblotted with the appropriate primary antibody. The secondary antibody was horseradish peroxidase-conjugated for autoradiographic detection by ECL Plus (Amersham Pharmacia, Piscataway, NJ). Antibodies for p27 and HO-1 were purchased from Santa Cruz Biotechnology (Dallas, TX). The antibody for p21 was acquired from Abcam (Cambridge, MA). To verify equal protein loading, membranes were stripped (0.2 M NaOH for 5 min) and reprobed with monoclonal anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO) for cytosolic and membrane proteins and with anti-lamin-B antibody (Santa Cruz) for nuclear proteins. Densitometric analysis was performed by ImageJ Software (National Institutes of Health, Bethesda, MD). For the indirect assessment of renal collagen content, a modification of a previously described method was used involving precipitation of collagen with picro-Sirius red (34, 38). Briefly, 100 μl of cytosolic fraction obtained from kidney homogenization was added to 1 ml of picro-Sirius red solution (NovaUltra Sirius Red Stain Kit, IHC World, Woodstock, MD) and agitated for 45 min followed by centrifugation at 10,000 g for 10 min. The Sirius red dye was released from the pellet with alkali reagent (1 N NaOH) and spectrophotometric readings were taken at 540 nm on a microplate reader (Molecular Devices, Sunnyvale, CA). Results were expressed relative to WT controls.
Reverse transcription and real-time PCR.
Whole kidney RNA was prepared with the RNeasy Plus Mini Kit and cDNA was prepared with the Superscript II First Strand Synthesis System. For quantification, specific primers were used with Power SYBR Green PCR Master Mix (10 min at 95°C with 50 cycles of 15 s at 95°C and 1 min at 60°C) in an AB7300 Real Time PCR System (Applied Biosystems, Foster City, CA). For some genes, we used Taqman PCR Universal Mastermix and primers to improve specificity and sensitivity of these reactions (Applied Biosystems) (see Table 1 for primer details). Relative fold increases were calculated using the Pfaffl technique of relative quantification, which accounts for real-time efficiencies (18). Each experiment was performed in triplicate.
Table 1.
Real-time PCR primers used
| Target | Forward | Reverse |
|---|---|---|
| SGLT1 | Mm00451203_m1 (Applied Biosystems) | |
| SGLT2 | Mm00453831_m1 (Applied Biosystems) | |
| GLUT1 | AACATGGAACCACCGCTACG | GTGGTGAGTGTGGTGGATGG |
| GLUT2 | ATCGCCCTCTGCTTCCAGTAC | GAACACGTAAGGCCCAAGGA |
| PEPCK | Mm01247058_m1 (Applied Biosystems) | |
| NFkB | Mm00476361_m1 (Applied Biosystems) | |
| CCL2 | Mm00441242-m1 (Applied Biosystems) | |
| CCL5 | Mm01302428-m1 (Applied Biosystems) | |
| CD14 | Mm00438094_g1 (Applied Biosystems) | |
| TIMP2 | Mm00441825_m1 (Applied Biosystems) | |
| IL6 | Mm00446190-m1 (Applied Biosystems) | |
| TGF-β | Mm01178820-m1 (Applied Biosystems) | |
| NOX4 | Mm00479246_m1 (Applied Biosystems) | |
| NOX2 | Mm01287743_m1 (Applied Biosystems) |
Determination of adipocyte size.
Epididymal white adipose tissue (WAT) was excised during terminal anesthesia, immediately fixed with PFA for 24 h, and embedded in paraffin. Paraffin embedded fat tissue was cut into 5-μm sections and stained with PAS. Pictures were taken on an Olympus IX81 microscope and adipocyte cross-sectional area was determined using ImageJ software. At least 300 adipocytes were analyzed per mouse.
Screening for urinary tract infection.
Twenty to fifty microliters bladder urine (collected at the time animal was euthanized) was streaked onto cystine-lactose-electrolyte-deficient medium plates (Thermo Scientific Remel, Lenexa, KS) which were incubated overnight at 37°C. The number of colony-forming units (CFUs) per milliliter of urine was determined. A density of >105 CFU/ml was considered to indicate ascending urinary tract infection (as recommended by manufacturer).
Statistical analysis.
Data are shown as means ± SE. ANOVA and unpaired Student's t-test were performed to analyze for statistical differences between groups. Regression analysis was performed to test for a linear relationship between blood glucose concentrations and various parameters. P < 0.05 was considered statistically significant.
RESULTS
Empagliflozin treatment of nondiabetic mice and Akita diabetes increased renal membrane expression of SGLT2 and reduced SGLT1 expression.
Renal membrane SGLT2 expression was increased in Akita/+ vs. WT mice by 38%, associated with a trend for higher SGLT2 mRNA expression (P = 0.087) (Fig. 1, A–C). Empagliflozin enhanced the renal protein expression of SGLT2 in WT mice by 47% without upregulation of mRNA (Fig. 1, A–C). Renal SGLT2 expression in empagliflozin-treated Akita/+ was similar to vehicle-treated Akita/+ or empagliflozin-treated WT.
Fig. 1.
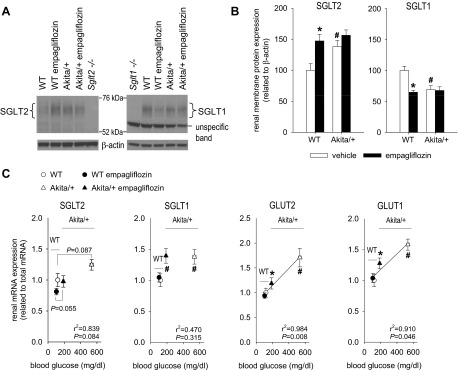
Empagliflozin treatment of nondiabetic mice and Akita-diabetes increased renal membrane protein expression of SGLT2 and reduced SGLT1 protein expression. Empagliflozin attenuated the diabetes-induced increase in renal mRNA expression of GLUT1 and GLUT2 but not SGLT1. Divergent effects on mRNA and membrane protein expression of SGLT2 and SGLT1 indicate complex translational and posttranslational regulation. Empagliflozin (300 mg/kg of diet) or repelleted diet (vehicle) were given to Akita/+ and wild-type (WT) mice. A and B: renal membrane SGLT2 and SGLT1 protein expression. Kidneys from knockout mice served as negative controls (Sglt2−/−, Sglt1−/−). C: renal mRNA expression of glucose transporters. For Sglt2 and Sglt1 mRNA analysis, kidneys from knockout mice confirmed specificity of primers (data not shown). *P < 0.05 vs. vehicle treatment in same genotype; #P < 0.05 vs. WT; ANOVA and unpaired Student's t-test and linear regression analysis. Linear regression lines were included when statistical significance was achieved; n = 10–13 per group.
Renal membrane protein expression of SGLT1 provided a mirror image of the changes in SGLT2. Akita diabetes, empagliflozin, and their combination reduced the renal membrane protein expression of SGLT1 by 33–37% (Fig. 1, A and B). In comparison, renal mRNA expression of SGLT1 was increased in Akita/+ compared with WT and not affected by empagliflozin treatment (Fig. 1C), indicating divergent effects of Akita diabetes on renal SGLT1 mRNA and membrane protein expression. The specificity of the analyses for SGLT2 and SGLT1 protein and mRNA was confirmed by the use of kidneys from gene knockout animals.
In WT mice, empagliflozin did not affect renal mRNA expression of GLUT1 or GLUT2 (Fig. 1C). Akita/+ increased renal mRNA expression of both transporters, and these responses were attenuated by empagliflozin.
Effects of empagliflozin on glucose excretion, plasma glucose, food intake, and body weight.
In WT mice, empagliflozin induced sustained increases in urinary glucose/creatinine ratios (Fig. 2B) and absolute urinary glucose concentrations (470 ± 34 vs. 4 ± 1 mM, P < 0.05), and modestly lowered blood glucose levels (Fig. 2A). The increased loss of glucose calories into the urine of empagliflozin-treated WT mice was partially offset by increased food (and fluid) intake (Fig. 2, D and E), consistent with previous reports in nondiabetic mice lacking Sglt2 (36). The compensatory increase in food intake is deemed to be “partial” because the body weight of the glucosuric WT mice was lower than in their nonglucosuric counterparts (Fig. 2F).
Fig. 2.
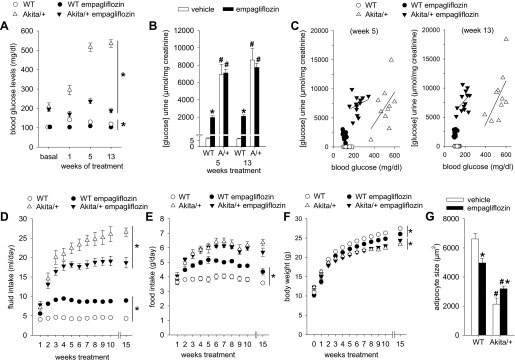
Empagliflozin strongly lowered hyperglycemia in diabetic Akita/+ mice. Empagliflozin (300 mg/kg of diet) or repelleted diet (vehicle) were given to Akita/+ and WT mice. A: empagliflozin lowered blood glucose levels in WT and much stronger in Akita/+ mice. B: empagliflozin increased urinary glucose/creatinine ratios in WT but not in Akita/+ mice. C: for a given level of hyperglycemia, urinary glucose/creatinine ratios were greater in empagliflozin-treated compared with vehicle-treated Akita/+ mice. D–G: in WT, empagliflozin lowered body weight and adipocyte size and increased food and fluid intake. In Akita/+ mice, empagliflozin did not affect food intake and modestly increased body weight and adipocyte size, respectively. *P < 0.05 vs. vehicle-treatment in same genotype; #P < 0.05 vs. WT. ANOVA and unpaired Student's t-test. A and D–F: statistical analysis refers to last time point shown; n = 10–13 per group for A–F; n = 3–5 for G.
In Akita/+ mice, mean plasma insulin concentration was less by 90% while blood glucose concentration was elevated 5-fold relative to WT (Figs. 2A and 3A). Empagliflozin reduced blood glucose concentration by 60% in hyperglycemic Akita/+ while further lowering plasma insulin levels, indicating presence of ∼10% residual beta islet function in Akita/+ that remains sensitive to changes in plasma glucose within the range of 200–600 mg/dl. Inhibition of glucose reabsorption by empagliflozin in Akita/+ was well-matched to the resulting decline in the filtered load of glucose, as indicated by the finding that treated and untreated animals wound up excreting similar amounts of glucose in their urine after coming into balance with disparate blood glucose concentrations (Fig. 2, A–C). This was associated with similar food intake (measured in regular cages) (Fig. 2E). Similar food intake and urinary glucose/creatinine ratios and absolute urinary glucose concentrations (urinary calorie loss) were previously reported between STZ-diabetic Sglt2−/− and control mice (36). Up to 10 wk of treatment did not significantly alter body weight in Akita/+ mice, but empagliflozin modestly increased body weight following 15 wk of treatment (Fig. 2F), i.e., over the course of the study the SGLT2 inhibitor induced opposite effects on body weight in WT vs. Akita/+ mice. The observed changes in body weight in response to diabetes and empagliflozin were accompanied by parallel changes in epididymal fat adipocyte size, i.e., empagliflozin reduced mean adipocyte size in WT and increased adipocyte size in Akita/+ mice (Fig. 2G).
Fig. 3.

Empagliflozin further lowered plasma insulin in diabetic Akita/+ mice and prevented diabetes-induced increase in renal mRNA expression of phosphoenolpyruvate carboxykinase (PEPCK), systolic blood pressure, and hematocrit. Empagliflozin (300 mg/kg of diet) or repelleted diet (vehicle) were given to Akita/+ and WT mice. Depicted are results for plasma insulin in nonfasted mice (A), renal PEPCK mRNA (B), plasma aldosterone (C), systolic blood pressure (D), heart rate (E), and hematocrit (F). *P < 0.05 vs. vehicle treatment in same genotype; #P < 0.05 vs. WT. ANOVA and unpaired Student's t-test; n = 10–13 per group.
Renal gluconeogenesis is upregulated in diabetic animal models and in humans with type 1 or type 2 diabetes, and may significantly contribute to hyperglycemia (for review see Ref. 10). Phosphoenolpyruvate carboxykinase (PEPCK) is a principal gluconeogenic enzyme, whose activity is directly and proportionally modulated by the rate of gene transcription and thus the amount of mRNA (21). We found renal PEPCK mRNA expression to be 60% elevated in Akita/+ vs. WT mice and normalized by empagliflozin (Fig. 3B). Empagliflozin did not affect the expression of PEPCK in WT.
Empagliflozin prevented the diabetes-induced modest increase in systolic blood pressure and hematocrit.
Systolic blood pressure (determined in trained and awake mice by an automatic tail cuff system) and hematocrit were modestly increased in Akita/+ vs. WT mice (Fig. 3, D and F). These increases in systolic blood pressure or hematocrit in Akita/+ were prevented with empagliflozin (Fig. 3, D and F). Heart rate and plasma concentrations of aldosterone were not affected by empagliflozin (Fig. 3, C and E). Note that aldosterone concentrations were elevated in Akita/+ compared with WT mice. Plasma Na+ concentration (not shown) was not modified by the treatment and similar between WT and Akita/+ mice.
Empagliflozin prevented the diabetes-induced increase in GFR and attenuated the increase in kidney weight and albuminuria in proportion to hyperglycemia.
In WT mice, empagliflozin modestly reduced GFR (Fig. 4A) while tending to increase kidney weight (Fig. 4B; P = 0.085) and significantly increasing the kidney-to-body weight ratio compared with vehicle treatment (12.0 ± 0.1 vs. 10.9 ± 0.2 mg/g body wt P < 0.001).
Fig. 4.

Empagliflozin prevented the diabetes-induced increase in glomerular filtration rate (GFR) and attenuated the increase in kidney weight, glomerular size, and albuminuria in proportion to hyperglycemia. Empagliflozin (300 mg/kg of diet) or repelleted diet (vehicle) were given to Akita/+ and WT mice. Depicted are results for GFR (A), kidney weight (B), urinary albumin/creatinine ratios (C), and glomerular size (D). *P < 0.05 vs. vehicle treatment in same genotype; #P < 0.05 vs. WT. ANOVA and unpaired Student's t-test and linear regression analysis. Linear regression lines were included when statistical significance was achieved. n = 10–13 per group for A–C, and n = 6–7 for D.
GFR was 30% greater among Akita/+ vs. WT mice, commensurate with diabetic glomerular hyperfiltration. Hyperfiltration was prevented by empagliflozin, which reduced GFR to the levels observed in WT (Fig. 4A). Empagliflozin attenuated increases in kidney weight and urinary albumin/creatinine ratio observed in Akita/+ vs. WT mice in proportion to lowering hyperglycemia (Fig. 4, B and C), and normalized glomerular size (Fig. 4D).
Empagliflozin attenuated diabetes-induced rise in renal expression of markers of kidney growth and inflammation.
Overexpression of the cyclin-dependent kinase inhibitors p27 and p21 has been implicated in the renal growth and G1 cell cycle arrest in the early diabetic kidney (35). Expression of p27 and p21 proteins was greater in renal nuclei from Akita/+ mice compared with WT (Fig. 5A). Renal content of the stress response protein, hemoxygenase-1 (HO-1), was also modestly increased in Akita/+ vs. WT. p21, p27, and HO-1 levels in Akita/+ mice were reduced by empagliflozin, although remaining slightly elevated. Empagliflozin did not suppress p21, p27, or HO-1 expression in WT.
Fig. 5.

Empagliflozin attenuated diabetes-induced rise in renal expression of markers of kidney growth and inflammation. Empagliflozin (300 mg/kg of diet) or repelleted diet (vehicle) were given to Akita/+ and WT mice and kidneys harvested after 15 wk. Depicted are results for renal nuclear expression of p27 and p21 and renal cytosolic expression of HO-1 (A), and renal mRNA expression of NFkB, CCL2, CD14, TIMP2, and IL6 (B). *P < 0.05 vs. vehicle treatment in same genotype; #P < 0.05 vs. WT. ANOVA and unpaired Student's t-test and linear regression analysis. Linear regression lines were included when statistical significance was achieved. n = 10–13 per group.
NFkB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a transcriptional regulator responsible for cytokine production, including inflammatory cytokines. Renal content of NFkB mRNA was elevated in Akita/+ as were mRNA for CCL2 (a cytokine that recruits proinflammatory cells), CD14 (a cell surface marker of macrophages), IL-6 (a dual pro- and anti-inflammatory cytokine), and TIMP2 (a tissue inhibitor of matrix metalloproteinases that arises in kidney injury). Empagliflozin reduced or normalized the renal mRNA content of all these inflammatory markers in Akita/+, without major effects on their expression in WT mice (Fig. 5B).
Several markers of oxidative stress and fibrosis were examined and found not to be different between the hyperfiltering Akita/+ mice with early diabetes and WT controls. Nor was there evidence that empagliflozin affected these markers. Markers included renal mRNA content of NADPH oxidases, NOX2 and NOX4, and the cytokine CCL5. Markers of fibrosis included TGFβ and renal collagen expression, as determined by Sirius red binding (data not shown). These results indicated that diabetes-induced renal changes in the current study were restricted to the early phase of renal hyperfunction (GFR), albuminuria, growth, and inflammation, rather than extensive oxidative stress or fibrosis.
Empagliflozin did not induce ascending urinary tract infection.
Glucosuria predisposes to urinary tract infection, but no animal in the present study contracted urinary tract infection as defined by >105 bacterial colony-forming units (CFUs) per milliliter of urine collected by direct bladder puncture under terminal anesthesia following 15 wk of treatment. Most samples from all groups produced no bacterial growth and no sample in any mouse produced more than >103 CFU/ml (n = 11–13). These findings are consistent with previous studies using nondiabetic and diabetic Sglt2−/− mice (36).
DISCUSSION
The main findings of the current study are that the selective SGLT2 inhibitor, empagliflozin, lowered blood glucose levels in diabetic Akita mice by ∼60% from >500 to ∼200 mg/dl and prevented glomerular hyperfiltration. Empagliflozin also attenuated, without completely eliminating, diabetes-associated increases in albuminuria, kidney weight, markers of renal growth, and several markers of inflammation. Finally, empagliflozin prevented the diabetes-induced overexpression of the gluconeogenic enzyme, PEPCK. Empagliflozin appeared to mitigate the effects of diabetes on kidney growth, albuminuria, and inflammation to the same extent that it mitigated hyperglycemia, whereas the mitigation of hyperfiltration by empagliflozin was complete. Together with previous findings discussed below, including studies in STZ-treated Sglt2−/− mice, we propose that inhibition of SGLT2 can suppress diabetic glomerular hyperfiltration independent of its blood glucose effect, consistent with the tubular hypothesis of diabetic glomerular hyperfiltration (35, 41). In comparison, the beneficial effect of SGLT2 inhibition on albuminuria, kidney growth, and inflammation in the early diabetic kidney may mostly be secondary to lower blood glucose.
The current results confirm that pharmacological inhibition of SGLT2 reduces renal glucose reabsorption thereby lowering blood glucose levels in diabetes mellitus. This is consistent with prior studies in STZ-diabetic mice lacking Sglt2 (38) or carrying a loss of function mutation in SGLT2 (15), and with studies using empagliflozin or other SGLT2 inhibitors in diabetic rats and humans (6, 7, 14, 17, 31, 40). Uptake experiments with a nonmetabolizable glucose analog in stable cell lines overexpressing human and mouse glucose transporters showed that empagliflozin inhibits SGLT2-mediated glucose uptake in a dose-dependent manner with an IC50 of 3.1 nM for human SGLT2 and 1.9 nM for mouse SGLT2, and is highly selective for SGLT-2 over SGLT-1 in humans (>2,500-fold) and mice (∼5,800-fold), respectively (12). Comparing multiple SGLT2 inhibitors, empagliflozin had the highest selectivity for SGLT-2 over SGLT-1 (12). Empagliflozin was given at a dose of 300 mg/kg of diet, which corresponded to 60–80 mg·kg body wt−1·day−1. Empagliflozin given in the diet at a dose of 30–35 mg per kilogram body weight and day induced a free plasma concentration of 1–2 nM in clearance studies performed during daytime in nondiabetic mice (25), which is close to the IC50 for SGLT2 in the mouse (12). Therefore, the dose of ∼60–80 mg·kg body wt−1·day−1 applied in the current study was expected to have inhibited SGLT2 function by >50% during daytime and possibly even stronger during nighttime when food and thus drug intake occurred, taking into consideration that the plasma half life of empagliflozin in the male mouse is ∼5.6 h.
Empagliflozin lowered blood glucose (measured during daytime) more potently in Akita/+ mice (by ∼300 mg/dl: from >500 to ∼200 mg/dl) than did knocking out the Sglt2 gene in STZ-diabetic mice (by ∼170 mg/dl: from ∼470 to ∼300 mg/dl) (38). One possible reason is the difference in renal SGLT2 expression. SGLT2 expression was upregulated in Akita/+ mice [similar to mice (38) and humans (22) with type 2 diabetes] and, therefore, the amount of transporter that can be inhibited by empagliflozin was increased. In contrast, STZ diabetes reduced the renal expression of SGLT2 and, thereby, the glucose reabsorption available to be suppressed by the knockout (38). SGLT2 expression was also increased by empagliflozin in WT mice, probably a compensatory response. Divergent effects on mRNA expression indicate that Akita diabetes and chronic SGLT2 inhibition may increase SGLT2 expression via different mechanisms. While the increase in SGLT2 expression in Akita/+ can contribute to a greater reduction or change in blood glucose levels in response to empagliflozin compared with the effect of the Sglt2 gene knockout in STZ diabetes, the differences in SGLT2 expression cannot explain why blood glucose was reduced to lower absolute levels in the current study.
Renal membrane SGLT1 protein expression provided a mirror image of the changes in SGLT2, i.e., Akita diabetes, empagliflozin, and their combination reduced renal membrane protein expression of SGLT1. Previous studies provided evidence for increased, unchanged, or reduced renal SGLT1 expression and/or activity in diabetes or under high glucose conditions (41). The effect of the SGLT2 inhibitor empagliflozin in nondiabetic mice mimicked the suppression of renal membrane SGLT1 protein expression reported in mice lacking Sglt2 (36). Glucose transport through SGLT1 is strongly increased when SGLT2 is deleted by gene targeting or inhibited by empagliflozin (25) or when luminal glucose delivery is increased in diabetes. Therefore, lowering renal membrane SGLT1 protein expression may serve to limit glucose reabsorption and possibly toxicity in the S2 and S3 segments of the proximal tubule when glucose delivery and uptake are increased. In vitro studies in renal proximal tubule cells indicated that high glucose-induced oxidative stress may reduce SGLT expression and Na+/glucose cotransport activity (12a). The observed divergent effects of Akita/+ and empagliflozin on mRNA and membrane protein expression of SGLT1 indicate a complex translational and posttranslation regulation. With regard to glucose homeostasis, downregulation of SGLT1 reduces its glucose transport and compensatory capacity and is expected to enhance the glucosuric and blood glucose-lowering effect of SGLT2 inhibition (20, 25).
In addition to inhibiting renal glucose reabsorption, empagliflozin attenuated, in proportion to hyperglycemia, the diabetes-associated increase in renal mRNA expression of PEPCK. This enzyme is a principal regulator of gluconeogenesis in the proximal tubule that is regulated at the mRNA level and upregulated in the diabetic kidney of rats and humans (9, 10, 21). This observed effect was specific for the diabetic kidney since empagliflozin did not affect the expression of PEPCK in WT kidneys. PEPCK expression by proximal tubules is mainly downregulated by insulin and upregulated by glucagon, although its expression also increases during chronic acidosis as part of the pathway for generating ammonium and bicarbonate (10, 21). The increase in PEPCK in Akita/+ was expected based on prior reports and known pathways (9, 10, 21). The mechanism whereby empagliflozin prevented this is not obvious. Effects of empagliflozin on plasma insulin levels cannot explain the effect on renal PEPCK expression in Akita/+ mice since insulin levels were modestly lower compared with vehicle-treated Akita/+ mice, consistent with lower blood glucose levels reducing the stimulus for insulin secretion. Further studies are needed to examine the basis for the novel relationship of PEPCK expression to SGLT2 and to determine the extent to which a reduction in renal PEPCK overexpression contributed to the blood glucose-lowering effect of empagliflozin.
Similar to the phenotype of knocking out the Sglt2 gene in nondiabetic mice (38), SGLT2 inhibition by empagliflozin increased food intake in WT mice, possibly to match the sustained urinary glucose and calorie loss. This was associated with lower body weight and smaller epididymal adipocytes, corroborating the notion that increased food intake among SGLT2-blocked mice is a compensatory response to a decrease in energy stores. The glucosuria in empagliflozin-treated WT mice and in nondiabetic Sglt2−/− mice (38) was sustained in steady state, indicating the reduction in glomerular filtration of glucose [due to lowering blood glucose levels and GFR (see below)] was too small to match the reduction in proximal tubular glucose reabsorption. In comparison, and as observed in STZ-diabetic Sglt2−/− mice (38), pharmacological inhibition of SGLT2 by empagliflozin in diabetic Akita/+ mice did not increase glucosuria after they reached steady state. Glucosuria did not increase because the reduction in glomerular filtration of glucose (due to lowering blood glucose and glomerular hyperfiltration) was large enough to nullify the impact of reduced tubular reabsorption. As a consequence of similar glucosuria, empagliflozin did not reduce body weight or fat stores (even small increases) or stimulate increased food intake in Akita/+ mice. This simple paradigm for the effects of SGLT2 blockade on body weight and food intake applies universally to mice, rats, and humans. Whatever differences may appear among various studies in humans, rats, and mice with respect to the effect of SGLT2 blockade on body weight can be accounted for by quantitative differences in the effect on glucosuria and the effect of glucosuria on blood glucose and the sensitivity of appetite to weight loss.
Hyperglycemia causes a primary increase in proximal tubular Na+ and fluid reabsorption, in part due to enhanced Na+-glucose cotransport. Enhanced reabsorption reduces the luminal Na-Cl-K concentration at the macula densa, and, via the physiology of tubuloglomerular feedback and a possible reduction in the hydrostatic pressure in Bowman space, increases GFR (35, 37). Empagliflozin slightly reduced GFR in WT mice, but normalized GFR in Akita/+ mice. This is consistent with attenuation of hyperreabsorption by SGLT2 inhibition, which through the described mechanisms lowers GFR, in particular in diabetic conditions (33, 35, 37). The stronger GFR response in diabetes is due to the greater tubular glucose delivery and, therefore, greater empagliflozin-inhibitable Na+-glucose cotransport in the proximal tubule. The described GFR-lowering mechanism of SGLT2 inhibition does not require a blood glucose-lowering effect (4, 33, 37, 38). Correcting hyperglycemia may, however, have additional effects on GFR, e.g., via inhibition of glomerular growth, assuming filtration pressure disequilibrium allows for an influence of glomerular capillary growth on GFR, which may be more relevant in humans than in rodents. If glomerular hyperfiltration is not only associated with but actually contributes to the development and progression of diabetic nephropathy, then lowering GFR by inhibiting SGLT2 has long-term nephroprotective potential (which may not become immediately obvious in short-term studies in rodents).
Diabetic kidney growth and its unique molecular signature have been linked to the development of diabetic nephropathy (35, 41). The tubular growth mechanism in diabetes includes an early, growth factor-driven hyperplastic phase which, through the activation of cyclin-dependent kinase inhibitors, including p27 and p21, is switched to a hypertrophic phase that shows aspects of senescence (28, 41). We reported previously that knockout of Sglt2 in STZ-diabetic mice, which lowered blood glucose levels from ∼470 to ∼300 mg/dl, did not limit the rise in albuminuria, kidney weight, or markers of kidney growth, inflammation, and injury (38), indicating that SGLT2-mediated glucose-reabsorption itself is not critical for these changes. Here we observed that empagliflozin given to Akita/+ mice induced a stronger blood glucose-lowering effect, from >500 to ∼200 mg/dl, and did not prevent but attenuated the rise in albuminuria and kidney growth, including the upregulation of p27 and p21, and reduced markers of renal inflammation in proportion to hyperglycemia. Together these studies indicate that inhibition of SGLT2 can have beneficial effects on the kidney beyond lowering GFR. The observed linear correlations between blood glucose levels and albuminuria, kidney weight, or markers of kidney inflammation in the current study would be expected if blood glucose was the only determinant, but the correlations cannot prove a causal relationship. Considering that the absence of SGLT2 itself or the associated reduction in BG to 300 mg/dl was not nephroprotective in STZ-diabetic rats (38), the current results are consistent with the notion that beneficial effects of SGLT2 inhibition on these parameters require and are mostly secondary to a strong blood glucose-lowering effect.
In nondiabetic mice, empagliflozin tended to increase kidney weight, and a small, numerical upregulation of the renal expression of p27 and HO-1 was observed. The changes did not reach statistical significance but are reminiscent of the reported modest increase in kidney weight and renal expression of p27 and HO-1 observed in nondiabetic Sglt2−/− vs. control mice (38). These changes may reflect tissue responses of growth and upregulation of protective mechanisms, respectively, when tubular glucose delivery downstream of the early proximal tubule is increased in the absence of hyperglycemia. Available data in humans with mutations in SGLT2 (SLC5A2), a rare and not well studied genetic disorder, indicate that no other complications, including chronic nephrologic complications, are known to be consistently associated with mutations of this gene (8, 16, 27, 29, 42). In this regard, the observed changes with SGLT2 inhibition in nondiabetic mice may reflect adaptive and minor changes, but further studies are necessary. In diabetic mice, empagliflozin attenuated but did not completely prevent kidney growth or p27 upregulation, which may reflect the fact that the SGLT2 inhibitor did not normalize blood glucose levels and/or did not reduce glucose delivery downstream of the early proximal tubule (see above). The finding that empagliflozin reduced kidney growth in Akita/+ mice without lowering urinary glucose excretion indicated that an increase in luminal glucose delivery downstream of the early proximal tubule alone is not sufficient to induce full diabetic kidney growth, arguing that additional factors (e.g., from glomerular filtrate and/or peritubular sites) also contribute.
Empagliflozin lowered blood glucose and diabetes-induced increases in GFR, kidney weight, albuminuria, markers of kidney inflammation, hematocrit, and blood pressure, yet the increase in plasma levels of aldosterone in Akita/+ mice was not affected by the SGLT2 inhibitor. This observation argues against a major pathophysiological role of enhanced aldosterone levels in these diabetes-induced changes. The results may also provide some clues to the mechanism(s) that enhance(s) plasma aldosterone levels in diabetes: a minor isotonic volume constriction, as indicated by higher hematocrit values in Akita/+ mice, may not be crucial since it was normalized by SGLT2 inhibition; in comparison and as discussed above, the enhanced glucose delivery downstream of the early proximal tubule, including the macula densa segment and the distal nephron, was not affected by empagliflozin in Akita/+ mice and can provide interactions with the renin-angiotensin-aldosterone system. Further studies are needed to better understand the nuances of enhanced luminal glucose concentrations along the nephron.
In summary, inhibition of SGLT2 by empagliflozin in diabetic Akita/+ mice strongly lowered blood glucose levels. This was the consequence of lowering renal glucose reabsorption, but empagliflozin also prevented the diabetes-induced upregulation of renal PEPCK mRNA expression, a prinicipal regulator of gluconeogenesis. Downregulation of renal membrane protein expression of SGLT1 in Akita/+ reduces SGLT1-mediated compensation and is expected to enhance the blood glucose-lowering effect of empagliflozin. Empagliflozin attenuated diabetes-induced albuminuria, kidney growth, and renal expression of molecular markers of kidney growth and inflammation. These modifications appear to depend on blood glucose reduction. In contrast, SGLT2 inhibition prevented glomerular hyperfiltration, and this effect can occur independent of lowering blood glucose (due to tubular Na and fluid transport inhibition). In addition, empagliflozin prevented the increase in systolic blood pressure observed in Akita/+ mice, similar to findings described in diabetic hypertensive patients. These results suggest that inhibition of SGLT2, with empagliflozin, could affect the development or progression of diabetic nephropathy by blood glucose-dependent and -independent mechanisms. Further studies are needed to confirm the putative nephroprotective effects of SGLT2 inhibitors in clinical practice.
GRANTS
The authors were supported by National Institutes of Health Grants R01-DK-56248, R01-HL-94728, the UAB/UCSD O'Brien Center of Acute Kidney Injury NIH-P30-DK-079337 (to V. Vallon and S. C. Thomson) (including Pilot and Feasiblity Grant to T. Rieg); the American Heart Association (Scientist Development Grant 10SDG2610034 to T. Rieg); a Carl W. Gottschalk Research Grant of the American Society of Nephrology to T. Rieg; a fellowship of the Manpei Suzuki Diabetes Foundation to T. Masuda; the Department of Veterans Affairs; and by Boehringer Ingelheim, Biberach an der Riss (to V. Vallon).
DISCLOSURES
V. Vallon has received within the past 12 months research grant support for basic science studies from Boehringer Ingelheim Pharma GmbH & KG and Amylin. E. Mayoux is employed by Boehringer Ingelheim.
AUTHOR CONTRIBUTIONS
Author contributions: V.V., M.G., M.R., E.M., S.C.T., and T.R. conception and design of research; V.V., M.G., M.R., T.M., J.S., and T.R. performed experiments; V.V., M.G., M.R., T.M., J.S., and T.R. analyzed data; V.V., M.G., M.R., T.M., J.S., H.K., S.C.T., and T.R. interpreted results of experiments; V.V., M.G., and M.R. prepared figures; V.V. drafted manuscript; V.V., M.G., M.R., T.M., J.S., E.M., H.K., S.C.T., and T.R. approved final version of manuscript; M.G., M.R., T.M., J.S., E.M., H.K., S.C.T., and T.R. edited and revised manuscript.
REFERENCES
- 1.Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, Sabolic I. Revised immunolocalization of the Na+-d-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295: C475–C489, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Baumgartl HJ, Sigl G, Banholzer P, Haslbeck M, Standl E. On the prognosis of IDDM patients with large kidneys. Nephrol Dial Transplant 13: 630–634, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Bognetti E, Zoja A, Meschi F, Paesano PL, Chiumello G. Relationship between kidney volume, microalbuminuria and duration of diabetes mellitus. Diabetologia 39: 1409, 1996 [PubMed] [Google Scholar]
- 4.Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Fagan NM, Woerle HJ, Johansen OE, Broedl UC, von Eynatten M. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. [E pub before print] [DOI] [PubMed] [Google Scholar]
- 5.Christiansen JS, Gammelgaard J, Frandsen M, Parving HH. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia 20: 451–456, 1981 [DOI] [PubMed] [Google Scholar]
- 6.DeFronzo RA, Davidson JA, del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 14: 5–14, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A Phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab 15: 721–728, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Francis J, Zhang J, Farhi A, Carey H, Geller DS. A novel SGLT2 mutation in a patient with autosomal recessive renal glucosuria. Nephrol Dial Transplant 19: 2893–2895, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Gatica R, Bertinat R, Silva P, Carpio D, Ramirez MJ, Slebe JC, San MR, Nualart F, Campistol JM, Caelles C, Yanez AJ. Altered expression and localization of insulin receptor in proximal tubule cells from human and rat diabetic kidney. J Cell Biochem 114: 639–649, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 24: 382–391, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H. Na+-d-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61: 187–196, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab 14: 83–90, 2012 [DOI] [PubMed] [Google Scholar]
- 12a.Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol Renal Physiol 288: F988–F996, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Lawson ML, Sochett EB, Chait PG, Balfe JW, Daneman D. Effect of puberty on markers of glomerular hypertrophy and hypertension in IDDM. Diabetes 45: 51–55, 1996 [DOI] [PubMed] [Google Scholar]
- 14.Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab 14: 601–607, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Ly JP, Onay T, Sison K, Sivaskandarajah G, Sabbisetti V, Li L, Bonventre JV, Flenniken A, Paragas N, Barasch JM, Adamson SL, Osborne L, Rossant J, Schnermann J, Quaggin SE. The Sweet Pee model for Sglt2 mutation. J Am Soc Nephrol 22: 113–123, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magen D, Sprecher E, Zelikovic I, Skorecki K. A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int 67: 34–41, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab 95: 34–42, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pill J, Kraenzlin B, Jander J, Sattelkau T, Sadick M, Kloetzer HM, Deus C, Kraemer U, Gretz N. Fluorescein-labeled sinistrin as marker of glomerular filtration rate. Eur J Med Chem 40: 1056–1061, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, Doree D, Jeter-Jones S, Mseeh F, Rodriguez LA, Harris A, Buhring L, Platt KA, Vogel P, Brommage R, Shadoan MK, Sands AT, Zambrowicz B. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab 304: E117–E130, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Quinn PG, Yeagley D. Insulin regulation of PEPCK gene expression: a model for rapid and reversible modulation. Curr Drug Targets Immune Endocr Metabol Disord 5: 423–437, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54: 3427–3434, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Rasch R. Tubular lesions in streptozotocin-diabetic rats. Diabetologia 27: 32–37, 1984 [DOI] [PubMed] [Google Scholar]
- 24.Rieg T. A high-throughput method for measurement of glomerular filtration rate in conscious mice. J Vis Exp 75: e50330, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt KA, Powell DR, Thomson SC, Koepsell H, Vallon V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacologicical SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol (First published November 13, 2013). 10.1152/ajprenal.00518.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rigalleau V, Garcia M, Lasseur C, Laurent F, Montaudon M, Raffaitin C, Barthe N, Beauvieux MC, Vendrely B, Chauveau P, Combe C, Gin H. Large kidneys predict poor renal outcome in subjects with diabetes and chronic kidney disease. BMC Nephrol 11: 3, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santer R, Calado J. Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 5: 133–141, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Satriano J, Mansoury H, Deng A, Sharma K, Vallon V, Blantz RC, Thomson SC. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am J Physiol Cell Physiol 299: C374–C380, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scholl-Burgi S, Santer R, Ehrich JH. Long-term outcome of renal glucosuria type 0: the original patient and his natural history. Nephrol Dial Transplant 19: 2394–2396, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Seyer-Hansen K, Hansen J, Gundersen HJ. Renal hypertrophy in experimental diabetes. A morphometric study. Diabetologia 18: 501–505, 1980 [DOI] [PubMed] [Google Scholar]
- 31.Thomas L, Grempler R, Eckhardt M, Himmelsbach F, Sauer A, Klein T, Eickelmann P, Mark M. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab 14: 94–96, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest 107: 217–224, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302: R75–R83, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urich D, Eisenberg JL, Hamill KJ, Takawira D, Chiarella SE, Soberanes S, Gonzalez A, Koentgen F, Manghi T, Hopkinson SB, Misharin AV, Perlman H, Mutlu GM, Budinger GR, Jones JC. Lung-specific loss of the laminin alpha3 subunit confers resistance to mechanical injury. J Cell Sci 124: 2927–2937, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10: 2569–2576, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol 111: 30–38, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vallon V, Sharma K. Sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Curr Opin Nephrol Hypertens 19: 425–431, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74: 351–375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Heuvel LP, Assink K, Willemsen M, Monnens L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 111: 544–547, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Zerbini G, Bonfanti R, Meschi F, Bognetti E, Paesano PL, Gianolli L, Querques M, Maestroni A, Calori G, Del MA, Fazio F, Luzi L, Chiumello G. Persistent renal hypertrophy and faster decline of glomerular filtration rate precede the development of microalbuminuria in type 1 diabetes. Diabetes 55: 2620–2625, 2006 [DOI] [PubMed] [Google Scholar]