

Abstract
In the kidney, the sodium-glucose cotransporters SGLT2 and SGLT1 are thought to account for >90 and ∼3% of fractional glucose reabsorption (FGR), respectively. However, euglycemic humans treated with an SGLT2 inhibitor maintain an FGR of 40–50%, mimicking values in Sglt2 knockout mice. Here, we show that oral gavage with a selective SGLT2 inhibitor (SGLT2-I) dose dependently increased urinary glucose excretion (UGE) in wild-type (WT) mice. The dose-response curve was shifted leftward and the maximum response doubled in Sglt1 knockout (Sglt1−/−) mice. Treatment in diet with the SGLT2-I for 3 wk maintained 1.5- to 2-fold higher urine glucose/creatinine ratios in Sglt1−/− vs. WT mice, associated with a temporarily greater reduction in blood glucose in Sglt1−/− vs. WT after 24 h (−33 vs. −11%). Subsequent inulin clearance studies under anesthesia revealed free plasma concentrations of the SGLT2-I (corresponding to early proximal concentration) close to the reported IC50 for SGLT2 in mice, which were associated with FGR of 64 ± 2% in WT and 17 ± 2% in Sglt1−/−. Additional intraperitoneal application of the SGLT2-I (maximum effective dose in metabolic cages) increased free plasma concentrations ∼10-fold and reduced FGR to 44 ± 3% in WT and to −1 ± 3% in Sglt1−/−. The absence of renal glucose reabsorption was confirmed in male and female Sglt1/Sglt2 double knockout mice. In conclusion, SGLT2 and SGLT1 account for renal glucose reabsorption in euglycemia, with 97 and 3% being reabsorbed by SGLT2 and SGLT1, respectively. When SGLT2 is fully inhibited by SGLT2-I, the increase in SGLT1-mediated glucose reabsorption explains why only 50–60% of filtered glucose is excreted.
Keywords: proximal tubule, glucose reabsorption, glucose transport, sodium glucose cotransport inhibitor, diabetes mellitus
about 180 g of glucose are filtered daily by the kidney and enter the renal tubular system in a healthy normoglycemic subject. Glucose in urine is absent or at very low concentrations in healthy adults due to near complete reabsorption along the nephron segments, primarily in the proximal tubule. The renal Na+-glucose cotransporter SGLT2 (SLC5A2) is localized to the early proximal tubule and thought to mediate the bulk of tubular glucose uptake across the apical membrane of the kidney (14, 17, 20). Studies in mice lacking Sglt2 demonstrated that SGLT2 mediates all glucose reabsorption in the early proximal tubule and most of overall glucose reabsorption by the kidney (17). In comparison, low-capacity SGLT1 (SLC5A1) “cleans up” most of the remaining luminal glucose in further distal parts of the proximal tubule (1, 3, 14, 20). Studies in mice lacking Sglt1 (Sglt1−/−) revealed a fractional glucose excretion of 3% compared with 0.2% in wild-type (WT) (3). Whether glucose transporters other than SGLT2 and SGLT1 contribute in a measurable extent to renal glucose reabsorption across the luminal membrane of the renal epithelia has never been tested. Potential candidates include a low-affinity Na+-d-glucose cotransporter cloned from the rat named NaGLT1, which has been proposed to be expressed in the apical membrane of the proximal tubule, but its quantitative contribution to glucose reabsorption, if any, is not known (6). Pharmacological inhibitors of the renal Na+-glucose cotransporter SGLT2 (SLC5A2) are new antidiabetic drugs that inhibit the renal reabsorption of glucose, thereby lowering blood glucose levels (2, 11, 19). Despite a proposed dominant contribution of SGLT2 to renal glucose reabsorption, previous studies in humans estimated, based on absolute renal glucose excretion data, that application of selective SGLT2 inhibitors increase fractional glucose excretion to only ∼50–70%, and thus a fractional glucose reabsorption (FGR) of 30–50% was maintained (5, 8, 15). Significant compensation by downstream SGLT1 appears likely and is consistent with a high maximal glucose transport rate of hSGLT1 (7). A recent study showed that absolute urine glucose excretion (UGE) was significantly greater in mice lacking both Sglt1 and Sglt2 vs. mice lacking only Sglt2 (by at least 67%) (12), suggesting that SGLT1 can compensate when Sglt2 is absent. These studies, however, did not measure absolute or fractional glucose reabsorption, and therefore the exact extent of compensation by SGLT1 and whether there is renal glucose reabsorption in the kidney beyond SGLT1 and SGLT2 were not determined. An alternative hypothesis proposed that the SGLT2 inhibitors may not reach their target in the early proximal tubule at high enough concentrations (9). The observation that mean FGR in euglycemic mice lacking Sglt2 (Sglt2−/−) was ∼36% (17), and thus very similar to the values observed in humans treated with SGLT2 inhibitors, provided first evidence against this hypothesis, but more specific studies were lacking. The latter studies in Sglt2−/− mice indicated that the mouse models may be suitable to shed further light on these issues.
To determine whether SGLT1 and SGLT2 account for all glucose reabsorption in the kidney, and whether the increase in SGLT1-mediated transport fully explains the limited glucosuric response to SGLT2 inhibition in euglycemia, we used pharmacological inhibition of SGLT2 in the presence and absence of SGLT1 in mice and compared renal glucose reabsorption with mice lacking both SGLT1 and SGLT2 (Sglt1/Sglt2−/−). These studies also provided insights into the question of whether pharmacological SGLT2 inhibitors reach SGLT2 in the early proximal tubule at high enough concentrations to fully inhibit SGLT2 in vivo.
METHODS
Animals.
All animal experimentation was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD) and was approved by the local Institutional Animal Care and Use Committee. Sglt1−/− or Sglt1/Sglt2−/− mice were used and compared with age- and gender-matched WT mice (3, 17). All mice were fed a low-glucose diet (in %: 52.5 protein, 11.3 fat, 19.9 fiber, 6.2 ash, 0.9 starch, 0.3 sugar, 13.8 MJ ME/kg; ssniff Spezialdiäten, Soest, Germany) to prevent glucose/galactose malabsorption and subsequent diarrhea due to the absence of SGLT1. Mice were housed in the same animal room with a 12:12-h light-dark cycle and free access to tap water.
Acute and chronic glucosuric responses to selective SGLT2 inhibition in Sglt1−/− and WT mice.
Empagliflozin is a selective SGLT2 inhibitor with an IC50 of 3.1 nM for human SGLT2 and 1.9 nM for mouse SGLT2, is highly selective for SGLT2 over SGLT1 in humans (>2,500-fold) and mice (∼5,800-fold), respectively (4), and was used as a pharmacological tool to inhibit SGLT2. Empagliflozin was provided by Boehringer Ingelheim, Biberach, Germany. The following three series of studies were performed.
First, WT and Sglt1−/− mice were treated by oral gavage with vehicle or empagliflozin (0.1–30 mg/kg) together with a water load (30 μl/g body wt) to facilitate subsequent quantitative urine collection in metabolic cages over 3 h.
Second, mice were treated with empagliflozin (300 mg/kg of diet) for 3 wk while body weight, urine glucose/creatinine ratios, blood glucose levels, and food and fluid intake were measured. Food and fluid intake was determined while the mice were maintained in their regular cages. Urine was obtained at the same time of day by picking up the mice to elicit reflex urination and holding them over a clean petri dish for sample collection. For paired glucose measurements, blood was collected by tail snip immediately after urine collection in awake mice.
Third, following 3 wk of treatment with empagliflozin in the diet as described above, inulin clearance studies were performed to determine glomerular filtration rate (GFR), filtered glucose, urinary glucose excretion (UGE), and FGR under terminal anesthesia as previously described (13, 17). Briefly, mice were anesthetized with thiobutabarbital (100 mg/kg ip, 2 μl/g body wt; Sigma-Aldrich, St. Louis, MO) and ketamine (100 mg/kg im, 2 μl/g body wt; Butler, Dublin, OH). The jugular vein was cannulated for continuous infusion of 2.25% bovine serum albumin in 0.85% NaCl at a rate of 0.4 ml·h−1·30−1 g body wt. For assessment of two-kidney GFR by inulin clearance, [3H]inulin was added to the infusion to deliver 5 μCi·h−1·30−1 g body wt. Urinary excretion of glucose and [3H]inulin was assessed by quantitative urine collection via a bladder catheter in 30-min periods. Blood samples (50 μl) were drawn midway through each period from an arterial catheter, which was also used to monitor blood pressure and heart rate. Concentrations of [3H]inulin in plasma and urine were measured by liquid scintillation counting.
Renal clearance studies in mice lacking Sglt1/Sglt2.
Renal clearance studies were performed under terminal anesthesia following the procedures described above.
Blood and urine analysis.
Blood glucose in awake mice was determined using an Ascensia Elite XL glucometer (Bayer, Mishawaka, IN). Plasma glucose in clearance studies and all urine glucose were determined by the hexokinase/glucose-6-phosphate dehydrogenase method (Infinity, Thermo Electron, Louisville, CO). Total concentrations of empagliflozin in plasma were determined by liquid chromatography-tandem mass spectrometry. We found that mean fractional protein binding of empagliflozin in mouse plasma is constant over a wide range (up to >50 μM) at 88.1 ± 0.5%. We used the latter information to calculate free plasma concentrations.
Statistical analysis.
Data are shown as means ± SE. ANOVA and paired or unpaired Student's t-tests were performed to analyze for statistical differences in paired data sets and between groups, respectively. P < 0.05 was considered statistically significant.
RESULTS
Acute glucosuric effect of SGLT2 inhibitor is doubled in Sglt1−/− vs. WT mice.
In metabolic cage studies, UGE was greater in Sglt1−/− compared with WT mice following vehicle application (Fig. 1A), consistent with previous results (3). Empagliflozin dose dependently increased UGE in WT mice (Fig. 1B): from 0.1 nmol/[min × g] in response to vehicle to a maximum of ∼30 nmol/[min × g] observed at doses of 3–10 mg/kg per os (po) (ED50 ∼1.2 mg/kg). Compared with WT, the empagliflozin-induced UGE was shifted leftward and the maximum response doubled in Sglt1−/− mice: from 1.2 with vehicle to a maximum of ∼60 nmol/[min × g] observed at doses of 3–30 mg/kg (ED50 of ∼0.4 mg/kg).
Fig. 1.

Maximum acute glucosuric response to sodium-glucose cotransporter SGLT2 inhibitor is enhanced 2-fold in Sglt1−/− vs. wild-type (WT) mice in metabolic cage studies. A: urinary glucose excretion (UGE) was greater in Sglt1−/− compared with WT mice following vehicle application. B: empagliflozin dose dependently increased UGE in WT. Please note different scale of y-axis vs. A. Compared with WT, the empagliflozin-induced UGE was shifted leftward and the maximum response doubled in Sglt1−/− mice. The difference between dose-response curves, which reflects the glucose reabsorption mediated via SGLT1 in WT mice, reached a maximum at a dose of ∼0.4 mg/kg (indicated at the left of the vertical lines) and was maintained (all vertical lines have same length) for higher doses up to 10 mg/kg, indicating a high selectivity of the SGLT2 inhibitor vs. SGLT1 in this dose range. Note that empagliflozin began to increase glucose excretion in WT when reabsorption via SGLT1 reached its maximum; n = 4–8/dose and genotype. *P < 0.05 vs. WT by ANOVA and unpaired Student's t-test.
Chronic glucosuric effect of SGLT2 inhibitor is enhanced 1.5- to 2-fold in Sglt1−/− vs. WT mice, associated with greater increases in food and fluid intake.
Treatment with empagliflozin (300 mg/kg of diet) induced a rapid (within the first day) increase in urine glucose/creatinine ratios to 1.5- to 2-fold higher levels in Sglt1−/− vs. WT mice, which persisted for the duration of the treatment (3 wk) (Fig. 2A). The associated reduction in blood glucose was enhanced in Sglt1−/− vs. WT mice after 24 h (−33 ± 5 vs −11 ± 5%; P < 0.05), whereas blood glucose was similar between groups after 1–3 wk of treatment (Fig. 2B). Empagliflozin increased food and fluid intake in both groups, and these responses were enhanced in Sglt1−/− compared with WT mice (Fig. 2, C and D). Absolute fluid intake was also higher in Sglt1−/− vs. WT mice during SGLT2 inhibition (e.g., at 21 days: 10.9 ± 0.2 vs. 9.2 ± 0.2 ml/day, P < 0.05). The greater increase in food intake in Sglt1−/− mice in response to SGLT2 inhibition was not sufficient to yield a higher absolute food intake compared with WT mice at the end of the study, due to a lower basal food intake in Sglt1−/− vs. WT mice before the start of empagliflozin [2.2 ± 0.1 vs. 2.6 ± 0.1 g/day, P < 0.05; basal fluid intake was not significantly different between genotypes: 5.4 ± 0.2 vs. 6.0 ± 0.2 ml/day, not significant (NS)]. The SGLT2 inhibitor modestly reduced body weight in both groups, and this response was similar between genotypes (Fig. 2E). Based on the values for food intake described above, 300 mg/kg of empagliflozin in the diet translated to a daily dose of ∼30–35 mg/kg body wt.
Fig. 2.
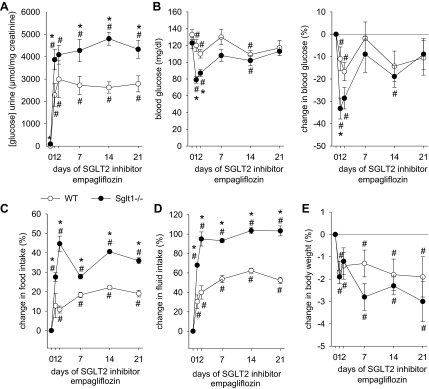
Chronic glucosuric response to SGLT2 inhibitor is enhanced 1.5- to 2-fold in Sglt1−/− vs. WT mice, associated with greater increases in food and fluid intake. Depicted are effects of empagliflozin application (300 mg/kg of diet) for 3 wk on urinary glucose/creatinine ratios (A), absolute levels of and changes in blood glucose (B), and changes in food intake (C), fluid intake (D), and body weight (E). Spontaneous urine collections were made, and blood was collected by tail snip in awake mice; food and fluid intake was measured in regular cages; n = 9–11/group. *P < 0.05 vs. WT by ANOVA and unpaired Student's t-test. #P < 0.05 vs. day 0 same group by paired Student's t-test.
SGLT2 inhibitor reduces renal FGR to 44% in WT and eliminates glucose reabsorption altogether in Sglt1−/− mice.
Following 3 wk of treatment with empagliflozin in the diet (see above), renal glucose handling was studied under terminal anesthesia in the late morning. Filtered glucose was computed as the product of plasma glucose and [3H]inulin clearance and compared with glucose excretion. Figure 3 depicts the relationship between filtered glucose and absolute renal glucose excretion (Fig. 3A), absolute renal glucose reabsorption (Fig. 3B), and FGR (Fig. 3C) in both genotypes. Chronic treatment with the SGLT2 inhibitor empagliflozin in the diet established values for FGR of 64 ± 2% in WT and 17 ± 2% in Sglt1−/− mice (P < 0.001). These effects were associated with free empagliflozin plasma concentrations of 1–2 nM in both genotypes (Fig. 3D), indicating early proximal luminal concentrations close to the reported IC50 value of 1.9 nM for this SGLT2 inhibitor in expression systems (4). To determine the ceiling for the glucosuric effect of empagliflozin, an additional single dose of empagliflozin (10 mg/kg ip) was given to some animals 1 h before the study. This additional dose increased the free plasma concentration of empagliflozin to 20–22 nM (Fig. 3D) and reduced FGR to 44 ± 3% in WT and to −1 ± 3% in Sglt1−/− mice (Fig. 3C). These values with high dose pharmacological SGLT2 inhibition in WT were similar to the mean FGR of 36 ± 8% previously reported in Sglt2−/− mice (17).
Fig. 3.
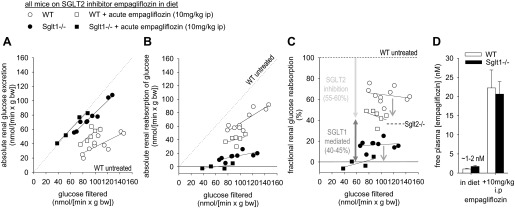
SGLT2 inhibitor empagliflozin reduces fractional renal glucose reabsorption to 44% in WT and completely prevented renal glucose reabsorption in Sglt1−/− mice. Inulin clearance studies were performed under terminal anesthesia in the late morning following empagliflozin application (300 mg/kg of diet) for 3 wk. Depicted is the relationship between filtered glucose and absolute renal glucose excretion (A), absolute renal glucose reabsorption (B), and fractional glucose reabsorption (C) in Sglt1−/− and WT mice. Each dot represents 1 clearance experiment period; n = 4–9/group. In A and B, the line of identity is included as a dashed line for ease of interpretation. Based on previous studies (3, 17), values for WT untreated mice are expected close to 0 for absolute renal glucose excretion (A), close to the line of identity for absolute renal glucose reabsorption (B), and close to 100% for fractional glucose reabsorption (C). D: free plasma concentrations of empagliflozin, corresponding to early tubular concentrations, were similar to reported IC50 for mouse SGLT2 (∼1–2 nM) (4), when the drug was given “in the diet.” Additional application of empagliflozin 1 h before the study increased free plasma concentrations to 20–22 nM and reduced fractional glucose reabsorption in WT by ∼55–60%, similar to the phenotype of untreated Sglt2−/− mice (17), with the remaining glucose reabsorption (∼40–45%) being mediated by SGLT1.
Combined gene knockout of Sglt1 and Sglt2 completely prevented renal glucose reabsorption.
Spontaneous collections of urine and blood revealed that absolute urinary glucose concentrations and urinary glucose/creatinine ratios were increased whereas blood glucose levels were reduced in Sglt1/2−/− vs. WT mice (Table 1). The urinary glucose and calorie loss in Sglt1/2−/− mice was associated with lower body weight, and greater food and fluid intake compared with WT mice. The increase in food intake and the previously reported lower serum levels of insulin (12) may contribute to the only modest reduction in body weight and blood glucose levels in Sglt1/2−/− mice. Higher values for hematocrit, plasma concentrations of sodium and aldosterone, as well as urine vasopressin/creatinine ratios provided indirect evidence for hemoconcentration in Sglt1/2−/− compared with WT mice (Table 1).
Table 1.
Plasma and urine parameters in mice deficient for both SGLT2 and SGLT1
| WT | Sglt1/2−/− | |
|---|---|---|
| Body weight, g | 29.6 ± 0.6 | 26.6 ± 0.9# |
| Urinary [glucose], mM | 1 ± 0.1 | 402 ± 34# |
| Urinary [glucose]/[creatinine], μmol/mg | 4 ± 1 | 4,906 ± 573# |
| Blood glucose, mg/dl | 115 ± 5 | 83 ± 3# |
| Food intake, g/24 h | 3.0 ± 0.1 | 4.3 ± 0.1# |
| Fluid intake, ml/24 h | 5.9 ± 0.1 | 15.1 ± 0.2# |
| Hematocrit, % | 48.1 ± 0.5 | 49.8 ± 0.3# |
| Plasma [Na+], mM | 153 ± 1 | 158 ± 2# |
| Plasma [aldosterone], pg/ml | 276 ± 42 | 1,311 ± 133# |
| Urine [vasopressin]/[creatinine], nmol/mmol | 110 ± 20 | 192 ± 34# |
Values are means ± SE; n = 9/group. WT, wild-type; SGLT, sodium-glucose cotransporter. Brackets indicate concentration.
P < 0.05 vs. WT.
Clearance studies were performed to determine the relationship between filtered glucose and absolute renal glucose excretion (Fig. 4A) and reabsorption (Fig. 4B) as well as FGR (Fig. 4C) in male and female Sglt1/Sglt2−/− mice. The clearance experiments revealed that glucose reabsorption was eliminated altogether when both SGLT2 and SGLT1 were absent.
Fig. 4.
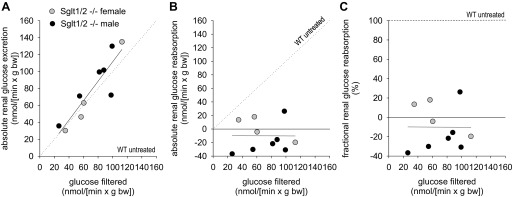
Combined gene knockout of Sglt1 and Sglt2 completely prevented renal glucose reabsorption. Inulin clearance studies were performed under terminal anesthesia. Depicted is the relationship between filtered glucose and absolute renal glucose excretion (A), absolute renal glucose reabsorption (B), and fractional renal glucose reabsorption (C) in male and female Sglt1/Sglt2 double knockout mice. Each dot represents 1 clearance experiment period. In A and B, the line of identity is included as a dashed line for ease of interpretation. SGLT2 and SGLT1 explain renal glucose reabsorption. Values expected for WT untreated mice are depicted for comparison (see legend for Fig. 3 for details).
DISCUSSION
The main findings of the current study are that SGLT2 and SGLT1 together can account for all glucose reabsorption in the kidneys of euglycemic mice. Consistent evidence is provided by pharmacological and gene knockout studies. Considering the presented data together with published results for Sglt1−/− and Sglt2−/− mice (3, 17), we propose that in kidneys of untreated nondiabetic mice, 97 and 3% of filtered glucose is reabsorbed via SGLT2 and SGLT1, respectively. During SGLT2 inhibition, the glucose load to the SGLT1-expressing S2/S3 segments of the proximal tubule is enhanced and a compensatory increase in SGLT1-mediated transport occurs. This explains why FGR is maintained at 40–50% in response to an SGLT2 inhibitor in euglycemic conditions.
When SGLT2 is inhibited, there is compensation by SGLT1. The compensation by SGLT1 was apparent with both acute and chronic application of the SGLT2 inhibitor. In metabolic cages, the compensation by SGLT1 in response to acute SGLT2 inhibition was indicated by the leftward shift and doubling of the maximum glucosuric response to empagliflozin in mice lacking SGLT1 compared with WT mice. These studies further revealed that the difference between the dose-response curves in Sglt1−/− and WT mice, which reflects the glucose reabsorption mediated via SGLT1 in WT mice, reached a maximum at an empagliflozin dose of ∼0.4 mg/kg po. This maximal difference was maintained at higher doses up to 10 mg/kg, consistent with a high selectivity of the inhibitor for SGLT2 vs. SGLT1 in this dose range (a reduction in SGLT2 to SGLT1 selectivity at higher doses would increase glucosuria in WT relative to Sglt1−/− mice and move the WT curve closer toward the Sglt1−/− curve). The compensation by SGLT1 with chronic application of the SGLT2 inhibitor was indicated by sustained 1.5- to 2-fold higher urinary glucose/creatinine ratios in Sglt1−/− compared with WT mice. The clearance studies showed that the differences in glucosuria in response to empagliflozin were due to differences in renal glucose reabsorption, which was consistently lower in Sglt1−/− compared with WT mice. The difference in FGR between Sglt1−/− and WT mice, i.e., the SGLT1-mediated FGR in WT, was ∼45% during inhibition of SGLT2 with empagliflozin. This difference was observed with chronic empagliflozin treatment in the diet as well as when additional empagliflozin was superimposed 1 h before clearance studies. These maneuvers induced free plasma, and thus early proximal tubule concentrations of empagliflozin of 1–2 nM (close to IC50 for SGLT2) and ∼20 nM, respectively, indicating that SGLT1 reabsorbed ∼45% of filtered glucose when SGLT2 inhibition was close to half-maximal or higher. The data are consistent with a high selectivity of the inhibitor for SGLT2 vs. SGLT1 in the doses and plasma concentrations tested and indirectly indicate that SGLT1 transport is saturated when SGLT2 is inhibited by 50% or more. High-dose empagliflozin lowered FGR in WT mice to 44%, which is close to the mean FGR of 38% previously reported in mice lacking SGLT2 (17). Mimicking the results obtained in Sglt1/Sglt2 double knockout mice, application of high-dose empagliflozin to Sglt1−/− mice completely prevented net renal glucose reabsorption, providing the first pharmacological and genetic evidence that SGLT2 and SGLT1 together account for all renal glucose reabsorption, at least under euglycemic conditions. The results further indicate that empagliflozin can reach and inhibit in vivo all SGLT2 that contributes to renal glucose reabsorption.
The current results, although performed in nondiabetic mice, support the concept that pharmacological inhibition of SGLT2 reduces renal glucose reabsorption, thereby lowering blood glucose levels, which is of therapeutic value in diabetes mellitus (2, 11, 19). This is consistent with recent genetic approaches in streptozotocin-diabetic mice, which showed that the gene knockout of SGLT2 (18) or a loss-of-function mutation in SGLT2 (10) mitigates hyperglycemia. In accordance with previous findings in nondiabetic Sglt2 gene knockout mice (17, 18), the SGLT2 inhibitor empagliflozin enhanced food and fluid intake in nondiabetic WT mice, possibly to compensate for the urinary glucose, caloric, and fluid loss, thereby maintaining fluid homeostasis and stabilizing body weight. We further observed that the enhanced glucosuric effect of SGLT2 inhibition in the absence of SGLT1 was associated with a stronger initial blood glucose-lowering effect and greater increases in food and fluid intake that presumably limited the body weight loss compared with WT mice. These results are consistent with the concept that combined renal inhibition of SGLT2 and SGLT1 is more glucosuric than inhibition of SGLT2 alone and confirm previous studies in diabetic Sglt1/Sglt2 double knockout mice (12). An increase in glucosuria will also increase diuresis, which likely contributed to the observed indirect evidence for hemoconcentration in nondiabetic Sglt1/2−/− compared with WT mice. Recent studies in streptozotocin-diabetic mice lacking Sglt2 (18) and in Akita diabetic mice treated with empagliflozin (16) illustrated and discussed the remarkably different effect of SGLT2 inhibition on glucosuria and, therefore, body weight and food intake in diabetic vs. nondiabetic conditions.
In summary, we show for the first time that SGLT2 and SGLT1 together can account for renal glucose reabsorption in euglycemic mice. Considering the presented data together with published results for Sglt1−/− and Sglt2−/− mice (3, 17), we conclude that in kidneys of untreated euglycemic mice, 97 and 3% of filtered glucose is reabsorbed via SGLT2 and SGLT1, respectively. During genetic or pharmacological inhibition of SGLT2, a compensatory increase in SGLT1-mediated transport occurs, which explains why renal FGR is maintained at 40–50%. The studies also provided evidence that the SGLT2 inhibitor empagliflozin can reach and inhibit in the kidney all SGLT2 that contributes to glucose reabsorption.
GRANTS
The authors were supported by National Institutes of Health Grants R01DK56248, R01HL94728, the UAB/UCSD O'Brien Center of Acute Kidney Injury NIH-P30DK079337 (to V. Vallon and S. C. Thomson and including a Pilot and Feasibility Grant to T. Rieg); the American Heart Association (Scientist Development Grant 10SDG2610034 to T. Rieg); a Carl W. Gottschalk Research Grant of the American Society of Nephrology to T. Rieg; a fellowship of the Manpei Suzuki Diabetes Foundation to T. Masuda; the Department of Veterans Affairs; and by Boehringer Ingelheim (to V. Vallon).
DISCLOSURES
V. Vallon has received within the past 12 months research grant support for basic science studies from Boehringer Ingelheim and Amylin. E. Mayoux is employed by Boehringer Ingelheim. K. Platt and D. R. Powell are employed by Lexicon.
AUTHOR CONTRIBUTIONS
Author contributions: T.R., T.M., M.G., E.M., K.A.P., D.R.P., S.C.T., H.K., and V.V. provided conception and design of research; T.R., T.M., M.G., and V.V. performed experiments; T.R., T.M., and V.V. analyzed data; T.R., T.M., S.C.T., and V.V. interpreted results of experiments; T.R. and V.V. prepared figures; T.R., T.M., M.G., E.M., K.A.P., D.R.P., S.C.T., H.K., and V.V. edited and revised manuscript; T.R., T.M., M.G., E.M., K.A.P., D.R.P., S.C.T., H.K., and V.V. approved final version of manuscript; V.V. drafted manuscript.
REFERENCES
- 1.Balen D, Ljubojevic M, Breljak D, Brzica H, Zlender V, Koepsell H, Sabolic I. Revised immunolocalization of the Na+-d-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295: C475–C489, 2008 [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA, Davidson JA, del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 14: 5–14, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H. Na+-d-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion Diabetes 61: 187–196, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab 14: 83–90, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Heise T, Seewaldt-Becker E, Macha S, Hantel S, Pinnetti S, Seman L, Woerle HJ. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks' treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab 15: 613–621, 2013 [DOI] [PubMed] [Google Scholar]
- 6.Horiba N, Masuda S, Takeuchi A, Takeuchi D, Okuda M, Inui K. Cloning and characterization of a novel Na+-dependent glucose transporter (NaGLT1) in rat kidney. J Biol Chem 278: 14669–14676, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Hummel CS, Lu C, Loo DF, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/d-glucose co-transporters. Am J Physiol Cell Physiol 300: C14–C21, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komoroski B, Vachharajani N, Boulton D, Kornhauser D, Geraldes M, Li L, Pfister M. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther 85: 520–526, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Liu JJ, Lee T, DeFronzo RA. Why Do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in humans? Diabetes 61: 2199–2204, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ly JP, Onay T, Sison K, Sivaskandarajah G, Sabbisetti V, Li L, Bonventre JV, Flenniken A, Paragas N, Barasch JM, Adamson SL, Osborne L, Rossant J, Schnermann J, Quaggin SE. The sweet pee model for Sglt2 mutation. J Am Soc Nephrol 22: 113–123, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab 95: 34–42, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, Doree D, Jeter-Jones S, Mseeh F, Rodriguez LA, Harris A, Buhring L, Platt KA, Vogel P, Brommage R, Shadoan MK, Sands AT, Zambrowicz B. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab 304: E117–E130, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Rieg T, Gerasimova M, Murray F, Masuda T, Tang T, Rose M, Drucker DJ, Vallon V. Natriuretic effect by exendin-4, but not the DPP-4 inhibitor alogliptin, is mediated via the GLP-1 receptor and preserved in obese type 2 diabetic mice. Am J Physiol Renal Physiol 303: F963–F971, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santer R, Calado J. Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 5: 133–141, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, Shalayda K, Demarest K, Rothenberg P. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab 13: 669–672, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol (First published November 13, 2013). 10.1152/ajprenal.00520.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vallon V, Sharma K. Sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Curr Opin Nephrol Hypertens 19: 425–431, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011 [DOI] [PubMed] [Google Scholar]