

Abstract
Farnesoid X receptor (FXR) is a bile acid nuclear receptor described through mouse knockout studies as a tumor suppressor for the development of colon adenocarcinomas. This study investigates the regulation of FXR in the development of human colon cancer. We used immunohistochemistry of FXR in normal tissue (n = 238), polyps (n = 32), and adenocarcinomas, staged I–IV (n = 43, 39, 68, and 9), of the colon; RT-quantitative PCR, reverse-phase protein array, and Western blot analysis in 15 colon cancer cell lines; NR1H4 promoter methylation and mRNA expression in colon cancer samples from The Cancer Genome Atlas; DNA methyltransferase inhibition; methyl-DNA immunoprecipitation (MeDIP); bisulfite sequencing; and V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) knockdown assessment to investigate FXR regulation in colon cancer development. Immunohistochemistry and quantitative RT-PCR revealed that expression and function of FXR was reduced in precancerous lesions and silenced in a majority of stage I-IV tumors. FXR expression negatively correlated with phosphatidylinositol-4, 5-bisphosphate 3 kinase signaling and the epithelial-to-mesenchymal transition. The NR1H4 promoter is methylated in ∼12% colon cancer The Cancer Genome Atlas samples, and methylation patterns segregate with tumor subtypes. Inhibition of DNA methylation and KRAS silencing both increased FXR expression. FXR expression is decreased early in human colon cancer progression, and both DNA methylation and KRAS signaling may be contributing factors to FXR silencing. FXR potentially suppresses epithelial-to-mesenchymal transition and other oncogenic signaling cascades, and restoration of FXR activity, by blocking silencing mechanisms or increasing residual FXR activity, represents promising therapeutic options for the treatment of colon cancer.
Keywords: bile acids, colon cancer, farnesoid X receptor, nuclear receptor, promoter methylation
colon cancer is the third most common cancer and the third leading cause of cancer-related deaths in the US. Two major risk factors for colon cancer development are high-fat and/or low-fiber diet (15, 19, 41). Bile acids (BAs) are amphipathic molecules essential for digestion and absorption of fats. High-fat diet increases BA load in the intestine whereas low dietary fiber prolongs gastrointestinal transit time, thus collectively increasing the level and time of BA exposure (2, 33). Although BAs are essential for lipid absorption, high concentration of BAs is linked to increased colon tumorigenesis (2, 17, 27). Indeed, patients with colorectal cancer increased fecal BA excretion (11, 43).
Farnesoid X receptor (FXR) is a ligand-activated transcription factor belonging to the nuclear receptor superfamily. FXR is highly expressed in liver and intestine and BAs are its endogenous ligands (28). FXR critically regulates the homoeostasis of BAs, including BA synthesis, transport, and intestinal reabsorption, as well as the free intracellular concentration of BAs to prevent their accumulation to cytotoxic levels (9, 12, 26, 39, 47). FXR deficiency in mice promotes the development of intestinal tumors (21, 25), implicating FXR as a tumor suppressor. Furthermore, overexpression of a constitutively active form of FXR decreases tumor size in mouse xenograft models (25). The mechanism of tumor-suppressor effects of FXR is not defined but may be mediated through the protection of colonic epithelium from inflammation and BA toxicity by upregulating intracellular BA binding proteins and efflux transporters while downregulating influx transporters and de novo synthesis of BAs (8). However, FXR also has antitumorigenic functions independent of its regulation of BA homeostasis (25). For example, FXR deficiency increases susceptibility to colon cancer development by increasing epithelial permeability to bacteria, promoting WNT/β-catenin signaling, increasing intestinal inflammation, and protecting against genotoxic compounds (8, 13, 25, 44).
Studies, on small samples sizes, have shown that FXR was silenced in later stages of colon cancer, implicating FXR as a marker of tumor malignancy (6, 16). Although polymorphisms within the FXR gene have been associated with decreased function in intrahepatic cholestasis of pregnancy (ICP) (23), no clinically known mutations exist within the FXR gene to explain decreased FXR expression or function in human colon cancer. The present study was conducted to identify 1) the timing of FXR silencing during colon cancer development in humans and 2) the mechanism(s) of FXR silencing. The results of this study suggest a potential therapeutic strategy for preventing and/or inhibiting colon cancer promotion by suppressing colon cancer-associated FXR silencing or activation of remnant FXR in surrounding healthy tissues.
MATERIALS AND METHODS
Human colon cancer samples.
Immunohistochemistry (IHC) analysis was done on paraffin-embedded normal colon tissue, polyps, and adenocarcinomas obtained with IRB approval from University of Texas MD Anderson Cancer Center [MDACC; Protocol Numbers ID99-296(38) and LAB09-0373] and University of Arizona Gastrointestinal SPORE (P50 CA95060; protocol no. 10-696-01). Tissue cores consisted of “real normal” (right and left tissues from patients with no history of cancer; n = 47), polyps with “matched normal” (n = 32), and adenocarcinomas stage I (n = 43), II (n = 39), III (n = 68), and IV (n = 9) with matched normal (n = 114). Standard IHC was performed by use of an automated IHC Leica Bond-Max system (Leica Microsystems, Buffalo Grove, IL). Anti-FXR mouse monoclonal antibody (PP-A9033A, R&D Biosystems) was used and labeling was detected by use of a Bond Polymer Refined Detection Kit (DS9800, Leica Microsystems). Staining was quantified by the following equation:
β-Catenin IHC analysis of polyps (n = 9) and adenocarcinomas (n = 2) was done with anti-β-catenin mouse monoclonal antibody (PA0083, Leica Microsystems) and the same detection system listed above. All IHC slides were determined positive or negative for nuclear FXR and β-catenin labeling and validated by a gastrointestinal pathologist (D. Maru).
cDNA from normal and cancerous colons (n = 5–18) was obtained from OriGene Technologies (Rockville, MD; http://www.origene.com; HCRT501). mRNA levels of FXR, organic solute transporter (OST)α, and OSTβ were measured and normalized to hβ-actin.
TCGA analysis.
Clinical patient data, NR1H4 (FXR gene name) expression, NR1H4 promoter methylation, microsatellite instability (MSI), V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation, hypermutated tumor status, and CpG island methylator phenotype (CIMP) were downloaded from published Cancer Genome Atlas (TCGA) Network work (4) and reanalyzed for correlation analysis. Full methylation of the FXR CpG island is designated by β-value > 0.6.
Cell line study.
FXR expression was studied in a panel of colon cancer cells lines representing transition from highly differentiated to poorly differentiated, along with the levels of E-cadherin and vimentin, markers of epithelial-to-mesenchymal transition (EMT). NCI-H508 (duplicates), NCI-H716, NCI-H747, Colo320DM, DLD-1, HCT-15, and Colo201 cells were grown in RPMI-1640 medium; HT-29 and HCT-116 (duplicates) were grown in McCoy's 5A modified medium; Caco-2, SK-CO-1, and LS174T were grown in Eagle's minimum essential medium; LoVo cells were grown F-12K medium; SW620, SW48, SW480, SW948, SW403 (duplicates), SW1116, and SW1417, were all grown in Leibovitz's L-15 medium. All media were supplemented with 10% FBS without antibiotics.
HT-29 and SW620 cells were used for azacytidine (AZA) treatment, methyl-DNA immunoprecipitation (MeDIP) analysis, and DNA methyltransferase (DNMT) siRNA experiments. Cells were cultured in media listed above, supplemented with both 10% FBS and 1% penicillin-streptomycin. These experiments were done before the recommendation for removal of antibiotics from complete media.
HT-29, SW620, SW1116, SK-CO-1, Colo201, and Colo320DM cells were used for small interfering KRAS (siKRAS) experiments with cells were cultured as described above with 10% FBS and without antibiotics.
RPPA.
Proteins and phosphoproteins from a panel of colon cancer cell lines were extracted and quantified by reverse-phase protein array (RPPA), as previously described (42).
The RPPA cluster heat map of 15 cell lines based on the 172 proteins was generated with Partek Genomics Suite v 6.6 in which each column is a protein marker and each row a colon cancer cell line annotated with phosphatidylinositol-4,5-bisphosphate 3 kinase (PI3K) catalytic subunit α isoform (PI3KCA), and KRAS mutation status. Both rows and columns were clustered by Ward's method based on rank (Spearman) dissimilarity (44a).
Azacytidine treatment.
HT-29 and SW620 cells were plated at 106 cells in 10-cm plates and treated with or without 2 μg/ml AZA (Sigma, St. Louis, MO), a DNMT inhibitor, for 3 days, with fresh solutions prepared each day (n = 3). RNA was extracted as described below. COL1A2 and FXR mRNA levels were determined by RT-quantitative PCR (RT-qPCR). COL1A2 gene encodes for the collagen-1α2 protein and has been shown to be methylated in colon cancer cell lines, SW620 (36). Therefore, the expression of this gene in response to AZA treatment was used as a positive control.
MeDIP assay.
MeDIP assay was performed as described (45). Briefly, ∼4 μg of sheared DNA was incubated overnight at 4°C with either the 5-methylated-cytosine (5-mC) antibody (Diagenode, MAb-335MEC-100), or mouse IgG negative control. Then DNA samples with antibody were incubated with prewashed Dynabeads at 4°C for 5 h with rotation. The DNA-antibody-beads complex was washed and DNA were eluted. Immunoprecipitated DNA was phenol/chloroform extracted and dissolved in Tris-EDTA (TE) buffer.
A CpG island located roughly 3 kb upstream of the FXR gene transcription start site (TSS) was identified by MethPrimer (Fig. 1; Ref. 18). This region was assessed for DNA methylation by MeDIP analysis. DNA precipitated by the 5-mC antibody was analyzed by qPCR using SYBR green chemistry (n = 2). Primers were designed to amplify a methylated CpG islands within the FXR promoter and the COL1A2 promoter (positive control; Ref. 36). Primer sequences are listed in Table 1. Primers designed to amplify a nonmethylated housekeeping gene (UBE2B) were used as a negative control and have been previously reported (40).
Fig. 1.
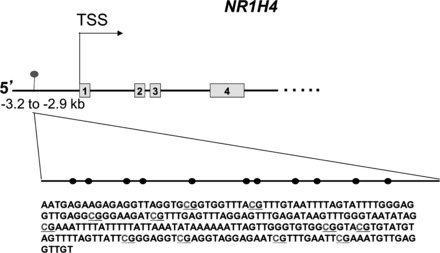
Predicted CpG island within the human farnesoid X receptor (FXR) (NR1H4) promoter. There is a predicted CpG island within the NR1H4 promoter determined by MethPrimer CpG island predictor software. This island is located approximately −3.2 to −2.9 kb upstream of the transcription start site (TSS). The CpG island is illustrated as a dashed line and CpG sites as black circles. There are 11 predicted CpG sites within this CpG island. The predicted sequence of this island after bisulfite sequencing has been determined (data not shown). The Cancer Genome Atlas (TCGA) methylation data confirm this region as a CpG site.
Table 1.
Primers for MeDIP and RT-qPCR
| Primer Name | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| MeDIP primers | ||
| FXR CpG | GTTTGAGACAAGCCTGGGCAACAT | ATTTCGGGTTCAAGCGGTTCTCCT |
| COL1A2 CpG | TGCAGACAACGAGTCAGAGTTTCC | GGGCTGGCTTCTTAAATTGGTTCC |
| RT-qPCR primers | ||
| FXR | TGCATTGAAGTTGCTCTCAGGT | CGCCTGACTGAATTACGGACA |
| OSTα | CTACACCTGGGTGAGCAGAA | AGAGGAATAGGGAGGCGAAC |
| OSTβ | GCAGCTGTGGTGGTCATTAT | TAGGCTGTTGTGATCCTTGG |
| DNMT1 | TGTACCGAGTTGGTGATGGTGTGT | TGCTGCCTTTGATGTAGTCGGAGT |
| DNMT 3b | ATTGTTTGATGGCATCGCGACAGG | ACAGCAATGGACTCCTCACACACT |
| KRAS | GGGGAGGGCTTTCTTTGTGTA | GTCCTGAGCCTGTTTTGTGTC |
RT-qPCR, RT-quantitative PCR; meDIP, methyl-DNA immunoprecipitation; FXR, farnesoid X receptor; OSTα, organic solute transporter α; OSTβ, organic solute transporter β; DNMT, DNA methyltransferase; KRAS, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog.
siRNA knockdown.
DNMT1 and 3B are enzymes associated with aberrant DNA methylation (34). Therefore, siRNA knockdown of DNMT1 and/or 3B in SW620 cells was done. SMARTpool siRNAs to knock down expression of DNMT1 or DNMT3B, and nontargeting siRNA, were obtained from Dharmacon (Lafayette, CO). SW620 cells were plated at 30% confluence in a six-well plate (n = 2) and reverse transfected with siDMNT 1, siDNMT 3B, or nontargeting siRNA with use of Turbofect siRNA transfection reagent (Fermentas, Glen Burnie, MD). After 96 h, total RNA was extracted and mRNA of DMNT 1, DMNT 3B, FXR, and COL1A2 (positive control) were measured by RT-qPCR analysis.
HT-29, SW620, SW1116, SK-CO-1, Colo201, and Colo320DM cells (106 cells/well in 6-well plates) were reverse transfected with 40 nM siKRAS or nontargeting siRNAs (Dharmacon). After 72–96 h, total RNA was extracted and mRNA of FXR, KRAS, and actin were measured by RT-qPCR analysis.
Western blot.
Thirty micrograms of whole cell lysates from a panel of colon cancer cell lines listed above were used for analysis. Primary anti-FXR mouse monoclonal antibody (R&D Biosystems), anti-E-cadherin mouse monoclonal antibody (Cell Signaling, Boston, MA), anti-vimentin rabbit monoclonal antibody (XP; Cell Signaling) and anti-actin polyclonal goat antibody (Santa Cruz Biotechnology, Dallas, TX) and secondary anti-rabbit, -mouse, or -goat horseradish peroxidase-conjugated antibodies (Santa Cruz Biotechnology) were used for protein labeling. The Western Lighting Plus ECL (PerkinElmer, Waltham, MA) was used for protein detection. Western blot images were scanned and band densities quantified by AlphaInnotech's AlphaView image analysis software (ProteinSimple, Santa Clara, CA).
RNA extraction and RT-qPCR.
Complimentary DNA prepared from human colon samples (OriGene) were used to measure mRNA levels of FXR, OSTα, and/or OSTβ. Total RNA from colon cancer cells line experiments was done with TRIzol reagent (Invitrogen, Carlsbad, CA) and cDNA prepared by standard RT-PCR methods with random primers (Fermentas). cDNA prepared from AZA-treated cells, DNMT 1 and 3B siRNA experiments, and siKRAS experiments were used to measure mRNA levels of FXR, COL1A2, and/or KRAS. The primer sequences are listed in Table 1. The primer sequences for COL1A2 are as previously reported (36). All real-time qPCR reactions were done using standard SYBR green chemistry and an ABI Prism 7900 Detection system (Applied Biosystems, Foster City, CA). The mRNA levels of these genes were normalized to hβ-actin or hGAPDH mRNA levels.
gDNA extraction.
Genomic DNA (gDNA) was extracted from HT-29 and SW620 for MeDIP analysis. Briefly, trypsinized cancer cells lysed in DNA lysis buffer (10 mM Tris pH 7.5, 10 mM EDTA, 10 mM NaCl, 0.5% sodium dodecyl sulfate) plus 1 mg/ml proteinase K, and incubated at 55°C for 5 h to overnight, and ethanol precipitated. GDNA was further purified by standard phenol/chloroform extraction methods. Purified gDNA was briefly sonicated to 200–1,000 bp, and column purified by use of standard PCR purification kits (Fermentas).
Statistical analysis.
Quantitative PCR data are expressed as means ± SE. One-way ANOVA was used with adjustment for multiple comparisons within model using Tukey's method. Unpaired two-way Student's t-test was used when comparing two groups.
FXR, E-cadherin, vimentin, and Western blot bands were plotted as a ratio over actin loading control. E-cadherin and vimentin ratios are plotted against FXR ratios. Spearman correlation analysis was used to assess the correlation between continuous variables.
All TCGA data points are plotted with single lines representing group means. Statistical analysis comparing the means between two groups was done by Student's t-test and Mann-Whitney U-test.
The expression levels of FXR and 172 other proteins in 15 colorectal cancer cell lines were profiled RPPA. The correlations between FXR densitometry ratio and 172 other proteins were assessed by using Spearman correlations on a protein-by-protein basis. To account for multiple testing, we estimated the false discovery rate (FDR) by the beta-uniform mixture method (32).
Linear mixed models with fixed effect of tissue groups were used to assess the differences in the expression of FXR between and among groups as determined by IHC analysis. A subject/individual-level random effect was included in the linear model where appropriate to account for the correlation between the matched pairs of normal and tumor or polyp for the same samples. We also examine for batch effects of three sets of data (MDACC colorectal cancer, Arizona colorectal cancer, MDACC polyp) and found no significant batch effect. Pairwise comparisons between groups were carried out by least squares estimate of the means with adjustment for within-model multiple comparisons by Tukey's method. We used quantile-quantile plots to examine the normality assumption of the residuals of the linear models. IHC analysis was converted to log scale for statistical analysis but reported as arithmetic values in figures. All the analyses were performed with SAS 9.3 and GraphPad Prism 6.01.
RESULTS
FXR expression decreased in human colon cancers.
FXR labeling is less in the left colon compared with the right, in polyps compared with matched normal colon, and in both stage II adenocarcinomas compared with matched normal samples (Fig. 2A). There was no statistical difference in FXR expression between normal human colons (real normal) and matched normal tissues of polyps and adenocarcinomas (data not shown). Therefore, data from real normal and matched normal tissues were combined for analysis. The expression of FXR decreased down the colonic tract with the cecum (highest FXR expression), ascending colon, and transverse colon having statistically higher levels of FXR expression than their immediate distal segments (Fig. 2B; P < 0.05). The trend of FXR downregulation between normal tissue, polyps, and stage I, II, II, and IV adenocarcinomas was similar between the right and left colon (data not shown). Therefore, data from right and left colon were combined for analysis.
Fig. 2.
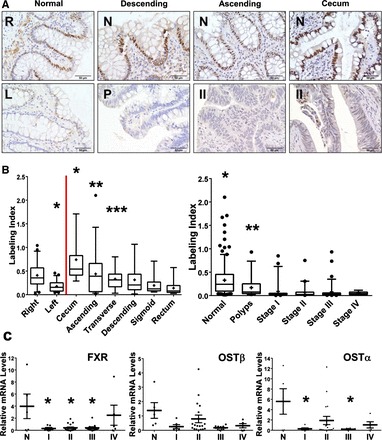
Expression of FXR and FXR-target genes in human colon cancer samples. A: FXR immunohistochemistry (IHC) images of right (R) and left (L) real normal colon (no colon cancer history) from same patient; polyp (P) of descending colon with matched normal (N) tissue, and stage II (II) adenocarcinomas of the ascending colon and cecum and their matched normal (N) tissues. B: quantification of FXR labeling from IHC analysis. The % positively labeled nuclei plus intensity of nuclei staining, reported as the labeling index. Theoretical maximum labeling index would be 3 and minimum 0.0001. ●, Outliers from 5–95% percentile. C: relative mRNA levels of FXR, organic solute transport (OST)α, and OSTβ in human colon cancer samples (n = 5–18). Data are expressed as means ± SE. ●, Outliers from range defined by Tukey's multiple comparison test. *P < 0.05.
To associate FXR expression with development of polyps, a low sample number of polyps and two colon tumors (positive control) IHC samples were labeled for β-catenin nuclear localization. Two of nine (22.2%) polyp samples stained positively for nuclear β-catenin, compared with two of two colon tumors (100%; Table 2A). Only one of nine polyps showed both a decrease in FXR expression (defined by decreased expression > 50% from normal) and positive nuclear localization of β-catenin (Table 3).
Table 2.
β-Catenin-positive polyp and tumor samples
| Polyps | Tumor | |
|---|---|---|
| Samples with positive nuclear β-catenin | 2 | 2 |
| Total samples | 9 | 2 |
| % Positive | 22.2% | 100% |
Table 3.
FXR LI and positive/negative β-catenin of colon polyps
| Patient | Normal FXR LI | Polyp FXR LI | % of Normal | Polyp Nuclear β-Catenin |
|---|---|---|---|---|
| 1 | 0.586 | 0.069 | 12 | + |
| 2 | 0.59 | 0.042 | 7 | − |
| 3 | 0.449 | 0.932 | 208 | − |
| 4 | 0.072 | 0.63 | 875 | − |
| 5 | 0.744 | 0.164 | 22 | − |
| 6 | 0.302 | 0.073 | 24 | − |
| 7 | 0.378 | 0.254 | 67 | + |
| 8 | 0.769 | 0.349 | 45 | − |
| 9 | 0.536 | 0.287 | 54 | − |
LI, labeling index.
FXR mRNA levels were reduced 6- to 10-fold stages I, II, and III colon adenocarcinoma (Fig. 2C), indicating that FXR silencing occurs at the transcriptional level. FXR mRNA in stage IV samples was not reduced compared with normal, likely because of small sample size. Reduction of mRNA levels of FXR target genes OSTα and OSTβ was only significant for OSTα mRNA for stage I and III. There was an overall decreased trend in these target genes all four stages, but most were found not to be statistically significant, likely because of the low sample number.
FXR expression is negatively correlated to vimentin and PI3K signaling.
There was no statistically significant positive or negative correlation between FXR, E-cadherin, or vimentin expression in a panel of colon cancer cell lines (Fig. 3, A and B). However, there is a subset of vimentin (≥0.25 ratio of vimentin/actin) samples that negatively correlated to FXR expression. It is clear that many of the colon cancer cell lines labeled negative for vimentin expression (Fig. 3A; designated < 0.25 vimentin/actin ratio). Therefore, using samples that label positive for vimentin (≥0.25 ratio of vimentin/actin; Fig. 3C), FXR was significantly and negatively correlated with vimentin expression (r = −0.864, P = 0.001).
Fig. 3.
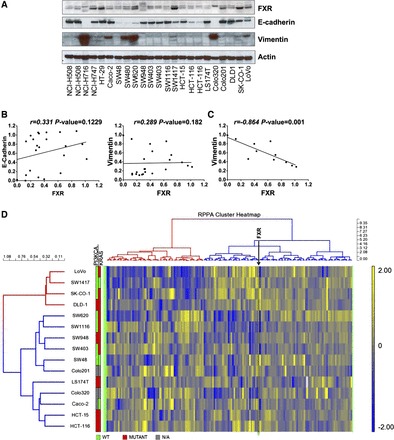
FXR Western blot and reverse-phase protein array (RPPA) cluster analysis and in human colon cancer cell lines. A: Western blot analysis of FXR, E-cadherin, vimentin, and actin in a panel of colon cancer cell lines. B: densitometry ratios of E-cadherin or vimentin over actin were plotted on the y-axis against FXR ratios on the x-axis. Correlation coefficients and P values are from linear regression and Spearman correlation analysis. C: a subset of samples (≥0.25 vimentin/actin ratio) appeared to have a linear and negative correlation to FXR expression. Correlation analysis was redone by using samples with ≥0.25 vimentin/actin ratio. The cells included in this third analysis were NCI-H716, HT-29, Caco-2, SW480, SW620, SW1417, Colo320, SK-CO-1, and LoVo. D: FXR/actin ratios were used for cluster analysis and heat map generation with RPPA data from colon cancer cell lines. Blue is relatively low (−2) and yellow high (+2) values. Cells cluster as high FXR (cluster red), including LoVo, SW1417, SK-CO-1, and DLD-1 cells, and low FXR (cluster blue), SW620, SW1116, SW948, SW403, SW48, Colo201, LS174T, Colo320, Caco-2, HCT-15, and HCT-116 cells. RPPA genes also clustered into cluster 1 (red) and cluster 2 (blue). The FXR column is indicated by ↓ within cluster 2. Mutational analysis of phosphatidylinositol-4, 5-bisphosphate 3 kinase (PI3K) catalytic subunit α isoform (PI3KCA) and V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) for each cell line is also shown in this figure. Red, green, and gray boxes indicate mutant, wild-type, or unavailable genotypes.
RPPA data from colon cancer cells were used for cluster analysis with FXR expression measured by Western blot. The colon cancer cells clustered into a high-FXR group (cluster red), including LoVo, SW1417, SK-CO-1, and DLD-1 cells, and low FXR group (cluster blue), including Colo320, Caco-2, HCT-116, HCT-15, SW948, SW403, SW1116, LS174T, SW48, and SW620 cells (Fig. 3D). Furthermore, RPPA genes separated into two clusters: cluster 1 (red) and cluster 2 (blue). The FXR column lies within cluster 2 (indicated by ↓ and labeled FXR). Mutational status (green = WT and red = mutant genotypes) of PI3KCA and KRAS, two commonly mutated genes in colon cancer (1), showed that four of eight cells (with PI3KCA mutation status) in cluster 2 (low FXR expression) carried a mutation in their PI3KCA gene compared with only one of four cell lines in cluster 1. There appeared to be no difference in KRAS mutation status between these two clusters. Spearman correlation analysis comparing FXR protein levels measured by RPPA analysis identified 23 and 5 proteins that significantly correlated, either positively (positive r value) or negatively (negative r value), respectively, with FXR expression when using a FDR cutoff values of 20 and 10% (Table 4). The most statistically significant positively and negatively correlated genes were BCL-x and eEF2. It should be noted that many of the mediators of PI3K signaling, including PI3KCA (PI3Kp110α) and mammalian target of rapamycin, negatively and significantly correlated to FXR expression.
Table 4.
Correlation of FXR expression with signaling pathways in colon cancer cell lines by RPPA analysis
| Signaling Protein | R | P value | FDR 0.1 | FDR 0.2 |
|---|---|---|---|---|
| Bcl.X | 0.6821 | 0.0065 | 0 | 1 |
| Bid | 0.6714 | 0.0077 | 0 | 1 |
| HER2 pY1248 | 0.6286 | 0.0142 | 0 | 1 |
| Collagen VI | 0.6036 | 0.0195 | 0 | 1 |
| HER2 | 0.6000 | 0.0204 | 0 | 1 |
| EphA2 | 0.5857 | 0.0243 | 0 | 1 |
| HER3 | 0.5714 | 0.0286 | 0 | 1 |
| ACC1 | −0.5607 | 0.0323 | 0 | 1 |
| FAK | −0.5893 | 0.0232 | 0 | 1 |
| X4EBP1 pT37_T46 | −0.5893 | 0.0232 | 0 | 1 |
| p27 | −0.6071 | 0.0187 | 0 | 1 |
| eEF2K | −0.6179 | 0.0163 | 0 | 1 |
| AMPKα | −0.6179 | 0.0163 | 0 | 1 |
| ACC pS79 | −0.6357 | 0.0129 | 0 | 1 |
| X4E.BP1 | −0.6429 | 0.0117 | 0 | 1 |
| PDK1 pS241 | −0.6500 | 0.0106 | 0 | 1 |
| mTOR | −0.6643 | 0.0086 | 0 | 1 |
| p70S6K | −0.6786 | 0.0069 | 0 | 1 |
| Cyclin E1 | −0.7179 | 0.0036 | 1 | 1 |
| PI3K.p110.α | −0.7536 | 0.0018 | 1 | 1 |
| Tuberin | −0.7571 | 0.0016 | 1 | 1 |
| JNK2 | −0.7571 | 0.0016 | 1 | 1 |
| eEF2 | −0.9571 | 0.0000 | 1 | 1 |
RPPA, reverse-phase protein array.
DNA methylation regulates FXR expression.
Mutation of the NR1H4 gene was first investigated as a potential cause of FXR silencing in three different cell lines: HT-29, Caco-2, and SW620. These cells show different baseline expression of FXR and no mutations within the entire gene and 5-kb promoter to account for FXR silencing in colon cancer (data not shown). TCGA data shows only two colon cancer samples have a NR1H4 gene variant and no samples have loss of NR1H4 copy number indicating the conservation of the NR1H4 gene locus in colon tumors (4). Sequence analysis of the FXR promoter revealed a putative CpG island located in the promoter of NR1H4 gene (Fig. 1). This island is located ∼3 kb upstream of the NR1H4 gene TSS and has 11 predicted CpG islands. Complete methylation of this site was observed in ∼12% TCGA colon cancer samples (4).
A panel of cDNAs prepared from 11 different colon cancer cell lines treated with a DNMT inhibitor, AZA, was used to measure mRNA levels of FXR. This preliminary screen revealed 6 of 11 of the colon cancer cell lines showed over a 1.5-fold increase (relative threshold for significance) in FXR expression, ranging from 1.7- to 233-fold (1.2- and 50-fold for HT-29 and SW620 cells), after AZA treatment. To confirm this, the colon cancer cell lines HT-29 and SW620 were treated with AZA and mRNA levels of FXR and COL1A2 were measured (n = 3, Fig. 4A). AZA treatment significantly increased mRNA levels of both FXR and COL1A2 in SW620 cells six- and eightfold, respectively (*P < 0.05), but not in HT-29 cells.
Fig. 4.
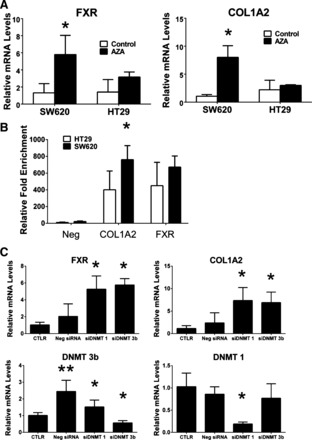
FXR expression in response to inhibition of DNA methylation and methyl-DNA immunoprecipitation (MeDIP) analysis of FXR promoter. A: relative mRNA levels of FXR and COL1A2 in HT-29 and SW620 cells treated with 2 μg/ml azacytidine (AZA) for 3 days (n = 3). B: immunoprecipitation of genomic DNA (gDNA) from HT-29 and SW620 cells with antibody against 5-methylated-cytosine (5-mC) followed by quantitative PCR analysis of MeDIP. A CpG island located in COL1A2 promoter (COL1A2 no. 1) was analyzed as a positive control and an unmethylated region (UBE2B) for the negative control (Neg). Values were recorded from replicate PCR reactions and are reported as fold over IgG (y-axis). C: relative mRNA levels of FXR and COL1A2 in SW620 cells after siRNA knockdown of DNMT 1 and 3B compared with negative siRNA controls. Messenger RNA levels of DNMT 1 and DNMT 3B were nearly 20% that of the nonspecific siRNA control. CTLR, nontransfected control. Data are expressed as means ± SE. *P < 0.05.
Methylation of the FXR promoter CpG island was assessed by MeDIP analysis on gDNA isolated from HT-29 and SW620 cells. Identification of a methylated CpG island near the TSS of COL1A2 gene was first confirmed (36). A housekeeping gene region that is not methylated (UBE2B) was used as a negative control for this assay (40). The results confirmed methylation COL1A2 promoter and the FXR gene promoter in HT-29 and SW620 cells, with relative enrichment levels of 400- and 600-fold over IgG, but not negative control region (Fig. 4B, *P < 0.05).
SW620 cells have lower basal levels of FXR expression compared with HT-29 cells and responded to AZA treatment (Fig. 4A); therefore, these cells were used for DNMT siRNA knockdown to determine the molecular machinery responsible for FXR downregulation. Knockdown of DNMT 1 and 3B by >80% (Fig. 4C; *P < 0.05) was sufficient to increase mRNA levels of FXR 6.7- and 7.3-fold, respectively, and positive control gene COL1A2 7- and 6.6-fold, respectively (Fig. 4C; *P < 0.05).
Methylation of FXR promoter in clinical colon tumors.
As noted previously, TCGA data revealed that roughly 12% of colon tumors have a fully methylated FXR promoter (determined by a methylation β value > 0.6). Clinical methylation and expression of NR1H4, KRAS mutational status, MSI, hypermutated status, and CIMP were obtained from previous report (4). Data analysis showed the methylation of the FXR promoter does not correlate with FXR expression (Fig. 5A), and there was no statistically significant difference in FXR expression in tumors with fully methylated promoters (Fig. 5B). FXR expression also did not significantly change in tumors with KRAS mutations, MSI-high (MSI-H) status, hypermutated status, or CIMP (Fig. 6, A–D).
Fig. 5.
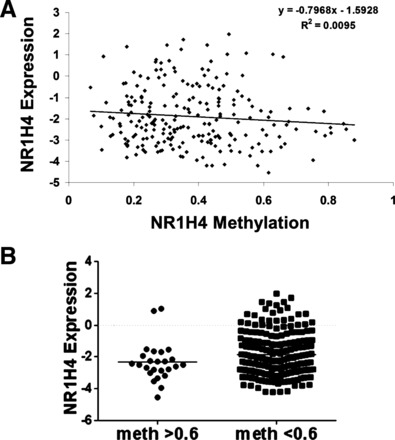
TCGA data analysis of the correlation of FXR promoter methylation and expression. A: correlation analysis of NR1H4 promoter methylation (x-axis) and NR1H4 expression (y-axis). There was no correlation between FXR promoter methylation and expression. B: comparison of FXR expression in tumors that had completely methylated FXR promoters (β-value > 0.6) or partially methylated/unmethylated. There is no statistically significant difference between these 2 groups.
Fig. 6.
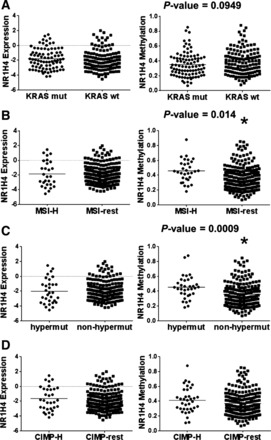
TCGA analysis of the levels of NR1H4 expression and promoter methylation in different tumor subtypes. Levels of NR1H4 expression and promoter methylation in KRAS WT (A), microsatellite instability (MSI)-high (MSI-H), and non-MSI-high (MSI-rest) (B), and hypermutated (hypermut) and nonhypermutated (non-hypermut) colon tumors (C). All sample values are plotted in these graphs and the line indicates the mean of all the samples. D: levels of NR1H4 expression and promoter methylation in CpG island methylator phenotype (CIMP)-high (CIMP-H) and nonCIMP-H (CIMP-rest) colon tumors. *P < 0.05 by Mann-Whitney comparisons.
TCGA data showed no statistically significant difference in the frequency of full FXR promoter methylation (β-value > 0.6) and KRAS mutation status, MSI-H status, hypermutated status, or CIMP status (Table 5). Partial methylation of CpG islands has been shown to have transcription inhibition effects (5, 10). Taking this into consideration, we assessed differences in the degree of FXR promoter methylation. Levels of methylation of FXR promoter was statistically higher in MSI-H and hypermutated but not KRAS mutant or CIMP tumors (Fig. 6, A–D). Interestingly, although not statistically significant, there was a decreased trend in FXR promoter methylation in KRAS mutant vs. nonmutant tumors (Fig. 6A).
Table 5.
Frequency of NR1H4 promoter methylation with different colon tumor subtypes
| NR1H4 meth β > 0.6 | NR1H4 meth β < 0.6 | Frequency β > 0.6 | Fisher Exact Test | |
|---|---|---|---|---|
| Total | 26 | 210 | 0.124 | |
| KRAS wt | 15 | 113 | 0.682 | 0.37 |
| KRAS mut | 7 | 84 | 0.574 | |
| N/A | 4 | 13 | ||
| MSI-H | 6 | 25 | 0.231 | 0.23 |
| MSI-L | 18 | 148 | 0.120 | |
| MSS | 2 | 36 | ||
| N/A | 0 | 1 | ||
| CIMP-H | 6 | 30 | 0.231 | 0.76 |
| CIMP-L | 7 | 46 | 0.143 | |
| Cluster3 | 8 | 69 | ||
| Cluster4 | 5 | 65 | ||
| N/A | 0 | 0 | ||
| Hypermut | 5 | 29 | 0.227 | 0.35 |
| Nonhypermut | 17 | 168 | 0.147 | |
| N/A | 4 | 13 |
meth, Methylation; wt, wild-type, mut, mutated; N/A, not applicable; MSI-H, microsatellite instability-high; MSI-L, microsatellite instability-low; MSS, microsatellite stability; CIMP-H, CpG island methylator phenotype-high; CIMP-L, CpG island methylator phenotype-low; hypermut, hypermutated; Nonhypermut, nonhypermutated.
Effects of KRAS signaling of FXR expression.
Because FXR expression in colon cancer cell lines negatively correlated with PI3K signaling in RPPA data, FXR expression was measured in PI3K isogenic cell lines (37). There was no difference in FXR expression in cells with no vs. high PI3K signaling (indicated by phospho-AKT; data not shown).
However, knockdown of KRAS signaling in PI3K isogenic cells lines resulted in dramatic increase in FXR levels in KRAS WT and mutant cells, measured by Western blot analysis (data not shown). Expanding this in more cells showed KRAS siRNA treatment significantly increased FXR mRNA levels in five of six colon cancer cell lines (Fig. 7A). Interestingly, this increase was greater in KRAS mutant cells (SW620, SK-CO-1, and SW1116) than KRAS WT cells (HT-29). KRAS mutational status of these cells can be found in Fig. 3D. HT-29 cells are WT for KRAS (31).
Fig. 7.
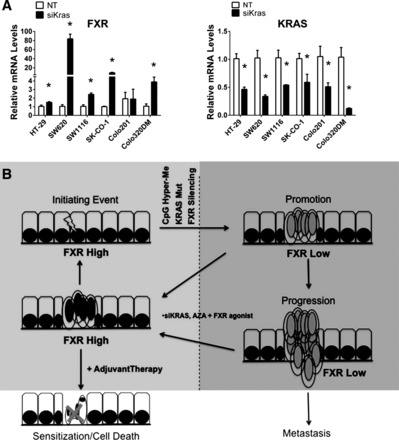
FXR and small interfering KRAS (siKRAS) expression in human colon cancer cell lines and the predicted role of FXR in colon cancer development. A: relative mRNA levels of FXR in colon cancer cell lines treated with siKRAS. B: relative mRNA levels of KRAS in colon cancer cell lines confirming knockdown of KRAS. Data are expressed as means + SE. *P < 0.05 compared with nontargeting (NT) siRNA controls. Hyper-Me, hypermethylation. C: colon cancer is initiated by an acquired mutation within genes involved in WNT signaling (APC or β-catenin) or DNA repair signaling. Promotion to an adenoma often occurs through CpG island hypermethylation and acquired KRAS mutations (22). Our results indicate that FXR is also silenced during this early period of adenoma formation and correlates with EMT and oncogenic signaling of PI3K, suggesting that FXR silencing contributes to colon cancer progression and/or metastasis. If FXR expression is restored by inhibition of DNA methylation or KRAS signaling and activated by synthetic FXR ligands, this may help restore normal cancer cell phenotype, slow cancer progression, and/or sensitize cancer cells to chemotherapy.
DISCUSSION
FXR is an adopted nuclear receptor responsible for regulating free BA levels in both liver and intestine and has been suggested to be a potential tumor suppressor for colon cancer development (7, 21, 25). Studies have shown that mice deficient in FXR have increased colonic tumorigenesis and that the FXR antitumorigenic effects are at least partially due to BA-independent mechanisms, namely by regulating intestinal integrity and inflammation and protection from genotoxic compounds (13, 21, 25, 44). FXR has also been suggested to suppress colon tumorigenesis by increasing apoptosis and suppressing epidermal growth factor receptor-mediated cell proliferation (21, 25, 30).
This study revealed that FXR is downregulated at the transcriptional level in colon adenomas and virtually silenced in all stages of adenocarcinoma. Previous reports have suggested FXR was downregulated in a stage-dependent manner (6, 16) compared with our findings that indicate that FXR was decreased in adenomas and almost completely silenced in stage I adenocarcinomas. Furthermore, FXR expression was positively and negatively correlated to expression of collagen IV and vimentin, markers for mesenchymal cell phenotype (14), and negatively correlated to PI3K, cyclin E1, and other oncogenic signaling mediators (46). This implicates that FXR is either a strong marker for cell differentiation or that FXR regulates these signaling pathways.
Mutations within WNT signaling cascade have been widely accepted as a major initiating event in colon cancer development. Our analysis showed that increased β-catenin nuclear localization was observed in only 22.2% of the polyps and adenomas we analyzed, compared with 66% (6 of 9) polyps having decreased FXR expression. The direct association between β-catenin activation and FXR silencing remains unclear and larger sample size and detailed analysis will be needed in the future to determine the degree of association.
Tumor suppressor genes can be silenced in cancer cells through acquired silencing mutations, epigenetic mechanisms, or transcriptional silencing through oncogenic signaling. We detected no genetic mutations within the FXR gene in colon cancer cell lines or clinical colon tumors (data not shown) that could account for the silencing and TCGA data support that genomic alterations of the FXR gene are a rare event in colon cancer. However, inhibition of DNMT 1/3B activity increased FXR mRNA levels in malignant colon cancer cell lines suggesting DNA methylation is a contributing mechanism for FXR silencing in colon cancer. We confirmed functional methylation of the FXR promoter within colon cancer cell lines and detected full methylation of FXR promoter in 12% of clinical colon tumors.
DNA methylation is clearly not the only mechanism of FXR silencing in colon tumors since 80–90% of tumors still have low levels of FXR expression in the absence of FXR promoter methylation. RPPA analysis revealed that PI3K signaling, a common molecular feature of colon cancer (46), was negatively correlated with FXR expression and could also play a role in FXR silencing. However, PI3K isogenic cell lines had no difference in basal expression of FXR. Conversely, silencing of KRAS signaling, another well-known oncogenic event in colon cancer (1), in multiple colon cancer cell lines increased FXR expression. Sequence analysis of the FXR promoter by use of MATCH 1.0 [which utilized TRANFAC Public 6.0 (24)] revealed a predicted binding site for activator protein 1 (AP-1; composed of c-Jun and c-Fos heterodimer), a transcriptional mediator of the c-Jun NH2-terminal kinase signaling pathway. This pathway can be involved in RAS-initiated tumor formation (3). Thus it is possible that AP-1 mediates the effects of KRAS and potentially other signaling pathways, to inhibit FXR expression in colon cancer.
TCGA data showed no molecular subtype, including FXR promoter methylation and KRAS mutation correlated with FXR expression. However, our results show that nearly all colon tumors, regardless of stage, had low to no FXR expression, illustrating the difficulty of detecting a molecular causal relationship. FXR promoter methylation was higher in MSI-H and hypermutated tumors and lower in KRAS mutant tumors. Hypermutated tumors often segregate with MSI-H tumors whereas KRAS tumors segregate with MSI-low and nonhypermutated (4, 35). Thus an inverse methylation pattern of FXR, higher methylation in MSI-H tumors and lower methylation in KRAS mutant tumors, suggests two distinct mechanisms of FXR silencing: 1) DNA methylation in MSI-H and 2) KRAS signaling in MSI-low tumors. No difference in FXR promoter methylation was detected between CIMP and non-CIMP tumors.
In conclusion, we have shown that FXR was downregulated very early in human colon cancer development, which was partly due to DNA methylation of the FXR promoter and increased KRAS signaling (Fig. 5B). Silencing of FXR alone is not sufficient to initiate colon cancer development, but activation of remnant FXR in healthy tissues may play an important role in preventing and inhibiting the promotion of colon cancer (21, 25, 44). Restoration of basal FXR expression through inhibition of DNA methylation or KRAS signaling, or through activation of residual FXR, might slow or prevent the progression of colon cancer either through direct antiproliferative or chemopreventative mechanisms. There are known FXR agonists, such as GW4064 and 6α-ethylchenodeoxycholic acid (6E-CDCA), that are currently in preclinical and clinical development for metabolic disorders (20, 29). It is conceivable that in situations where FXR is not completely absent these agents might be able to restore lost FXR activity in colon cancer resulting in inhibited tumor growth.
GRANTS
This work was supported by CA172670 (G. Powis), CPRIT Training Grants RP101502 to The University of Texas MD Anderson Cancer Center (A. M. Bailey), DK031343 (G. L. Guo), and DK090036 (G. L. Guo).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.M.B., L.Z., D.M., I.S., C.R.P., J.I., N.H., C.W., V.B., H.L., S.K., G.P., and G.L.G. conception and design of research; A.M.B., L.Z., and G.K. performed experiments; A.M.B., L.Z., D.M., C.R.P., G.K., C.W., V.B., H.L., G.P., and G.L.G. analyzed data; A.M.B., L.Z., D.M., I.S., G.K., J.I., N.H., C.W., V.B., H.L., S.K., G.P., and G.L.G. interpreted results of experiments; A.M.B., C.R.P., C.W., and G.L.G. prepared figures; A.M.B., C.W., H.L., and G.L.G. drafted manuscript; A.M.B., L.Z., D.M., I.S., C.R.P., G.K., J.I., N.H., C.W., V.B., H.L., S.K., G.P., and G.L.G. edited and revised manuscript; A.M.B., L.Z., D.M., I.S., C.R.P., G.K., J.I., N.H., C.W., V.B., H.L., S.K., G.P., and G.L.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We appreciate the help and discussion from Dr. Vadivel Ganapathy at the Medical College of Georgia, and for human colon cancer cell line cDNAs provided by his laboratory. A special thanks goes to the late Dr. Mani Maran Rengasamy for help with animal studies and scientific discussion. We also thank the University of Kansas Cancer Center and University of Kansas Cancer Center Biospecimen Core for pilot grants and human colon samples. We also thank Lynda Corley from the histology core facilities within the Department of Pathology at MD Anderson Cancer Center for assistance and guidance with the analysis and data acquisition of IHC slides. Furthermore, special thanks goes to Dr. Patricia Thompson at the University of Arizona Cancer Center for providing colon tissue microarrays used for this study and research pathologist Dr. Viren R. Patel at MD Anderson Cancer Center for extracting tissue microarrays information necessary for IHC data analysis.
REFERENCES
- 1.Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 16: 790–799, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 85: 863–871, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P, Jacks T, Davis RJ. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol 31: 1565–1576, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comprehensive molecular characterization of human colon, and rectal cancer. Nature 487: 330–337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol 22: 3157–3173, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Gottardi A, Touri F, Maurer CA, Perez A, Maurhofer O, Ventre G, Bentzen CL, Niesor EJ, Dufour JF. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig Dis Sci 49: 982–989, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Eloranta JJ, Kullak-Ublick G. The role of FXR in disorders of bile acid homeostasis. Physiology 23: 286–295, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S, Penna G, Laverny G, Adorini L, Moschetta A, van Mil SW. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60: 463–472, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Grober J, Zaghini I, Fujii H, Jones SA, Kliewer SA, Willson TM, Ono T, Besnard P. Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement of the farnesoid X receptor/9-cis-retinoic acid receptor heterodimer. J Biol Chem 274: 29749–29754, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Hsieh CL. Stability of patch methylation and its impact in regions of transcriptional initiation and elongation. Mol Cell Biol 17: 5897–5904, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imray CH, Radley S, Davis A, Barker G, Hendrickse CW, Donovan IA, Lawson AM, Baker PR, Neoptolemos JP. Faecal unconjugated bile acids in patients with colorectal cancer or polyps. Gut 33: 1239–1245, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2: 217–225, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA 103: 3920–3925, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivaska J. Vimentin: central hub in EMT induction? Small GTPases 2: 51–53, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kune GA. The Melbourne Colorectal Cancer Study: reflections on a 30-year experience. Med J Aust 193: 648–652, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Lax S, Schauer G, Prein K, Kapitan M, Silbert D, Berghold A, Berger A, Trauner M. Expression of the nuclear bile acid receptor/farnesoid X receptor is reduced in human colon carcinoma compared with nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int J Cancer 130: 2232–2239, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Lechner S, Muller-Ladner U, Schlottmann K, Jung B, McClelland M, Ruschoff J, Welsh J, Scholmerich J, Kullmann F. Bile acids mimic oxidative stress induced upregulation of thioredoxin reductase in colon cancer cell lines. Carcinogenesis 23: 1281–1288, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Macrae F. Wheat bran fiber and development of adenomatous polyps: evidence from randomized, controlled clinical trials. Am J Med 106: 38S–42S, 1999 [DOI] [PubMed] [Google Scholar]
- 20.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem 43: 2971–2974, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, Gao X, Zhang Y, Ganapathy V, Gonzalez FJ, Guo GL. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther 328: 469–477, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361: 2449–2460, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marzolini C, Tirona RG, Gervasini G, Poonkuzhali B, Assem M, Lee W, Leake BF, Schuetz JD, Schuetz EG, Kim RB. A common polymorphism in the bile acid receptor farnesoid X receptor is associated with decreased hepatic target gene expression. Mol Endocrinol 21: 1769–1780, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modica S, Murzilli S, Salvatore L, Schmidt DR, Moschetta A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res 68: 9589–9594, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Okuwaki M, Takada T, Iwayanagi Y, Koh S, Kariya Y, Fujii H, Suzuki H. LXR alpha transactivates mouse organic solute transporter alpha and beta via IR-1 elements shared with FXR. Pharm Res 24: 390–398, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates β-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 15: 2156–2163, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science 284: 1365–1368, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 45: 3569–3572, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Peng Z, Raufman JP, Xie G. Src-mediated cross-talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis. PLoS One 7: e48461, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perucho M, Goldfarb M, Shimizu K, Lama C, Fogh J, Wigler M. Human-tumor-derived cell lines contain common and different transforming genes. Cell 27: 467–476, 1981 [DOI] [PubMed] [Google Scholar]
- 32.Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics 19: 1236–1242, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Reddy BS. Diet and excretion of bile acids. Cancer Res 41: 3766–3768, 1981 [PubMed] [Google Scholar]
- 34.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416: 552–556, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, Leppert MF, Slattery ML. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol 158: 1517–1524, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sengupta PK, Smith EM, Kim K, Murnane MJ, Smith BD. DNA hypermethylation near the transcription start site of collagen α2(I) gene occurs in both cancer cell lines and primary colorectal cancers. Cancer Res 63: 1789–1797, 2003 [PubMed] [Google Scholar]
- 37.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260: 85–88, 1993 [DOI] [PubMed] [Google Scholar]
- 38.Shureiqi I, Chen D, Day RS, Zuo X, Hochman FL, Ross WA, Cole RA, Moy O, Morris JS, Xiao L, Newman RA, Yang P, Lippman SM. Profiling lipoxygenase metabolism in specific steps of colorectal tumorigenesis. Cancer Prev Res (Phila) 3: 829–838, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102: 731–744, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Sorensen AL, Jacobsen BM, Reiner AH, Andersen IS, Collas P. Promoter DNA methylation patterns of differentiated cells are largely programmed at the progenitor stage. Mol Biol Cell 21: 2066–2077, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tantamango YM, Knutsen SF, Beeson L, Fraser G, Sabate J. Association between dietary fiber and incident cases of colon polyps: the Adventist Health Study. Gastrointest Cancer Res 4: 161–167, 2011 [PMC free article] [PubMed] [Google Scholar]
- 42.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5: 2512–2521, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Tong JL, Ran ZH, Shen J, Fan GQ, Xiao SD. Association between fecal bile acids and colorectal cancer: a meta-analysis of observational studies. Yonsei Med J 49: 792–803, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaquero J, Briz O, Herraez E, Muntane J, Marin JJ. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim Biophys Acta 1833: 2212–2219, 2013 [DOI] [PubMed] [Google Scholar]
- 44a.Ward JH., Jr Hierarchical grouping to optimize an objective function. J Am Stat Assoc 58: 236–244, 1963 [Google Scholar]
- 45.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–862, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Roberts TM, Shivdasani RA. Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology 141: 50–61, 2011 [DOI] [PubMed] [Google Scholar]
- 47.Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, Fuchsbichler A, Halilbasic E, Denk H, Marschall HU, Trauner M. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-α/β in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol 290: G923–G932, 2006 [DOI] [PubMed] [Google Scholar]