

Abstract
Congenital tufting enteropathy (CTE) is a severe diarrheal disease of infancy characterized by villous changes and epithelial tufts. We previously identified mutations in epithelial cell adhesion molecule (EpCAM) as the cause of CTE. We developed an in vivo mouse model of CTE based on EpCAM mutations found in patients with the aim to further elucidate the in vivo role of EpCAM and allow for a direct comparison to human CTE. Using Cre-LoxP recombination technology, we generated a construct lacking exon 4 in Epcam. EpcamΔ4/Δ4 mice and CTE patient intestinal tissue integrity was analyzed by histology using both light immunohistochemistry and electron microscopy. EpcamΔ4/Δ4 mice demonstrate neonatal lethality and growth retardation with pathological features, including epithelial tufts, enterocyte crowding, altered desmosomes, and intercellular gaps, similar to human CTE patients. Mutant EpCAM protein is present at low levels and is mislocalized in the intestine of EpcamΔ4/Δ4 mice and CTE patients. Deletion of exon 4 was found to decrease expression of both EpCAM and claudin-7 causing a loss of colocalization, functionally disrupting the EpCAM/claudin-7 complex, a finding for the first time confirmed in CTE patients. Furthermore, compared with unaffected mice, mutation of Epcam leads to enhanced permeability and intestinal cell migration, uncovering underlying disease mechanisms.
Keywords: congenital tufting enteropathy, EpCam, chronic diarrhea, intestinal failure, mouse model
congenital tufting enteropathy (CTE) is one of several intractable diarrheal diseases of infancy that typically presents in the neonatal period with chronic watery diarrhea, electrolyte imbalances, and impaired growth. The diagnosis of CTE is made with the recognition of villous changes in the small intestinal epithelium. Typical findings include total or partial villous atrophy, crypt hyperplasia, and focal epithelial tufts in the small intestine (5, 29). These highly characteristic tufts are composed of enterocytes with rounding of the plasma membrane, resulting in teardrop-like configurations. This is a severe disease resulting in intestinal failure, and most patients are dependent on parenteral nutrition to acquire adequate caloric intake necessary for improved growth and development (14). Prolonged parenteral therapy is associated with a low quality of life and has inevitable complications such as liver disease, infection, and vascular complications (11, 16). Although small bowel transplant is a therapeutic option, it carries its own risks, including significant mortality (26).
We previously identified mutations in epithelial cell adhesion molecule (EpCAM) as the underlying cause of CTE (30). In normal adult tissues, EpCAM is expressed on the basolateral surface of simple, pseudostratified, and transitional epithelia in various tissues of the gastrointestinal tract, reproductive system, and respiratory tract. EpCAM was first recognized as an antigen overexpressed on human carcinoma cells, including tumors of the gastrointestinal system, breasts, thyroid, and kidneys (3). EpCAM has been reported to have roles in cell adhesion and proliferation, as well as pancreatic islet cell development; however, its function in the intestine has not been elucidated (8, 18, 24). Expression of EpCAM protein in CTE patients is significantly decreased, but not absent in the intestine, as seen by immunohistochemistry and Western blot analysis (30).
EpCAM is highly conserved in mammals. Both human and murine EpCAM consists of nine exons on chromosome 2p21 and 17, respectively (Fig. 1A). Murine EpCAM has 81% identity and 89% similarity to the human protein, with only one additional amino acid for a total 315 amino acids and the same predicted molecular weight of 35 kDa (2, 6). The pattern of murine Epcam mRNA expression in tissues is similar to human EpCAM, with the highest expression in the gut and lower levels in the kidneys, pancreas, mammary glands, lungs, and genitalia, consistent with its epithelial cell distribution (25). EpCAM protein has also been identified on the surface of mouse embryonic, neonatal, and adult germ cells, as well as embryonic stem cells (4, 12). In vivo roles for EpCAM have been investigated in two zebrafish knockout models, demonstrating functionality in proper epithelial morphogenesis integrity during epiboly and skin development and a decrease in proneuromast deposition (32, 33).
Fig. 1.

EpCAM is highly conserved in mammals. Construction of EpcamΔ4/Δ4 mouse model with neonatal lethality and phenotypic changes. A: human (top) and mouse (middle) EpCAM exon arrangement and mutant Epcam Δ4 construct (bottom). Exons are numbered above with number of base pairs in parentheses. The predicted open reading frame of human EpCAM is ∼1.5 kb, encoding a 314-amino acid protein with a predicted molecular weight of 35 kDa. Murine Epcam consists of nine coding exons spanning ∼17 kb on mouse chromosome 17. The predicted open reading frame is 79% identical to its human counterpart, with an additional three base pairs added to mouse exon 7. B: survival of EpcamΔ4/Δ4 mice is limited to less than 1 wk; n ≥ 10 per group. C: lack of weight gain in EpcamΔ4/Δ4 mice compared with EpcamWT/WT and EpcamWT/Δ4; n ≥ 10 per group. D: phenotypic comparison of EpcamΔ4/Δ4 mice to littermate controls at days 3 and 4 of life. E: full stomachs and shorter intestinal length in EpcamΔ4/Δ4 mice. Pictured at day 4 of life. Representative of n ≥ 10 mice per group.
In 2009, an Epcam knockout mouse was reported to be embryonically lethal; however, three groups recently generated viable Epcam knockout mice to study the function of EpCAM. The first model demonstrated EpCAM's contribution to the formation of functional tight junctions and recruitment of claudins (20, 25). Another viable knockout constructed by gene trapping, revealed severe hemorrhagic enteropathy and intracellular accumulation of E-cadherin and β-catenin (15). Additionally, a conditional Epcam knockout mouse of epithelial Langerhans cells demonstrated a role for these cells in motility and migration in the skin (10).
In this study, we generated an in vivo mouse model of CTE with a mutation that corresponds to a human EpCAM mutation found in patients. To date, several mutations of EpCAM have been reported in the extracellular portion of EpCAM in CTE patients. In particular, the homozygous G>A substitution at the donor splice site of exon 4 (∼491+1G>A) results in an mRNA splice form of EpCAM lacking exon 4 (1, 17, 27, 31). On the basis of these findings, a murine model with deletion of exon 4 in Epcam was used to further elucidate the in vivo role of EpCAM and allow for a direct comparison to human CTE. We hypothesized that Epcam mutations associated with CTE would result in an abrogated, but not a total loss of protein, as seen in CTE patients and that this viable model will demonstrate pathology mimicking human disease (30).
MATERIALS AND METHODS
Generation of targeting construct and mutant allele genotyping.
Our strategy was to develop a construct in which loxP sequences flank Epcam exon 4 and efficiently delete it through Cre-LoxP recombination, modeling an alternatively spliced shortened form observed in CTE patients. We used a pfloxFRTNeo cloning vector, containing two FRT sites surrounding a positive (neomycin) selection marker, two loxP sites with a multiple cloning site to insert the genomic region of the targeted exon, and a diptheria toxin-A (DTA)-negative selection marker (21). The construct (Fig. 1A) was configured with a 5′ DTA-negative selection marker, followed by a 4.4 kb 5′ homology arm, and a neomycin-resistant positive selection marker that is flanked by FRT sites, followed by loxP sites flanking the targeted region consisting of 95 bp of intron 3, exon 4 (66 bp), and 89 bp of intron 4, ending with a 3.9 kb 3′ homology arm. Mouse development and breeding were carried out, allowing for deletion of exon 4 (Δ4) in the early mouse embryo and efficient germ-line transmission of Δ4 to subsequent generations (19). Genotyping was performed using specific primers (primer 1: 5′-TTTGTGCTGGAGAGACAAGTGG-3′, primer 2: 5′-AGCCATCGGAGTGTCAGTCT-3′, and primer 3: 5′-GGGGTTTGCTCGACATTG-3′). Primers 1 and 2 were used to detect the wild-type allele (663 bp), the targeted allele after removal of Neo (754 bp), and the mutant Δ4 allele (475 bp). Primers 2 and 3 were used to detect the targeted allele before removal of the Neo (695 bp). Mice heterozygous for deletion of exon 4 (EpcamWT/Δ4) were then bred to each other, generating mutant mice homozygous for deletion of exon 4 (EpcamΔ4/Δ4).
Animals.
Mice were cared for in accordance with appropriate institutional guidelines. Experimental protocols were approved by the University of California, San Diego (UCSD) Institutional Animal Care and Use Committee. B6.Cg-Tg(ACTFLPe)9205Dym/J and FVB/N-Tg(EIIa-cre) C5379Lmgd/J mouse strains were used in this study, both acquired from Jackson Laboratories (Bar Harbor, ME).
Murine staining and histologic analysis.
Whole body and intestinal staining with hematoxylin and eosin (H&E) was performed according to published methods (22). Histological sections (5 μm) were prepared from selected tissues that had been fixed in 10% buffered formalin (intestine; Fisher Scientific, Waltham, MA) or Bouin's fixative solution (whole body; Ricca Chemical, Arlington, TX) for 24 h and embedded in paraffin. Pathological analysis was performed on embryonic days 17 and 19 and daily after birth. Intestines were not opened longitudinally because of the small nature of neonatal mice and the fragility of the tissue. Additionally, whole bodies of killed mice were collected to look for extraintestinal disease, specifically in the eyes and nares, based on associated signs from human CTE patients.
Electron microscopy.
Small intestinal tissue for electron microscopy (EM) was collected from EpcamΔ4/Δ4 mice, littermate controls, and CTE patients. All specimens obtained for microscopy and immunohistochemistry were analyzed with approval from the UCSD Institutional Review Board (IRB; first approval April 22, 2005) and consents provided from patients and families. Samples were immersed in modified Karnovsky's fixative (2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M sodium cacodylate buffer, pH 7.4) for at least 4 h and processed according to protocol by UCSD electron microscopy facility. Images were obtained at ×1400, ×2900, ×4800, and ×6800.
In vivo permeability, migration, and proliferation assays.
To assess barrier function, 600 mg/kg of FITC-dextran (FD4; Sigma, St. Louis, MO) was orally gavaged into 4-day-old EpcamΔ4/Δ4 and littermate control mice. Four hours after gavage, serum was collected following decapitation. Fluorescent intensity of each sample was measured on a TECAN Genios Pro plate reader. FITC-dextran concentrations were determined from standard curves generated by serial dilutions of FITC-dextran (34).
BrdU.
50 mg/kg of BrdU (BD Pharmingen, San Jose, CA) was injected intraperitoneally into 3- to 4-day old EpcamΔ4/Δ4 and EpcamWT/WT mice, which were then killed at time points of 4 and 24 h following injection. Histological sections were then prepared as earlier described and stained for BrdU using a BrdU in situ detection kit (BD Pharmingen, San Jose, CA) per the manufacturer's instructions. For proliferation analysis, 10–20 well-oriented villi per slide were identified, and the number and position of BrdU-labeled enterocytes on one side of each villus were independently counted by two observers blinded to the sample group. Each sample group included at least three mice, allowing for testing of approximately 60 discrete villi per group. Proliferation index was calculated as the number of BrdU-labeled cells out of the first 10 cells per crypt-villus. Because of the highly dysmorphic intestinal pathology and villous atrophy seen in EpcamΔ4/Δ4 mice, the positively stained cell ratio was limited to the first 10 enterocytes to normalize WT and mutant samples. To investigate migrational effects, absolute number of cells from the base of the crypt to the farthest traveled positively stained enterocyte at 24 h was obtained. Additionally, longitudinal distance (in micrometers) from base of the crypt to positive cell closest to the villus tip was measured.
RNA isolation and analysis.
RNA was isolated from whole flash-frozen murine small intestines and human duodenal biopsies using TRIzol (Invitrogen, Foster City, CA). Total RNA was reverse transcribed into cDNA with random hexamers (Roche Diagnostics, Indianapolis, IN). Transcription levels were measured by quantitative polymerase chain reaction (qPCR) using TaqMan gene expression assays (Applied Biosystems Instruments, New York, NY) (Epcam exons 3 and 4 Mm00493211_m1, Epcam exons 6 and 7 Mm00493214_m1, claudin-7 Mm00516817_m1, Hprt Mm00446968_m1, EpCAM exons 1 and 2 Hs00158980_m1, EpCAM exons 3 and 4 Hs00901885_m1), on a CFX96 real-time system (Bio-Rad, Hercules, CA). WT intestinal cDNA was used to generate a standard curve for all primer probe sets, and the relative standard curve method was used to quantify expression levels. All qPCR reactions were performed in triplicate on n = 3 biological samples for mice and n = 1 for human, CTE patient, and unaffected control. Graphs represent the mean ± SD.
Immunoblotting and immunoprecipitation.
Immunoblotting was performed according to a previously described protocol (30) using antibodies: E144 to EpCAM (ab32392; Abcam, Cambridge, UK), claudin-7 (ab27487; Abcam), and β-actin (1:20,000, sc 47778; Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies [1:10,000; enhanced chemiluminescence (ECL), anti-mouse IgG; NA931V; GE Healthcare, Chalfont St. Giles, UK] and (1:2,000, ECL, anti-rabbit IgG; NA9340V; GE Healthcare, Pittsburg, PA).
Immunoprecipitation was performed using HEK293 cells transiently transfected with COOH-terminal FLAG-tagged WT or ΔEpCAM in pcDNA3.1(+) (Invitrogen). Transfected HEK293 cells were lysed as described above. Anti-FLAG Sepharose beads (Sigma-Aldrich) were used to immunoprecipitate FLAG-tagged EpCAM from cleared lysates. Blots of the lysates and immunoprecipitates were then probed for EpCAM (ANTI-FLAG-HRP, A8592; Sigma-Aldrich), claudin-7 (ab27487; Abcam), and α-actinin (ab9465; Abcam) as a loading control. Quantification of bands on blots from three different experiments was performed using ImageJ software. Scale bars represent the means ± SE.
Intestinal immunohistochemical staining.
After IRB consent, immunofluorescent staining of available formalin-fixed, paraffin-embedded duodenal biopsy tissue was performed. Tissue from five patients with CTE and five normal patients was studied. Slides were stained for EpCAM using anti-EpCAM antibody (323/A3 Ab8601; AbCAM, Cambridge, England), as previously described (30). Isotype controls were performed. Subsequently, four patients with ulcerative colitis, four patients with Crohn's disease, six patients with celiac disease, two patients with microvillus inclusion disease, two patients with nonspecific duodenitis, three patients with giardiasis, and four normal age-matched controls were included.
Statistical analyses.
Statistical analyses and graphing were performed in Excel and GraphPad Prism programs with the unpaired Student's t-test.
RESULTS
Mice expressing mutant EpCAM show neonatal lethality.
We developed a novel mouse model allowing for efficient universal deletion of exon 4 in Epcam without causing a frameshift, paralleling what is observed in CTE patients carrying the ∼491+1G>A mutation (30). Survival of EpcamΔ4/Δ4 mice is limited to 5 to 7 days, and pups show minimal to no weight gain over their life spans compared with their wild-type (EpcamWT/WT) and heterozygous (EpcamWT/Δ4) littermates (Fig. 1, B and C). At an early age, postnatal day 3 (P3) EpcamΔ4/Δ4 mice could be distinguished phenotypically from littermates by runted appearance, as well as lethargy (Fig. 1D). EpcamWT/WT and EpcamWT/Δ4 mice are indistinguishable. A similar amount of milk is seen in stomachs of EpcamΔ4/Δ4 vs. EpcamWT/WT mice with no significant luminal bleeding present (Fig. 1E). Upon dissection, EpCAMΔ4/Δ4 mice demonstrate shorter intestinal length (Fig. 1E) compared with littermate controls (EpcamWT/WT and EpcamWT/Δ4).
EpcamΔ4/Δ4 mice show pathological changes resembling congenital tufting enteropathy.
Detailed characterization of intestinal abnormalities was thoroughly investigated, as this is the major feature in CTE patients. Necropsy of EpcamΔ4/Δ4 mice at embryonic days 17 and 19 did not reveal alterations of villous structure when compared with EpcamWT/WT or EpcamWT/Δ4 littermates (Fig. 2, A and B). Postnatal pathology of whole bodies revealed microscopic changes in the EpcamΔ4/Δ4 intestine throughout the duodenum, jejunum, and ileum. H&E staining revealed progressive changes over time, with abnormal invaginations between enterocytes seen by day 2, along with blunting of the intestinal villi, tufted villi, edema, crowding of enterocytes, and an inflammatory infiltrate, all manifesting by day 4. No alteration in goblet cells was noted. These intestinal abnormalities are very similar to pathologies seen in CTE. No pathologic changes were observed in EpcamWT/WT (Fig. 2A) or EpcamWT/Δ4 (data not shown). Other parts of the alimentary canal, including stomach and colon, were unaffected. No extra-intestinal manifestations were seen. Particularly, the pathology of eyes, kidneys, liver, gall bladder, and bones were normal.
Fig. 2.

Intestinal pathology. A: EpcamWT/WT with lack of pathology in the small or large intestine (proximal small and large intestine pictured). B: EpcamΔ4/Δ4 intestinal pathology. Prenatal (E17) and postnatal (D1–D6) intestinal pathology demonstrating progressive changes in small and large intestine with invaginations (asterisk), enterocyte crowding/tuft formation (arrowhead), and cell destruction (arrow). Pathology is representative of n ≥ 10 mice. Scale bars represent 50 μm.
Gross structural changes (crowding of enterocytes, loss of columnar shape) were appreciable by EM in EpcamΔ4/Δ4 mice and intestinal desmosomes were found to have sporadic irregularity with crowding and lengthening of the desmosomes (Fig. 3B). Intestinal desmosomes of EpcamWT/WT and EpcamWT/Δ4 mice were found to be intact and of normal length. Gaps in the intercellular space were also noted between EpcamΔ4/Δ4 mouse enterocytes, a finding also not seen in littermate controls. Tight junctions and adherens junctions of all mice appeared normal. EM of CTE patient duodenal biopsies revealed similar desmosome lengthening, and intercellular gaps were not present (Fig. 3C) in unaffected patient tissue (data not shown).
Fig. 3.

Electron microscopy of EpcamWT/WT (A) with normal ultrastructure compared with EpcamΔ4/Δ4 (B) mice showing intercellular gaps and increased length and number of desmosomes in EpcamΔ4/Δ4 mice. Gross structural changes, including loss of columnar shape (bottom right) were also seen. Images are representative of four EpcamΔ4/Δ4 mice and five littermate controls. C: electron microscopy of patients with CTE, demonstrating increased number and length of desmosomes (⌘) and intercellular gaps (❖).
EpcamΔ4/Δ4 mice show intestinal permeability defects and enhanced migration and proliferation.
Intestinal permeability was assessed using administration of FITC-dextran by gavage and subsequent serum detection. EpcamΔ4/Δ4 mice demonstrate significantly more FITC-dextran in their serum (1,464.5 ng FITC-dextran/ml serum) compared with littermate controls (38.2 ng FITC-dextran/ml serum), suggesting increased intestinal permeability allowing for the dextran to cross the intestinal barrier into the bloodstream (Fig. 4A, P < 0.0001).
Fig. 4.

Functional intestinal changes in EpcamΔ4/Δ4 mice. A: FITC-dextran administration reveals increased intestinal permeabililty in EpcamΔ4/Δ4 compared with littermate controls. n ≥ 5 mice per group measured in triplicate. B–D: proliferation index and migration markers of BrdU-stained enterocytes. n ≥ 3 mice per group. E: representative images of BrdU-stained intestine following injection of BrdU at 4- and 24-h timepoints. *P < 0.05, bars indicate standard errors of the mean.
Enterocyte proliferation was assessed using BrdU at a 4-h time point following intraperitoneal injection. EpcamΔ4/Δ4 mice demonstrate a significantly higher proliferation index than EpcamWT/WT at 4 h (Fig. 4B; P = 0.0003). To assess migration, BrdU-positive cells were assessed at 24 h. Though significant changes were not seen with traditional counting methods at 24 h (Fig. 4B; P = 0.3324), analyzing location (Fig. 4C, P < 0.0001 4 h, P = 0.0004 24 h) and the distance of farthest traveled BrdU-positive cells (Fig. 4D; P < 0.0001 4 h, P = 0.0013, 24 h), revealed that EpcamΔ4/Δ4 mice have significantly higher migration of BrdU-positive enterocytes compared with EpcamWT/WT (Fig. 4E).
Mutation of EpCAM causes disruption of the EpCAM/claudin-7 complex in mice and humans.
To confirm proper deletion of exon 4 in the mutant Epcam, RNA and protein studies were pursued. Using specific primer sets, qPCR revealed the complete absence of exon 4 in EpcamΔ4/Δ4 transcript and a significant reduction of transcript in the EpcamWT/Δ4 transcript relative to EpcamWT/WT (Fig. 5A; P = 0.0011). Equivalent expression levels between the three genotypes were observed further downstream between exon 6 and exon 7 (Fig. 5B). CTE patients with EpCAM mutation ∼491+1G>A similarly showed a significant loss of transcript between exon 3 and 4 compared with unaffected control specimens (Fig. 5C; P = 0.0051), while maintaining intact EpCAM transcript from exons 1 and 2 (Fig. 5D).
Fig. 5.
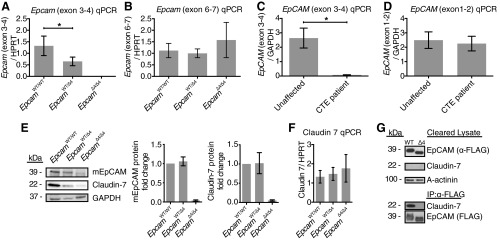
Protein and RNA expression. A and B: Epcam RNA levels by qPCR in small intestine of EpcamΔ4/Δ4 mice with decreased RNA expression from exons 3 and 4 and intact downstream from exons 6 and 7 compared with EpcamWT/WTand EpcamWT/Δ4 mice. n = 3 mice per group in triplicate. C and D: EpCAM RNA levels by quantitative PCR (qPCR) in the small intestine of the congenital tufting enteropathy (CTE) patient compared with an unaffected control, decreased from exons 3 and 4, intact from exons 1 and 2. n = 1 E: protein levels by Western blot analysis in small intestine of EpcamWT/WT, EpcamWT/Δ4, and EpcamΔ4/Δ4 mice demonstrate mutant EpCAM in EpcamΔ4/Δ4 and decreases of EpCAM and claudin-7 in EpcamΔ4/Δ4 and EpcamWT/Δ4 mice with protein quantification. F: claudin 7 RNA levels by qPCR in small intestine of EpcamWT/WT, EpcamWT/Δ4, and EpcamΔ4/Δ4 mice. n = 3 mice per group in triplicate. Bars indicate mean ± SD. G: immunoprecipitation of WT or Δ4 FLAG-tagged EpCAM from transfected HEK293 cells revealed coimmunoprecipitation of endogenous claudin-7 (expressed at near undetectable levels) with WT EpCAM and lack thereof with EpCAM Δ4.
Immunoblotting analysis was performed on EpcamWT/WT, EpcamWT/Δ4, and EpcamΔ4/Δ4 mice for EpCAM using antibody E144 on full-thickness, flash-frozen small and large intestine specimens. EpCAM was found to be truncated, and total protein levels were markedly reduced in EpcamΔ4/Δ4 mice compared with EpcamWT/WT and EpcamWT/Δ4 (Fig. 5E). Interestingly, a Western blot for claudin-7, a known EpCAM interacting protein, revealed a similar reduction of protein levels in both EpcamΔ4/Δ4 and EpcamWT/Δ4 mice, while claudin-7 RNA levels remained relatively equivalent (Fig. 5F).
Immunoprecipitation of WT or Δ4 FLAG-tagged EpCAM from transfected HEK293 cells revealed coimmunoprecipitation of endogenous claudin-7 with WT EpCAM, but not with EpCAM Δ4 (Fig. 5G).
EpcamΔ4/Δ4 mice immunohistochemistry (IHC) showed decreased EpCAM in the intestinal epithelium and mislocalization of mutant EpCAM within the cytoplasm rather than along the plasma membrane. A similar decrease and mislocalization of mutant EpCAM was seen in CTE patient tissue (Fig. 6, A and B). We also examined claudin-7 expression in mouse and human tissue by IHC. In EpcamWT/WT mice, EpCAM and claudin-7 were found to colocalize at the basolateral surface. Deletion of exon 4 in Epcam led to decreased expression of claudin-7 and a loss of colocalization (Fig. 6A). Fluorescent immunohistochemistry of human tissue from patients with CTE also exhibits decreased expression of EpCAM and claudin-7 with a disruption of colocalization at the basolateral surface compared with normal and non-CTE human intestine control specimens (Fig. 6B).
Fig. 6.

Mutation of EpCAM causes disruption of the EpCAM/claudin-7 complex. A: fluorescent immunohistochemistry demonstrating colocalization of EpCAM and claudin-7 in proximal small intestinal tissue from EpcamWT/WT mice and decreased/mislocalized EpCAM and claudin-7 in EpcamΔ4/Δ4 mice. B: fluorescent immunohistochemistry demonstrating colocalization of EpCAM and claudin-7 in tissue from unaffected/non-CTE subjects with decreased/mislocalized EpCAM and claudin-7 in proximal small intestine of CTE patients. Scale bars represent 50 μm in larger frame and 25 μm in inset.
EpCAM deficiency is specific to congenital tufting enteropathy.
Previously, a lack of EpCAM staining was noted in the duodenal tissue of human patients with congenital tufting enteropathy, while normal control tissue showed EpCAM staining in the intestinal epithelium (7), most prominent at the basolateral membrane (Fig. 7A). To determine whether the disruption of EpCAM occurs in other diarrheal diseases, immunohistochemical analysis was performed in tissue specimens from patients with celiac disease, Crohn's disease, ulcerative colitis, chronic granulomatous disease, giardiasis, and microvillus inclusion disease. Although staining showed some variability, EpCAM was found to be present at normal levels and location (Fig. 7B), suggesting EpCAM deficiency is specific to CTE.
Fig. 7.

EpCAM mutation yields deficiency in CTE patients. A: characteristic epithelial cell tufting seen in CTE. Fluorescent immunohistochemistry of CTE duodenal biopsies demonstrating minimal nonspecific staining of affected patient tissue and EpCAM's epithelial predominance in unaffected patient tissue. Asterisks denote tufts. B: fluorescent immunohistochemistry of duodenal biopsies in various intestinal disease processes with normal EpCAM expression.
DISCUSSION
We previously identified EpCAM as the causative gene in congenital tufting enteropathy (30). Similar to previously reported CTE patients and Epcam knockout mouse models (15, 20), our mutant mice with universal Epcam exon 4 deletion showed widespread epithelial dysplasia, further demonstrating the pivotal role of EpCAM in intestinal epithelial structure and integrity. In line with the severity of intestinal failure seen in CTE, EpcamΔ4/Δ4 mice have significantly limited life spans with a dramatic lack of weight gain compared with EpcamWT/WT and EpcamWT/Δ4 mice. The hallmark finding of enterocyte tufting in CTE was found to be present in our EpcamΔ4/Δ4 mouse model. These changes are appreciable as early as days 1 and 2 of life, although they are not seen in the embryonic period. The lack of pathology in embryos may suggest a role for microbial colonization or adaptation. EpCAM's normal function is likely enhanced in the perinatal period when mammals switch from placental to enteral nutrition. In newborn pigs, suckling induces rapid intestinal growth and changes digestive functions (35). Similarly, mouse and human EpCAM may be critical for the tremendous intestinal growth that occurs in the neonatal period.
Although EpCAM is expressed in numerous glandular tissues and organs, alterations in EpCAM leads to pathology confined mostly to the intestine. This phenomenon raises questions about the role of EpCAM and its possible relation to rapid turnover of the gut or biological redundancies that may occur in other tissues that are not present in the intestine. Likewise, most human CTE patients show little or no extraintestinal manifestations (14). In comparing pathological findings with affected human patients and previous models, our mutant mouse model shows epithelial changes throughout the small intestine, as opposed to more proximal changes noted in a previous study (20). Patients and EpcamΔ4/Δ4 mice do not demonstrate hemorrhage, as reported in Epcam knockout mice, which may be due to a lack of ulcerations or transmural changes in our model (15, 20). Interestingly, desmosomes in patients with CTE have been shown to be increased in length and number, a feature uniquely demonstrated in our mouse model and confirmed by direct comparison to CTE patient intestinal tissue (13). In previous EpCAM knockout mouse models, tight and adherens junction abnormalities have been described; however, neither our mouse model nor CTE patient tissue displayed either finding (15, 20).
Our mouse model expresses a mutant form of EpCAM at much lower levels than WT, as seen by both immunohistochemistry and Western blot analysis. The presence of mutant EpCAM, albeit reduced, may account for the structural and functional epithelial differences in our mutant mice compared with Epcam knockout models. As recently reported, a lack of exon 4 in in vitro models leads to mislocalization of mutant EpCAM to the endoplasmic reticulum, a finding consistent with our study, showing mislocalization from the plasma membrane to the cytoplasm in EpcamΔ4/Δ4 and CTE patient tissue (28). Although the functionality of mutant EpCAM remains unclear, mislocalization may be more applicable to human CTE than a complete loss of protein and may serve as the mechanism by which EpCAM functionality is impaired.
EpCAM is known to interact with claudin-7, and recently, a claudin-7 knockout mouse was shown to have severe intestinal defects similar to pathology observed in EpcamΔ4/Δ4 mice (9, 18). In our studies, we demonstrate that EpCAM Δ4 has a posttranscriptional effect on claudin-7, with mRNA levels of claudin-7 intact but protein expression levels decreasing. We confirmed that in WT intestines, claudin-7 colocalizes with EpCAM (20). This interaction was disrupted in EpcamΔ4/Δ4 mouse intestines, where decreased expression levels and mislocalization of claudin-7 were found, indicating a major role for EpCAM in stabilizing claudin-7. Similarly, overexpression of WT EpCAM in HEK293 cells resulted in the coimmunoprecipitation of endogenous claudin-7, whereas EpCAM Δ4 failed to pull down claudin-7. Deletion of exon 4 likely causes a misfolding and mislocalization of EpCAM, resulting in the disruption of the interacting surface and localization to the plasma membrane.
As opposed to decreased expression in CTE patients, overexpression of EpCAM is associated with changes in proliferation and carcinogenesis (23, 24). EpCAM is known to play a role in proliferation by enhancing cell cycle progression via the cyclin-regulated pathway and upregulating the proto-oncogene c-myc, which may explain its role in carcinomas (7, 24). Here, we demonstrate that mutation of EpCAM results in increased proliferation, with enhanced BrdU-positive cells seen in affected vs. normal mice at 4 h but not at 24 h following injection. Additionally, migration is enhanced perhaps to compensate for villous atrophy and destruction. This enhanced migration may also lead to the crowding of enterocytes and tufts that are characteristic of CTE. Enhanced proliferation was also seen in the claudin-7 knockout model, suggesting that disruption of claudin-7 or the EpCAM-claudin-7 complex likely mediates this process (9).
Decreases in claudin-7 and mislocalization were also demonstrated in patients and may explain the disruption of intestinal epithelial tissue integrity pathognomonic to CTE. This may also lead to the intercellular gaps noted in both our affected mice and human patients. Although we were not able to detect diarrhea in our mice due to neonatal lethality, we were able to demonstrate, for the first time in Epcam mutant or knockout mice, significantly increased in vivo intestinal permeability in our FITC-dextran gavage studies. Intestinal permeability experiments have not been reported in humans, although the impressive secretory diarrhea experienced by patients is evidence of increased permeability, likely due to the epithelial defect from mutant EpCAM expression.
Given our findings in this study, we propose a functional model of disease for congenital tufting enteropathy (Fig. 8). Although the mechanism of the permeability defect and epithelial pathology is speculative, we have provided direct evidence for EpCAM-claudin-7 interaction, barrier dysfunction, villous changes, and desmosome abnormalities. Mutation in EpCAM leads to decrease and mislocalization of EpCAM and disrupts the EpCAM/claudin-7 complex. Decreases of EpCAM and claudin-7 allow for the intercellular gap formation seen in human tissue and our mouse model. Because of instability of the epithelium, villous atrophy and tuft formation occur. This mechanism may explain the profuse diarrhea that is characteristic of CTE. Potentially, intestinal desmosomes are increased to preserve the integrity of the epithelial barrier. We propose that intestinal tissue compensates for epithelial dysfunction leading to enhanced proliferation and migration with crowding of enterocytes, which may act to reverse the villous atrophy.
Fig. 8.

Functional model of disease. Mutation of EpCAM and subsequent reduction of EpCAM and claudin-7 causes features of CTE, including tufting, intercellular gaps, increased desmosomes, and villous atrophy. This leads to functional consequences of increased proliferation, migration, and permeability with diarrhea in patients.
The diagnosis of CTE can be difficult to make and often requires repeated biopsies (14). This is because of the subtlety of pathological findings, as well as a lack of diffuse villous changes. Here, we demonstrate the ability to use EpCAM staining, given normal staining in other diarrheal diseases, to aid in the diagnosis of CTE. Decreased EpCAM staining is specific to CTE patients, while expression of EpCAM in other gut epithelial disorders, such as inflammatory bowel disease and microvillus inclusion disease, appears relatively normal. Therefore, staining for EpCAM may be a useful tool in the evaluation of congenital and chronic diarrheal diseases alongside histology and genetics.
Utilizing a novel mouse model of CTE, our studies confirm and broaden EpCAM's significant role in intestinal epithelial development and function. These studies were limited by size, age, and lethality of mice; therefore, adult-inducible disease models may also be helpful to further elucidate the effects of mutant EpCAM. Our murine model demonstrates pathology closely resembling human disease and will be essential for further elucidation of disease mechanisms and targeted treatment strategies important for modifying this devastating disorder.
GRANTS
This work was supported by grants to M. Sivagnanam from National Institutes of Health Grant K08 DK-078672-01A1, the Isenberg Fellowship and the George Ferry Young Investigator/CDHNF/Nestlé Nutrition Research Young Investigator Development Award. We acknowledge University of California, San Diego (UCSD), Neuroscience Microscopy Shared Facility Grant P30 NS047101 and UCSD Digestive Diseases Research Development Center Grant DK-80506.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.L.M. and M.S. conception and design of research; J.L.M., M.D.M., C.A.P., and M.S. performed experiments; J.L.M., M.D.M., C.A.P., and M.S. analyzed data; J.L.M., M.D.M., C.A.P., and M.S. interpreted results of experiments; J.L.M., M.D.M., C.A.P., and M.S. prepared figures; J.L.M., M.D.M., C.A.P., and M.S. edited and revised manuscript; J.L.M., M.D.M., C.A.P., and M.S. approved final version of manuscript; M.S. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank the patients and their families for participating in the study, as well as Hal Hoffman, Kim E. Barrett, Lars Eckmann, Declan McCole, and Richard Kolodner for intellectual support of this project.
REFERENCES
- 1.Al-Mayouf SM, Alswaied N, Alkuraya FS, Almehaidib A, Faqih M. Tufting enteropathy and chronic arthritis: a newly recognized association with a novel EpCAM gene mutation. J Pediatr Gastroenterol Nutr 49: 642–644, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Balzar M, Winter M, De Boer C. The biology of the 17–1A antigen (Ep-CAM). J Mol Med 77: 699–712, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17–1A antigen (Ep-CAM). J Mol Med 77: 699–712, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Basak S, Speicher D, Eck S, Wunner W, Maul G, Simmons MS, Herlyn D. Colorectal carcinoma invasion inhibition by CO17–1A/GA733 antigen and its murine homologue. J Natl Cancer Inst 90: 691–697, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Beck NS, Kang IS, Suh YL. Protracted diarrhea: results of the five-year survey in a tertiary hospital in Korea. J Korean Med Sci 16: 736–741, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergsagel PL, Victor-Kobrin C, Timblin CR, Trepel J, Kuehl W. A murine cDNA encodes a pan-epithelial glycoprotein that is also expressed on plasma cells. J Immunol 148: 590–596, 1992 [PubMed] [Google Scholar]
- 7.Chaves-Perez A, Mack B, Maetzel D, Kremling H, Eggert C, Harrus U, Gires O. EpCAM regulates cell cycle progression via control of cyclin D1 expression. Oncogene 32: 641–650, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Cirulli V, Ricordi C, Hayek A. E-cadherin, NCAM, and EpCAM expression in human fetal pancreata. Transplant Proc 27: 3335, 1995 [PubMed] [Google Scholar]
- 9.Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology 142: 305–315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaiser MR, Lammermann T, Feng X, Igyarto BZ, Kaplan DH, Tessarollo L, Germain RN, Udey MC. Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc Natl Acad Sci USA 109: E889–E897, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gambarara M, Diamanti A, Ferretti F, Papadatou B, Knafelz D, Pietrobattista A, Castro M. Intractable diarrhea of infancy with congenital intestinal mucosa abnormalities: outcome of four cases. Transplant Proc 35: 3052–3053, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez B, Denzel S, Mack B, Conrad M, Gires O. EpCAM is involved in maintenance of the murine embryonic stem cell phenotype. Stem Cells 27: 1782–1791, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Goulet O, Kedinger M, Brousse N, Cuenod B, Colomb V, Patey N, de Potter S, Mougenot JF, Canioni D, Cerf-Bensussan N. Intractable diarrhea of infancy with epithelial and basement membrane abnormalities. J Pediatr 127: 212–219, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Goulet O, Salomon J, Ruemmele F, de Serres NP, Brousse N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis 2: 20, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerra E, Lattanzio R, LaSorda R, Dini F, Tiboni GM, Piantelli M, Alberti S. mTrop1/Epcam knockout mice develop congenital tufting enteropathy through dysregulation of intestinal E-cadherin/B-catenin. PloS One 7: e49302, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeppesen PB, Langholz E, Mortensen PB. Quality of life in patients receiving home parenteral nutrition. Gut 44: 844–852, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko JS, Seo JK, Shim JO, Hwang SH, Park HS, Kang GH. Tufting enteropathy with EpCAM mutations in two siblings. Gut Liver 4: 407–410, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ladwein M, Pape UF, Schmidt DS, Schnolzer M, Fiedler S, Langbein L, Franke WW, Moldenhauer G, Zoller M. The cell-cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res 309: 345–357, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA 93: 5860–5865, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei Z, Maeda T, Tamura A, Nakamura T, Yamazaki Y, Shiratori H, Yashiro K, Tsukita S, Hamada H. EpCAM contributes to formation of functional tight junction in the intestinal epithelium by recruiting claudin proteins. Dev Biol 371: 136–145, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Liang X, Zhou Q, Li X, Sun Y, Lu M, Dalton N, Ross J, Jr, Chen J. PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol Cell Biol 25: 3056–3062, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moolenbeek C, Ruitenberg E. The Swiss roll: a simple technique for histological studies of the rodent intestine. Lab Anim 15: 57–59, 1981 [DOI] [PubMed] [Google Scholar]
- 23.Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69: 5627, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Munz M, Kieu C, Mack B, Schmitt B, Zeidler R, Gires O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene 23: 5748–5758, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Nagao K, Zhu J, Heneghan MB, Hanson JC, Morasso MI, Tessarollo L, Mackem S, Udey MC. Abnormal placental development and early embryonic lethality in EpCAM-null mice. PLoS One 4: e8543, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paramesh AS, Fishbein T, Tschernia A, Leleiko N, Magid MS, Gondolesi GE, Kaufman SS. Isolated small bowel transplantation for tufting enteropathy. J Pediatr Gastroenterol Nutr 36: 138–140, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Salomon J, Espinosa-Parrilla Y, Goulet O, Al-Qabandi W, Guigue P, Canioni D, Bruneau J, Alzahrani F, Almuhsen S, Cerf-Bensussan N. A founder effect at the EPCAM locus in congenital tufting enteropathy in the Arabic Gulf. Eur J Med Genet 54: 319–322, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Schnell U, Kuipers J, Mueller JL, Veenstra-Algra A, Sivagnanam M, Giepmans BN. Absence of cell surface EpCAM in congenital tufting enteropathy. Hum Mol Genet 22: 2566–2571, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr 38: 16–26, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Sivagnanam M, Mueller JL, Lee H, Chen Z, Nelson SF, Turner D, Zlotkin SH, Pencharz PB, Ngan BY, Libiger O, Schork NJ, Lavine JE, Taylor S, Newbury RO, Kolodner RD, Hoffman HM. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology 135: 429–437, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sivagnanam M, Schaible T, Szigeti R, Byrd RH, Finegold MJ, Ranganathan S, Gopalakrishna GS, Tatevian N, Kellermayer R. Further evidence for EpCAM as the gene for congenital tufting enteropathy. Am J Med Genet A 152A: 222–224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slanchev K, Carney TJ, Stemmler MP, Koschorz B, Amsterdam A, Schwarz H, Hammerschmidt M. The epithelial cell adhesion molecule EpCAM is required for epithelial morphogenesis and integrity during zebrafish epiboly and skin development. PLoS Genet 5: e1000563, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villablanca EJ, Renucci A, Sapde D, Lec Vr Soubiran F, Sandoval PC, DamblyChaudire C, Ghysen A, Allende ML. Control of cell migration in the zebrafish lateral line: Implication of the gene “tumour-associated calcium signal transducer,” tacstd. Dev Dyn 235: 1578–1588, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Fang CH, Hasselgren PO. Intestinal permeability is reduced and IL-10 levels are increased in septic IL-6 knockout mice. Am J Physiol Regul Integr Comp Physiol 281: R1013–R1023, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Malo C, Buddington RK. Suckling induces rapid intestinal growth and changes in brush border digestive functions of newborn pigs. J Nutr 127: 418–426, 1997 [DOI] [PubMed] [Google Scholar]