

Abstract
Poor cerebrovascular function in metabolic syndrome (MetSyn) likely contributes to elevated risk of cerebrovascular disease in this growing clinical population. Younger MetSyn adults without clinical evidence of cerebrovascular disease exhibit preserved hypercapnic vasodilation yet markedly impaired hypoxic vasodilation, but the mechanisms behind reduced hypoxic vasodilation are unknown. Based on data from rats, we tested the hypothesis that younger adults with MetSyn exhibit reduced cerebral hypoxic vasodilation due to loss of vasodilating prostaglandins. Middle cerebral artery velocity (MCAv) was measured with transcranial Doppler ultrasound in adults with MetSyn (n = 13, 33 ± 3 yr) and healthy controls (n = 15, 31 ± 2 yr). Isocapnic hypoxia was induced by titrating inspired oxygen to lower arterial saturation to 90% and 80% for 5 min each. Separately, hypercapnia was induced by increasing end-tidal CO2 10 mmHg above baseline levels. Cyclooxygenase inhibition (100 mg indomethacin) was conducted in a randomized double-blind, placebo controlled design. MCAv was normalized for group differences in blood pressure (healthy: 89 ± 2 mmHg vs. MetSyn: 102 ± 2 mmHg) as cerebrovascular conductance index (CVCi), and used to assess cerebral vasodilation. Hypoxia increased CVCi in both groups; however, vasodilation was ∼55% lower in MetSyn at SpO2 = 80% (P < 0.05). Indomethacin tended to decrease hypoxic vasodilation in healthy controls, and unexpectedly increased dilation in MetSyn (P < 0.05). In contrast to hypoxia, hypercapnia-mediated vasodilation was similar between groups, as was the decrease in vasodilation with indomethacin. These data indicate increased production of vasoconstrictor prostaglandins restrains hypoxic cerebral vasodilation in MetSyn, preventing them from responding appropriately to this important physiological stressor.
Keywords: metabolic syndrome, cerebral blood flow, cyclooxygenase
metabolic syndrome (MetSyn) afflicts 20–44% of Western populations (2, 11, 25) and contributes to the risk of cerebrovascular disease (21, 30, 31). Cerebrovascular disease is a leading cause of death worldwide (4) and encompasses clinical-grade conditions which develop as a result of cerebral blood vessel dysfunction. Newer evidence indicates preclinical impairment of cerebrovascular function is also linked to increased risk of cerebrovascular disease (14, 19, 26). Uncovering cerebrovascular dysfunction and the mechanisms responsible in conditions preceding type 2 diabetes, including obesity and MetSyn, will provide important information for understanding the development of cerebrovascular disease.
Given that both decreased cerebrovascular function and MetSyn are linked to cerebrovascular disease, it is not surprising both animal models of MetSyn (5, 22) and humans with MetSyn (12, 15) demonstrate decreased cerebrovascular reactivity. However, conclusions from human studies depend on the stressor, subject age, and comorbidities. While younger adults with MetSyn exhibit preserved cerebrovascular dilation to carbon dioxide (15), older adults with MetSyn exhibit an impaired response (12). Interestingly, in the younger adults with MetSyn (37 yr), although cerebral vasodilation to hypercapnia was preserved, cerebral vasodilation to hypoxia was impaired (15). Altogether, it is possible specific aspects of cerebrovascular function decline decades prior to clinical presentation of cerebrovascular disease, but it has not been investigated. Despite the associative connection between MetSyn and cerebrovascular disease, the physiological mechanisms responsible for impaired cerebrovascular function in MetSyn remain unexplored in humans, particularly the ability of relatively young MetSyn adults to respond to hypoxia.
Cyclooxygenase (COX) products maintain tonic cerebral blood flow (34, 35) and regulate hypercapnia-induced cerebral vasodilation in healthy humans (1, 6, 35). The role of COX in hypoxic vasodilation is not well explored in humans. The limited amount of human data suggests COX products are not important for hypoxic cerebral vasodilation (6). Animal data conversely indicate COX products are important for hypoxic vasodilation (3, 10) and MetSyn animals demonstrate a loss of vasodilating COX products (prostacyclin) (5). It remains unknown if decreased contribution of prostacyclin accounts for impaired hypoxia-mediated cerebral vasodilation demonstrated in MetSyn humans (15).
The purpose of this study was to determine the regulatory role of COX products in hypoxic cerebral vasodilation in younger adults with MetSyn. We hypothesized adults with MetSyn would exhibit impaired hypoxic vasodilation due to decreased contribution of vasodilating prostaglandins. We also studied hypercapnic vasodilation for comparison with previous findings and to investigate the vasodilatory capacity in younger adults with MetSyn. Finally, we focused on young adults (∼30 yr old) to determine whether MetSyn confers negative vascular consequences independent of the aging process associated with clinical cerebrovascular disease.
MATERIALS AND METHODS
Subjects
Thirteen adults with metabolic syndrome (MetSyn, 33 ± 3 yr) and fifteen lean, healthy controls (31 ± 2 yr) participated in this study. In addition to providing written informed consent, each subject completed a physical activity and healthy history questionnaire. All subjects were sedentary (<60 moderate-intensity aerobic exercise/wk) and free of overt cardiovascular, neurological, cerebrovascular, metabolic, pulmonary, gastrointestinal, renal, and all other chronic diseases (self-report). Subjects were not taking regular medication of any kind. Females were studied in the early follicular phase of their menstrual cycle (oral contraception allowed) and were required to take a urine pregnancy test to exclude pregnancy.
MetSyn participants met the NCEP-ATP III definition of metabolic syndrome as modified by the American Diabetes Association and International Diabetes Federation (13). Adults with MetSyn were required to meet three of the following five criteria: blood glucose ≥100 mg/dl, resting blood pressure ≥135/≥ 85 mmHg, waist circumference >102 cm men, >88 cm women, triglycerides ≥150 mg/dl, and/or HDL <40 mg/dl women, <50 mg/dl men. Healthy controls were excluded if they met any of the criteria for MetSyn.
Subjects reported to the lab after a 12 h fast, including abstaining from exercise, alcohol, NSAIDs, and caffeine for 18 h. All procedures adhered to the Declaration of Helsinki and were approved by the University of Wisconsin-Madison Health Science Institutional Review Board.
Measurements
Height and weight were measured to calculate body mass index (BMI, kg/m2). Waist and hip circumference were measured to confirm adiposity. Prior to undergoing any study conditions, blood was drawn from an antecubital vein and analyzed for glucose and cholesterol concentration to confirm the presence of MetSyn. Throughout the study visits, subjects were continuously monitored with a pulse oximeter (arterial oxygen saturation, SpO2), automatic sphygmomanometer (blood pressure, MBP), and three-lead electrocardiogram (heart rate, HR) (Datex-Ohmeda; Helsinki, Finland).
A 2-MHz transcranial Doppler ultrasound probe (Neurovision model 500M, Multigon Industries,; Yonkers, NY) was held in place over the temporal window by an adjustable headband to measure middle cerebral artery velocity (MCAv). A linear heated pneumotachometer (model 3813, Hans Rudolph, Shawnee, KS) measured respiratory flow and a GEMINI gas analyzer (CWE, Ardmore, PA) continuously measured inspiratory and expiratory gas concentration.
Protocol
Subjects reported to the laboratory on two separate occasions. The timeline of each visit is illustrated in Fig. 1. Using a double-blind, placebo-controlled design, we tested cerebral vasodilation to graded hypoxia and hypercapnia with acute oral administration of indomethacin to inhibit COX. Subjects were in the semirecumbent position throughout both study visits. After baseline data collection, subjects were given either indomethacin (100 mg) or placebo pills in randomized order. Maalox (20 ml) was administered with both sets of pills to minimize gastrointestinal discomfort. Hemodynamic and respiratory data were collected every 10 min in 5-min intervals following administration of the pharmaceutical. Ninety minutes following oral administration of the pharmaceutical, hypoxia and hypercapnia trials were conducted in randomized order.
Fig. 1.

Experimental timeline of study visits. Placebo and indomethacin were administered in a double-blind, randomized order. Hypoxia and hypercapnia were performed in randomized order each visit. Indo, Indomethacin; SpO2, pulse oximeter arterial saturation; FiCO2, fraction of inspired CO2; HR, heart rate; BP, blood pressure; MCAv, middle cerebral artery velocity; PetCO2, end-tidal partial pressure of carbon dioxide.
Hypoxia.
Hypoxia methods have been described earlier (15). After 5 min of baseline, room air breathing, graded isocapnic hypoxia was induced by titrating inspired oxygen (FiO2) to achieve and sustain 5 min of SpO2 = 90% followed by 5 min of SpO2 = 80%. An SpO2 = 90% is approximately an arterial Po2 = 60 mmHg and an SpO2 = 80% is approximately an arterial Po2 = 43 mmHg (32). The composition of inspired air was controlled using a gas mixer and a two-way non-rebreathing valve (2700 Series, Hans Rudolph) with the subject wearing a nose clip. Throughout the trial, end-tidal carbon dioxide (PetCO2) was maintained at baseline levels by adding CO2 to the blended gas to isolate the effects of lowered SpO2.
Hypercapnia.
The hypercapnia protocol is identical to previously reported research methods (15). Briefly, subjects breathed through a mouthpiece connected to a three-way sliding valve (model 2870, Hans Rudolph) while nasal airflow was occluded. A meteorological balloon filled with a hyperoxic (FiO2 = 0.40), hypercapnic (FiCO2 = 0.03) mix of gas was attached to the three-way sliding valve. The balloon was filled to a volume 1 liter above vital capacity as estimated based on sex, height, and age. After 5 min of steady-state room air breathing, subjects were switched to breathing the hyperoxic, hypercapnic mix of gas until PetCO2 reached 10 mmHg above baseline values (∼2 min). The trial was repeated and results were averaged.
Plasma Assay of COX Metabolites
Blood samples were drawn at baseline and 90 min after oral administration of indomethacin and placebo. Blood was centrifuged and plasma was drawn off and stored at −80°C. Commercial enzyme immunoassay kits (Cayman Chemical, Ann Arbor, MI) were used to measure plasma concentrations of 6-keto-prostaglandin F1α and 2,3-dinor thromboxane B2. Only plasma samples from the indomethacin day were used for analysis due to discontinuation of an assay kit.
Data Acquisition and Analysis
Continuous MCAv, heart rate, and pulse oximetry were collected along with breath-by-breath gas analysis. Data were digitized, recorded, and stored using PowerLab and LabChart (ADInstruments, Colorado Springs, CO) and analyzed offline. To investigate the effect of oral administration of the drugs over the 90 min of drug wash-in, the last 30 s of baseline and steady-state data were averaged. For hypoxia trials, the last 30 s of baseline and steady-state data were used for analysis. Hypercapnia data analysis included averaging the last 30 s of baseline and the last 10 s of hypercapnia to examine the peak response to CO2. Blood pressure was recorded during the last 30 s of each condition.
To account for group differences in perfusion pressure, MCAv was divided by MBP and is presented as cerebrovascular conductance index (CVCi = MCAv × 100/MBP) (15, 24). The primary analysis was to investigate whether cerebral vasodilation to hypoxia or hypercapnia was different between groups. Cerebral vasodilation was defined as a positive change of CVCi from baseline in response to hypoxia or hypercapnia (ΔCVCi = CVCiCondition − CVCiBaseline). Additionally, analysis was conducted to examine the contribution of COX products to both hypoxia and hypercapnia cerebral vasodilation. The contribution of COX products was quantified as a change in the ΔCVCi to both hypoxia and hypercapnia conditions (Change of ΔCVCi = ΔCVCiIndomethacin − ΔCVCiPlacebo).
Statistical analysis was performed with Minitab 16 (State College, PA). Subject characteristics were compared using an unpaired Student's t-test. Group differences in cerebral vasodilation to hypoxia and hypercapnia with and without indomethacin were analyzed using a repeated measures analysis of variance to determine the significance of the fixed effect of group (MetSyn, lean), condition (baseline, hypoxia or hypercapnia), and indomethacin (placebo, indomethacin) on dependent variables of interest. Based on our previous research showing decreased hypoxia-mediated cerebral vasodilation (15), a sample size of n = 11 is necessary to provide 80% power to detect a 20% group difference in ΔCVCi at α = 0.05. Data are presented as means ± standard error of the mean (SE). Significance was determined a priori at P ≤ 0.05. Bonferroni post hoc analyses were conducted when significance was detected.
RESULTS
Subject characteristics are listed in Table 1. Groups were well matched for age. MetSyn subjects displayed significantly greater weight, BMI, waist circumference, hip circumference, and waist-to-hip ratio. Despite similar clinically normal blood glucose values, MetSyn subjects exhibited greater insulin, LDL cholesterol, and triglyceride concentrations along with lower HDL cholesterol. Arterial blood pressure was also higher in MetSyn subjects (Table 1).
Table 1.
Subject characteristics
| Healthy | MetSyn | |
|---|---|---|
| n (M/F) | 15 (8/7) | 13 (9/4) |
| Age, yr | 31 ± 2 | 33 ± 3 |
| Height, cm | 172 ± 2 | 174 ± 3 |
| Weight, kg | 66 ± 2 | 110 ± 6* |
| BMI, kg/m2 | 23 ± 1 | 37 ± 2* |
| Waist, cm | 83 ± 2 | 117 ± 4* |
| Hip, cm | 98 ± 2 | 122 ± 4* |
| Waist/hip ratio | 0.85 ± 0.02 | 0.96 ± 0.02* |
| Glucose, mg/dl | 71 ± 2 | 79 ± 4 |
| Total cholesterol, mg/dl | 161 ± 7 | 173 ± 8 |
| HDL, mg/dl | 65 ± 5 | 43 ± 3* |
| LDL, mg/dl | 79 ± 7 | 101 ± 8* |
| TC/HDL ratio | 2.7 ± 0.2 | 4.2 ± 0.3* |
| Non-HDL/HDL ratio | 1.7 ± 0.2 | 3.2 ± 0.3* |
| Triglycerides, mg/dl | 87 ± 10 | 173 ± 24* |
| Systolic BP, mmHg | 116 ± 2 | 134 ± 3* |
| Diastolic BP, mmHg | 76 ± 1 | 86 ± 3* |
| Mean BP, mmHg | 89 ± 2 | 102 ± 2* |
| Insulin, μU/ml | 10 ± 1 | 17 ± 2* |
| PAQ, kcal/wk | 1098 ± 139 | 1694 ± 503 |
Values are means ± SE. MetSyn, metabolic syndrome; M, males; F, females; BMI, body mass index; HDL, high-density lipoprotein; LDL, low-density lipoprotein; TC/HDL ratio, total cholesterol-to-HDL ratio; Non-HDL/HDL ratio, non-HDL-to-HDL ratio; BP, blood pressure; PAQ, physical activity questionnaire score.
P < 0.05 vs. healthy.
Basal Brain Blood Flow: Effect of Indomethacin
Hemodynamic and gas exchange variables collected during the 90 min of drug wash-in are listed in Table 2. MetSyn adults displayed higher MBP, lower MCAv, and lower CVCi (Table 2). Placebo did not significantly change MBP, MCAv, or CVCi in either group (Table 2).
Table 2.
Hemodynamic and gas exchange variables before and 90 min after placebo or indomethacin
| Healthy |
MetSyn |
|||
|---|---|---|---|---|
| Placebo | Indo | Placebo | Indo | |
| MBP, mmHg* | ||||
| Baseline | 89 ± 2 | 89 ± 2 | 103 ± 2 | 101 ± 3 |
| 90 min | 90 ± 2 | 94 ± 2 | 104 ± 2 | 104 ± 3 |
| PetCO2, mmHg* | ||||
| Baseline | 37 ± 1 | 38 ± 1 | 37 ± 1 | 37 ± 1 |
| 90 min | 39 ± 1 | 38 ± 0 | 38 ± 1 | 37 ± 1 |
| SpO2, %* | ||||
| Baseline | 98 ± 0 | 98 ± 0 | 97 ± 0 | 97 ± 0 |
| 90 min | 98 ± 0 | 98 ± 0 | 98 ± 0 | 97 ± 0 |
| MCAv, cm/s*†‡§ | ||||
| Baseline | 72 ± 3 | 71 ± 3 | 65 ± 4 | 63 ± 4 |
| 90 min | 73 ± 4 | 45 ± 2 | 66 ± 5 | 42 ± 3 |
| ΔMCAv | ||||
| 90 min | −27 ± 2 | −22 ± 2 | ||
| %ΔMCAv | ||||
| 90 min | −37 ± 2 | −34 ± 2 | ||
| CVCi, cm·s−1 ·mmHg−1*†‡§ | ||||
| Baseline | 82 ± 4 | 81 ± 4 | 64 ± 4 | 63 ± 4 |
| 90 min | 82 ± 5 | 48 ± 3 | 64 ± 5 | 40 ± 3 |
| ΔCVCi* | ||||
| 90 min | −33 ± 3 | −23 ± 2 | ||
| %ΔCVCi | ||||
| 90 min | −41 ± 2 | −36 ± 3 | ||
Values are means ± SE.
Indo, indomethacin; MBP, mean blood pressure; PetCO2, end-tidal carbon dioxide; SpO2, pulse oximeter arterial saturation; MCAv, middle cerebral artery velocity; ΔMCAv, absolute change in MCAv; %ΔMCAv, relative change in MCA; CVCi, cerebrovascular conductance index; ΔCVCi, absolute change in CVCi; %ΔCVCi, relative change in CVCi.
Main effect of group,
main effect of time,
main effect of Indo,
Indo × time interaction (P < 0.05).
Indomethacin did not increase MBP; however, indomethacin decreased MCAv (Table 2). The absolute and relative decrease in MCAv was similar between groups (Table 2). Additionally, indomethacin decreased CVCi in both groups (Fig. 2, A and B). The absolute decrease in CVCi (ΔCVCi) was greater in healthy subjects, but the relative change (%ΔCVCi) was similar between groups (Table 2).
Fig. 2.

A: indomethacin (Indo) decreased baseline cerebrovascular conductance index (CVCi) in healthy subjects 30 min following administration. B: Indo also decreased baseline CVCi in metabolic syndrome (MetSyn) subjects 30 min following administration. C: Indo caused a greater absolute decrease of CVCi in healthy subjects. D: Indo caused a similar relative decrease in CVCi between groups. *P < 0.05 vs. 0 min; †P < 0.05 vs. placebo; ‡P < 0.05, main effect of group.
Hypoxia-Mediated Cerebral Vasodilation: Role of COX
Table 3 contains the hemodynamic and gas exchange variables collected during the hypoxia trials. By design, there was a significant main effect of hypoxia as determined by arterial pulse oximeter saturation (SpO2). Although MetSyn adults had higher MBP and lower end-tidal carbon dioxide (PetCO2) at baseline, hypoxia did not affect either variable in lean or MetSyn adults (Table 3). With placebo, ΔCVCi was higher in healthy subjects compared with MetSyn (P < 0.05, Fig. 3E).
Table 3.
Hemodynamic and gas exchange variables during hypoxia
| Healthy |
MetSyn |
|||
|---|---|---|---|---|
| Placebo | Indo | Placebo | Indo | |
| MBP, mmHg* | ||||
| Baseline | 90 ± 2 | 95 ± 2 | 102 ± 3 | 105 ± 2 |
| 90% | 94 ± 2 | 94 ± 2 | 105 ± 3 | 104 ± 3 |
| 80% | 92 ± 3 | 95 ± 2 | 105 ± 3 | 104 ± 4 |
| PetCO2, mmHg* | ||||
| Baseline | 39 ± 1 | 39 ± 0 | 38 ± 1 | 37 ± 1 |
| 90% | 39 ± 0 | 39 ± 0 | 38 ± 1 | 37 ± 1 |
| 80% | 39 ± 0 | 39 ± 0 | 38 ± 1 | 38 ± 1 |
| SpO2, %† | ||||
| Baseline | 98 ± 0 | 98 ± 0 | 98 ± 0 | 98 ± 0 |
| 90% | 90 ± 0 | 90 ± 0 | 90 ± 0 | 90 ± 0 |
| 80% | 80 ± 0 | 81 ± 0 | 80 ± 0 | 80 ± 0 |
| FiO2, %† | ||||
| Baseline | 19 ± 0 | 19 ± 0 | 20 ± 0 | 20 ± 0 |
| 90% | 12 ± 0 | 12 ± 0 | 13 ± 0 | 12 ± 0 |
| 80% | 9 ± 0 | 10 ± 0 | 10 ± 0 | 9 ± 0 |
| MCAv, cm/s*†‡ | ||||
| Baseline | 73 ± 5 | 48 ± 2 | 66 ± 4 | 44 ± 3 |
| 90% | 79 ± 5 | 51 ± 3 | 68 ± 5 | 48 ± 3 |
| 80% | 85 ± 5 | 55 ± 3 | 71 ± 5 | 54 ± 3 |
| ΔMCAv, cm/s†‡ | ||||
| 90% | 6 ± 1 | 3 ± 1 | 2 ± 1 | 4 ± 1 |
| 80% | 11 ± 2 | 8 ± 1 | 8 ± 2 | 12 ± 1 |
| Indo Change ΔMCAv* | ||||
| 90% | −3 ± 1 | 2 ± 1 | ||
| 80% | −4 ± 2 | 4 ± 2 | ||
| CVCi, cm·s−1 ·mmHg−1*†‡§ | ||||
| Baseline | 82 ± 5 | 51 ± 3 | 65 ± 5 | 42 ± 3 |
| 90% | 86 ± 6 | 55 ± 3 | 65 ± 5 | 46 ± 3 |
| 80% | 94 ± 7 | 59 ± 3 | 68 ± 5 | 53 ± 3 |
| ΔCVCi, cm·s−1 ·mmHg−1†‡§ | ||||
| 90% | 3 ± 1 | 4 ± 1 | 0 ± 1 | 4 ± 1 |
| 80% | 11 ± 2 | 8 ± 2 | 5 ± 2 | 13 ± 2 |
| Indo Change ΔCVCi* | ||||
| 90% | 0 ± 1 | 4 ± 1 | ||
| 80% | −3 ± 2 | 7 ± 3 | ||
Values are means ± SE. Indo, indomethacin; MBP, mean blood pressure; FiO2, fraction of inspired oxygen; Indo Change ΔMCAv, magnitude of change of ΔMCAv after Indo; Indo Change ΔCVCi, magnitude of change of ΔCVCi after Indo.
Main effect of group,
main effect of hypoxia,
main effect of Indo,
group × Indo interaction (P < 0.05).
Fig. 3.

CVCi vs. oxygen saturation (SpO2) with and without Indo in healthy (A) and MetSyn (B) adults. C: Indo tended to decrease dilation in healthy subjects at SpO2 = 80% (P = 0.13). D: Indo increased cerebral vasodilation in MetSyn adults. E: with placebo, ΔCVCi was lower in adults with MetSyn. F: Indo influenced cerebral vasodilation differently in healthy and MetSyn adults. *Main effect of group (P < 0.05), †main effect of Hypoxia, ‡main effect of Indo, §group × Indo interaction (P < 0.05).
Indomethacin did not significantly affect PetCO2, SpO2, or MBP (Table 3). Indomethacin significantly reduced both MCAv and CVCi (P < 0.05, Table 3). However, there was a group-specific effect on hypoxia-mediated cerebral vasodilation (ΔCVCi). Indomethacin significantly increased ΔCVCi in adults with MetSyn (P < 0.05, Fig. 3D) along with a trend for decreasing ΔCVCi in healthy controls at SpO2 = 80% (P = 0.13, Fig. 3C). The directionally different effect of indomethacin on ΔCVCi lead to a significant difference in the magnitude of change in cerebral vasodilation caused by indomethacin (P < 0.05, Fig. 3F).
Hypercapnia-Mediated Cerebral Vasodilation: Role of COX
The hemodynamic and gas exchange variables collected during hypercapnia trials are listed in Table 4. Hypercapnia significantly increased PetCO2 and SpO2 in both groups, while MBP was unchanged (Table 4). With placebo, there was no group difference in absolute ΔMCAv or ΔCVCi (Table 4, Fig. 4).
Table 4.
Hemodynamic and gas exchange variables during hypercapnia
| Healthy |
MetSyn |
|||
|---|---|---|---|---|
| Placebo | Indo | Placebo | Indo | |
| MBP, mmHg* | ||||
| Baseline | 91 ± 2 | 94 ± 2 | 102 ± 3 | 104 ± 3 |
| Hypercapnia | 94 ± 2 | 95 ± 2 | 105 ± 3 | 108 ± 4 |
| PetCO2, mmHg*† | ||||
| Baseline | 39 ± 0 | 38 ± 0 | 38 ± 1 | 37 ± 1 |
| Hypercapnia | 49 ± 0 | 48 ± 0 | 47 ± 1 | 47 ± 1 |
| SpO2, %*† | ||||
| Baseline | 98 ± 0 | 98 ± 0 | 98 ± 0 | 98 ± 0 |
| Hypercapnia | 100 ± 0 | 100 ± 0 | 99 ± 0 | 99 ± 0 |
| MCAv, cm/s†‡ | ||||
| Baseline | 71 ± 3 | 48 ± 3 | 65 ± 4 | 46 ± 3 |
| Hypercapnia | 87 ± 4 | 56 ± 3 | 84 ± 5 | 56 ± 3 |
| ΔMCAv, cm/s‡ | ||||
| Hypercapnia | 17 ± 1 | 8 ± 1 | 20 ± 2 | 10 ± 1 |
| Indo Change ΔMCAv | ||||
| Hypercapnia | −9 ± 1 | −9 ± 2 | ||
| CVCi, cm·s−1·mHg−1*†‡ | ||||
| Baseline | 80 ± 5 | 51 ± 3 | 64 ± 4 | 44 ± 2 |
| Hypercapnia | 94 ± 5 | 59 ± 4 | 81 ± 5 | 52 ± 3 |
| ΔCVCi, cm·s−1·mmHg−1‡ | ||||
| Hypercapnia | 15 ± 1 | 8 ± 1 | 16 ± 1 | 8 ± 1 |
| Indo Change ΔCVCi | ||||
| Hypercapnia | −8 ± 1 | −9 ± 1 | ||
Values are means ± SE.
Main effect of group,
main effect of hypercapnia,
main effect of Indo (P < 0.05).
Fig. 4.
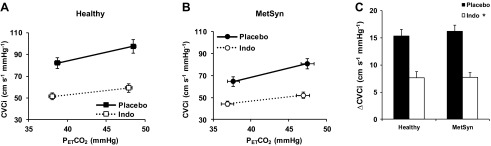
CVCi vs. PetCO2 with and without Indo in healthy (A) and MetSyn adults (B). C: ΔCVCi was similar between groups with and without Indo. *Main effect of Indo (P < 0.05).
Once again, indomethacin did not have a significant effect on PetCO2, SpO2, or MBP. Indomethacin significantly reduced MCAv and CVCi (Table 4) as well as the responses to hypercapnia: ΔMCAv (Table 4) and ΔCVCi (P < 0.05, Fig. 4). The indomethacin-mediated reduction in ΔMCAv (Table 4) and ΔCVCi (Table 4) was similar between groups.
Basal Plasma Concentration of COX Metabolites
Plasma concentrations of 6-keto prostaglandin F1α and 2,3-dinor thromboxane B2 are summarized in Table 5. MetSyn adults exhibited greater plasma concentration of 6-keto prostaglandin F1α (P < 0.05). Indomethacin decreased plasma concentration of 6-keto prostaglandin F1α (P < 0.05) by ∼50% in healthy adults and 47% in MetSyn adults. Plasma concentration of 2,3-dinor thromboxane B2 tended be higher in MetSyn adults (P = 0.10). Indomethacin decreased plasma concentration of 2,3-dinor thromboxane B2 (P < 0.05) by ∼89% in healthy adults and 90% in MetSyn adults.
Table 5.
Plasma concentration of arachidonic acid metabolites before and 90 min after indomethacin administration
| Healthy | MetSyn | |
|---|---|---|
| 6-keto-PGF1α, pg/ml*† | ||
| Baseline | 214 ± 31 | 372 ± 49 |
| 90 min | 109 ± 48 | 199 ± 64 |
| 2,3-dinor TxB2, pg/ml† | ||
| Baseline | 323 ± 85 | 476 ± 74 |
| 90 min | 36 ± 8 | 49 ± 14 |
Values are mean ± SE. For 6-keto-PGF1α, we assayed plasma from 12 healthy and 12 MetSyn adults. For 2,3-dinor TxB2, we assayed plasma from 11 healthy and 11 MetSyn adults. 6- keto-PGF1α, 6-keto prostaglandin F1α; 2,3-dinor TxB2, 2,3-dinor thromboxane B2.
Main effect of group,
main effect of time (P < 0.05).
DISCUSSION
The primary purpose of this study was to test the hypothesis that adults with MetSyn display impaired hypoxic cerebral vasodilation due to loss of COX vasodilator products. Several novel findings are worth highlighting. First, the role of COX-mediated prostaglandins in maintaining basal cerebral blood flow is preserved in MetSyn. Second, hypercapnia-mediated cerebral vasodilation is preserved in MetSyn, as is the role of vasodilator prostaglandins. Third, COX plays little to no role in mediating hypoxic cerebral vasodilation in healthy controls. Fourth, contrary to the hypothesis, COX inhibition increased hypoxic cerebral vasodilation in adults with MetSyn. Collectively, these findings indicate younger adults with MetSyn maintain the capacity to dilate the cerebral vasculature to hypercapnia, but a distinct shift in COX signaling restrains cerebral vasodilation in response to hypoxia. This profound change in COX signaling in young adults with MetSyn supports the concept of preclinical vascular impairments developing decades prior to clinical manifestation of cerebrovascular disease, providing an opportunity to intervene and restore vascular function.
Hypoxia
This is the first study to investigate a potential mechanism responsible for decreased hypoxia-mediated cerebral vasodilation in MetSyn humans. Based on previous research, it was hypothesized adults with MetSyn would display reduced hypoxia-mediated cerebral vasodilation and reduced contribution of vasodilating prostaglandins. Importantly, this study confirms the MetSyn-dependent impairment of hypoxia-mediated cerebral vasodilation in humans (15) and animals (22). Contrary to the hypothesis, the present data indicate prostaglandins do not contribute to hypoxic cerebral vasodilation in healthy controls (Fig. 3). Moreover, the increase in ΔCVCi with indomethacin in MetSyn suggests COX predominately produces a vasoconstrictor product in this population. These findings parallel data from hypertensive adults in whom COX products limit acetylcholine-mediated vasodilation in the forearm (27). We speculate the most likely candidate is hypoxia-induced production of thromboxane A2, a powerful vasoconstrictor and platelet aggregator (9).
Literature indicates the contribution of COX products to hypoxia-mediated cerebral vasodilation is variable and dependent on the severity and duration of hypoxia, indomethacin dosage, measurement technique, and model studied. Animal data indicate COX is important for hypoxia-mediated increases cerebral blood flow in newborn piglets (3), as well as dilating isolated rat middle cerebral arteries during severe hypoxia (10). In contrast, recent evidence in humans indicates indomethacin does not alter hypoxia-mediated cerebral vasodilation (6). To a certain degree, this is in line with our control group where indomethacin tended to decrease vasodilation at SpO2 = 80% (P = 0.13). The present results differ slightly from Fan et al. (6), most likely due to our longer duration of hypoxia exposure. The present study held 5 min of steady-state hypoxia at SpO2 = 80%, whereas Fan et al. (6) terminated the hypoxia trial upon briefly reaching SpO2 = 80%. Although animal data indicate COX is an important contributor to hypoxic cerebral vasodilation (3, 10), human data indicate little (present data) or no role (6) for COX-derived vasodilator prostaglandins in mediating hypoxia-induced cerebral vasodilation in healthy humans, leaving alternate mechanisms to be explored.
We speculate our findings establish inducible thromboxane production as a likely mechanism responsible for impaired hypoxic vasodilation in MetSyn, but the signal which induces thromboxane production was not tested in this study. Correlative analysis (data not shown) indicates the collective accumulation of MetSyn criteria, and not an individual risk factor, is responsible for the decreased hypoxia-mediated cerebral vasodilation. We further speculate the combined impact of cardiovascular risk factors likely induces a low-grade vascular inflammation and oxidative stress in the MetSyn adults (29), which may enhance the production of eicosanoids and modify their effects from vasodilation to vasoconstriction (8). Although we did not measure inflammation and oxidative stress in the current cohort, MetSyn adults from another study in our lab (18) demonstrate greater levels of C-reactive protein (2.0 mg/dl) compared with healthy controls (0.4 mg/dl), which is consistent with early, subclinical cardiovascular disease. While the mechanistic origin driving this change is not apparent, our data clearly demonstrate COX-derived vasoconstriction limits hypoxic cerebral vasodilation in young adults with MetSyn.
Although it is assumed the main target of indomethacin is endothelial COX, it is possible indomethacin affects vascular smooth muscle. Research conducted in a rat model of MetSyn indicates cerebral vascular smooth muscle produces vasoconstricting prostaglandins (thromboxane) when mitochondrial ATP-sensitive potassium channels are activated (16), which could potentially be activated by hypoxia (28). Thus our findings cannot specify the exact source of vasoconstrictor, but these observations are consistent with the conclusion that hypoxia induced preferential production of vasoconstricting prostanoids in MetSyn.
In summary, current results indicate adults with MetSyn exhibit impaired hypoxia-mediated cerebral vasodilation. While this is consistent with an animal model of MetSyn, novel findings demonstrate the mechanism of impaired hypoxic cerebral vasodilation is different in humans. Rather than a loss of vasodilating COX products, the present findings indicate hypoxia elicits production of the vasoconstrictor thromboxane in MetSyn adults, which limits cerebral vessel dilation.
Hypercapnia
MetSyn adults displayed preserved hypercapnia-mediated cerebral vasodilation, confirming previous findings (15) and advancing our understanding that MetSyn adults maintain the ability to utilize vasodilator prostaglandins to mediate ∼50% of hypercapnia responses. Impaired hypoxic cerebral vasodilation in the face of preserved hypercapnic vasodilation suggests these younger adults with MetSyn retain the capacity to dilate the cerebral vasculature, but MetSyn negatively impacts the functional vascular responses to hypoxia. Further, the similar reduction in vasodilation to hypercapnia (Fig. 4) with indomethacin indicates COX-mediated vasodilation is preserved, but MetSyn promotes activation of a vasoconstrictor pathway during hypoxia. In other words, both groups retain the enzymatic capacity to activate vasodilator prostaglandins in cerebral vessels, but MetSyn alters endothelial signaling during hypoxia such that a vasoconstrictor prostaglandin signal dominates. Thromboxane is a well-known vasoconstrictor and a potent thrombus activator; thus an increase in inducible thromboxane signaling may help explain the increased risk of ischemic stroke in MetSyn humans.
In summary, COX products have consistently been shown to contribute to hypercapnia-mediated cerebral vasodilation (1, 7, 35) and we show the contribution of COX products is preserved in younger adults with MetSyn. These findings indicate adults with MetSyn retain the capacity to dilate to hypercapnia and the functional decline is specific to hypoxia. These are also the first data demonstrating the role of COX mediating vasodilation is fundamentally different between hypercapnia and hypoxia vasodilation in the same human volunteers.
Experimental Considerations
The current research approach has several factors worthy of consideration. Principally, measuring MCAv with TCD is an estimate of blood flow through the MCA. Since the cross-sectional area of the MCA remains constant during the severities of hypoxia and hypercapnia used in this study (23, 33), changes in MCAv accurately reflect changes in cerebral blood flow. Newer evidence suggests the diameter of the MCA may change with more severe alterations in blood gases than used in this study (33). Second, SpO2 and PetCO2 were used as quantitative surrogate measures of arterial blood gas values to minimize invasiveness of the study and should not limit the conclusions as there is no reason to suspect the oxyhemoglobin dissociation curve is altered in MetSyn, and since PetCO2 has been commonly used (1, 15, 35) as an accurate and reliable estimate of arterial CO2 (20). Third, a standard oral dose of indomethacin (100 mg) (6, 7, 34, 35) resulted in healthy subjects receiving a significantly greater relative dose of indomethacin (1.5 mg/kg), raising the possibility that MetSyn subjects were not adequately dosed to inhibit COX (0.9 mg/kg). However, data from the onset of action of indomethacin (Fig. 2), similar decreases in COX metabolites (Table 5), and similar decreases in hypercapnia-mediated responses (Fig. 4) indicate indomethacin adequately inhibited COX to the same extent in both groups. Given the directional effect of indomethacin during hypoxia was opposite to the hypothesis, underdosing in MetSyn may underestimate the contribution of COX restricting hypoxic cerebral vasodilation. Finally, we did not collect plasma samples during hypoxia to test for increased thromboxane production during hypoxia. We feel this is a minor limitation because our assay of resting plasma samples verifies robust inhibition of thromboxane synthesis and it seems unlikely systemic concentration of thromboxane would accurately reflect local cerebrovascular production of COX metabolites. Combining the plasma assay results (Table 5) with the directionally opposite hypoxic responses in MetSyn (Fig. 3) supports our conclusion the vasoconstrictor COX product is thromboxane A2.
Clinical Implications
The novel mechanistic findings of this study are important for understanding the pathophysiology of cerebrovascular disease closer to its induction. In the context of older MetSyn adults (12), our contrasting findings between hypoxic and hypercapnic vasodilation critically draw attention to the fact that a single vascular function test would fail to identify early functional decline in the cerebral circulation of this young diseased population. When studying early disease processes, it may be prudent to include multiple physiological testing to maximize physiological insight into disease progression at its inception.
Previous studies indicate cerebrovascular responses to CO2 decline with advancing age (1), particularly in the presence of MetSyn (12). Taken together, evidence indicates aging with MetSyn may be more detrimental to cerebrovascular function than healthy aging. This concept is consistent with the observation that the existence of MetSyn in middle-aged men nearly doubles the risk of stroke (17). Finally, detection of cerebrovascular function in ∼30-yr-old MetSyn adults opens a substantial window of opportunity to prevent further functional decline, and the indomethacin results point to a potential target to restore function.
Conclusion
In conclusion, young adults with MetSyn demonstrate marked reductions in hypoxia-mediated cerebral vasodilation. In contrast to our hypothesis, inhibition of COX improved cerebral vasodilation in MetSyn, indicating COX products restrain hypoxia-mediated cerebral vasodilation. In contrast to hypoxia, cerebrovascular reactivity to hypercapnia is preserved in MetSyn, as is the contribution of COX vasodilators. The collective interpretation is, decades prior to clinical manifestation of cerebrovascular disease, younger adults with MetSyn produce a COX-derived vasoconstrictor signal, likely thromboxane A2, which limits cerebral vasodilation to hypoxia. These mechanistic findings have important implications for understanding the progression of cerebrovascular disease as this rapidly growing clinical population ages, providing an opportunity to intervene and restore vascular function.
GRANTS
Funding for this project was provided by the American Heart Association (11PRE7390038, J. W. Harrell), National Institutes of Health (HL-105820, W. G. Schrage), and the University of Wisconsin School of Medicine and Public Health Shapiro Summer Research Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.W.H. and W.G.S. conception and design of research; J.W.H. performed experiments; J.W.H. analyzed data; J.W.H. and W.G.S. interpreted results of experiments; J.W.H. prepared figures; J.W.H. drafted manuscript; J.W.H. and W.G.S. edited and revised manuscript; J.W.H. and W.G.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all participants. Additionally, we thank P. A. Yanke, G. L. Peltonen, C. L. Rousseau, R. E. Johansson, M. K. Crain, G. P. Barton and the Wisconsin National Primate Research Center (insulin assay) for technical assistance.
All experiments were conducted in the Bruno Balke Biodynamics Lab on campus at the University of Wisconsin-Madison.
REFERENCES
- 1.Barnes JN, Schmidt JE, Nicholson WT, Joyner MJ. Cyclooxygenase inhibition abolishes age-related differences in cerebral vasodilator responses to hypercapnia. J Appl Physiol 112: 1884–1890, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boden-Albala B, Sacco RL, Lee HS, Grahame-Clarke C, Rundek T, Elkind MV, Wright C, Giardina EG, DiTullio MR, Homma S, Paik MC. Metabolic syndrome and ischemic stroke risk: Northern Manhattan Study. Stroke 39: 30–35, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coyle MG, Oh W, Petersson KH, Stonestreet BS. Effects of indomethacin on brain blood flow, cerebral metabolism, and sagittal sinus prostanoids after hypoxia. Am J Physiol Heart Circ Physiol 269: H1450–H1459, 1995 [DOI] [PubMed] [Google Scholar]
- 4.Di Carlo A. Human and economic burden of stroke. Age Ageing 38: 4–5, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Erdos B, Miller AW, Busija DW. Impaired endothelium-mediated relaxation in isolated cerebral arteries from insulin-resistant rats. Am J Physiol Heart Circ Physiol 282: H2060–H2065, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Fan JL, Burgess KR, Thomas KN, Peebles KC, Lucas SJ, Lucas RA, Cotter JD, Ainslie PN. Influence of indomethacin on the ventilatory and cerebrovascular responsiveness to hypoxia. Eur J Appl Physiol 111: 601–610, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Fan JL, Burgess KR, Thomas KN, Peebles KC, Lucas SJ, Lucas RA, Cotter JD, Ainslie PN. Influence of indomethacin on ventilatory and cerebrovascular responsiveness to CO2 and breathing stability: the influence of Pco2 gradients. Am J Physiol Regul Integr Comp Physiol 298: R1648–R1658, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Feletou M, Cohen RA, Vanhoutte PM, Verbeuren TJ. TP receptors and oxidative stress hand in hand from endothelial dysfunction to atherosclerosis. Adv Pharmacol 60: 85–106, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feletou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol 164: 894–912, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fredricks KT, Liu Y, Rusch NJ, Lombard JH. Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. Am J Physiol Heart Circ Physiol 267: H580–H586, 1994 [DOI] [PubMed] [Google Scholar]
- 11.Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, Montori VM. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol 49: 403–414, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Giannopoulos S, Boden-Albala B, Choi JH, Carrera E, Doyle M, Perez T, Marshall RS. Metabolic syndrome and cerebral vasomotor reactivity. Eur J Neurol 17: 1457–1462, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112: 2735–2752, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, Vinters HV, Holtzman DM, Rosenberg GA, Wallin A, Dichgans M, Marler JR, Leblanc GG. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37: 2220–2241, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Harrell JW, Morgan BJ, Schrage WG. Impaired hypoxic cerebral vasodilation in younger adults with metabolic syndrome. Diab Vasc Dis Res 10: 135–142, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katakam PV, Domoki F, Snipes JA, Busija AR, Jarajapu YP, Busija DW. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol 296: R289–R298, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurl S, Laukkanen JA, Niskanen L, Laaksonen D, Sivenius J, Nyyssonen K, Salonen JT. Metabolic syndrome and the risk of stroke in middle-aged men. Stroke 37: 806–811, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Limberg JK, Harrell JW, Johansson R, Eldridge MW, Proctor LT, Sebranek JJ, Schrage WG. Microvascular function in younger adults with obesity and metabolic syndrome: role of oxidative stress. Am J Physiol Heart Circ Physiol 305: H1230–H1237, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marshall RS, Rundek T, Sproule DM, Fitzsimmons BF, Schwartz S, Lazar RM. Monitoring of cerebral vasodilatory capacity with transcranial Doppler carbon dioxide inhalation in patients with severe carotid artery disease. Stroke 34: 945–949, 2003 [DOI] [PubMed] [Google Scholar]
- 20.McSwain SD, Hamel DS, Smith PB, Gentile MA, Srinivasan S, Meliones JN, Cheifetz IM. End-tidal and arterial carbon dioxide measurements correlate across all levels of physiologic dead space. Respir Care 55: 288–293, 2010 [PMC free article] [PubMed] [Google Scholar]
- 21.Panza F, Frisardi V, Capurso C, Imbimbo BP, Vendemiale G, Santamato A, D'Onofrio G, Seripa D, Sancarlo D, Pilotto A, Solfrizzi V. Metabolic syndrome and cognitive impairment: current epidemiology and possible underlying mechanisms. J Alzheimers Dis 21: 691–724, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R522–R530, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Poulin MJ, Robbins PA. Indexes of flow and cross-sectional area of the middle cerebral artery using Doppler ultrasound during hypoxia and hypercapnia in humans. Stroke 27: 2244–2250, 1996 [DOI] [PubMed] [Google Scholar]
- 24.Reichmuth KJ, Dopp JM, Barczi SR, Skatrud JB, Wojdyla P, Hayes D, Jr, Morgan BJ. Impaired vascular regulation in patients with obstructive sleep apnea: effects of continuous positive airway pressure treatment. Am J Respir Crit Care Med 180: 1143–1150, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez-Colon SM, Mo J, Duan Y, Liu J, Caulfield JE, Jin X, Liao D. Metabolic syndrome clusters and the risk of incident stroke: the atherosclerosis risk in communities (ARIC) study. Stroke 40: 200–205, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Silvestrini M, Vernieri F, Pasqualetti P, Matteis M, Passarelli F, Troisi E, Caltagirone C. Impaired cerebral vasoreactivity and risk of stroke in patients with asymptomatic carotid artery stenosis. JAMA 283: 2122–2127, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Taddei S, Virdis A, Mattei P, Salvetti A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 21: 929–933, 1993 [DOI] [PubMed] [Google Scholar]
- 28.Tomiyama Y, Brian JE, Jr, Todd MM. Cerebral blood flow during hemodilution and hypoxia in rats: role of ATP-sensitive potassium channels. Stroke 30: 1942–1947; discussion 1947–1948, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Van Guilder GP, Hoetzer GL, Greiner JJ, Stauffer BL, Desouza CA. Influence of metabolic syndrome on biomarkers of oxidative stress and inflammation in obese adults. Obesity (Silver Spring) 14: 2127–2131, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Vieira JR, Elkind MS, Moon YP, Rundek T, Boden-Albala B, Paik MC, Sacco RL, Wright CB. The metabolic syndrome and cognitive performance: the Northern Manhattan Study. Neuroepidemiology 37: 153–159, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wannamethee SG, Shaper AG, Lennon L, Morris RW. Metabolic syndrome vs. Framingham Risk Score for prediction of coronary heart disease, stroke, and type 2 diabetes mellitus. Arch Intern Med 165: 2644–2650, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Willie CK, Macleod DB, Shaw AD, Smith KJ, Tzeng YC, Eves ND, Ikeda K, Graham J, Lewis NC, Day TA, Ainslie PN. Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol 590: 3261–3275, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson MH, Edsell ME, Davagnanam I, Hirani SP, Martin DS, Levett DZ, Thornton JS, Golay X, Strycharczuk L, Newman SP, Montgomery HE, Grocott MP, Imray CH. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia—an ultrasound and MRI study. J Cereb Blood Flow Metab 31: 2019–2029, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie A, Skatrud JB, Barczi SR, Reichmuth K, Morgan BJ, Mont S, Dempsey JA. Influence of cerebral blood flow on breathing stability. J Appl Physiol 106: 850–856, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie A, Skatrud JB, Morgan B, Chenuel B, Khayat R, Reichmuth K, Lin J, Dempsey JA. Influence of cerebrovascular function on the hypercapnic ventilatory response in healthy humans. J Physiol 577: 319–329, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]