

Abstract
Pulmonary arterial hypertension is a severe progressive disease with marked morbidity and high mortality in which right ventricular (RV) failure is the major cause of death. Thus knowledge of the mechanisms underlying RV failure is an area of active interest. Previous studies suggest a role of NADPH oxidase in cardiomyocyte dysfunction in the left heart. Here we postulate that acute pressure overload induced by pulmonary artery banding (PAB) leads to a Nox4-initiated increase in reactive oxygen species (ROS) in mouse RV that may lead to feed-forward induction of Nox2. To test our hypothesis, ROS production was measured in RV and left ventricle homogenates. The data show that hydrogen peroxide (H2O2), but not superoxide anion (O2·−), was increased in the early phases (within 6 h) of PAB in RV and that this increase was diminished by catalase and diphenyleneiodonium chloride but not by SOD, Nω-nitro-l-arginin methyl ester, febuxostat, or indomethacin. H2O2 production in RV was not attenuated in Nox2 null mice subjected to 6 h PAB. Moreover, we observed an upregulation of Nox4 mRNA after 1 h of PAB and an increase in mitochondrial Nox4 protein 6 h post-PAB. In contrast, we observed an increase in Nox2 mRNA 1 day post-PAB. Expression of antioxidant enzymes SOD, catalase, and glutathione peroxidase did not change, but catalase activity increased 6 h post-PAB. Taken together, these findings show a role of mitochondria-localized Nox4 in the early phase of PAB and suggest an involvement of this isozyme in early ROS generation possibly contributing to progression of RV dysfunction and failure.
Keywords: NADPH oxidase, reactive oxygen species, pulmonary artery banding, right ventricle
pulmonary arterial hypertension (PAH) is a severe progressive disease characterized by a sustained increase in the pulmonary vascular resistance leading to right ventricular (RV) failure and death (13, 17). Although many drugs are available for the treatment of this disease, mortality remains high principally due to the RV failure, whose mechanisms are not fully known. Emerging evidence reveals that reactive oxygen species (ROS), generated in cardiomyocytes by different sources among which the most important are mitochondria, NADPH oxidase (Nox), and nitric oxide synthases (4), may be a driver of cardiac disease progression.
Many studies showed the importance of Nox-derived ROS in the progression of cardiac hypertrophy (8, 12, 21, 28, 29). The two Nox isoforms reported to be most expressed and have functional effects in left heart cardiac myocytes have been Nox2 and Nox4 (36). In general, Nox2 is normally quiescent and is acutely activated by stimuli such as G protein-coupled receptor agonists (e.g., angiotensin II, endothelin-1), growth factors, and cytokines (39) to generate superoxide (O2·−), which, in turn, is rapidly dismuted to hydrogen peroxide (H2O2) through the activity of SOD (7, 25). In contrast, Nox4 is constitutively active, and seems to be regulated largely by changes in transcription and translation and/or post-translational modifications (7, 10, 25, 34, 40). In this respect, Nox4 may be regarded as an inducible isoform (39). Additionally, Nox4 appears to generate predominantly H2O2 rather than O2·−, although this is still somewhat controversial and may involve close association of the isoform with SOD (4, 24). Moreover, the intracellular locations of the two isoforms in cardiomyocytes are distinct (39). There is broad consensus that activated Nox2 is found predominantly on the plasma membrane, whereas Nox4 is found in multiple organelles intracellularly (7, 24). Nox2 may be activated by several different cardiac stressors, and an increasing number of studies suggest important pathophysiological roles for this isoform in cardiac disease (8, 12, 31, 39). Nox4 expression increases during stress such as hypoxia (30, 39), mitochondrial dysfunction (18, 41), and in in vivo left ventricle (LV) pressure overload (12, 24, 42), and recent studies have shown both a deleterious and a protective role of Nox4 in response to chronic LV pressure overload (33, 42).
Much less is known about the role of Nox2 and Nox4 in the RV. In this study, we investigate the role of Nox isoforms using pulmonary artery (PA) banding (PAB), a surgical model of RV pressure overload, and hypothesize that PAB results in an increase in ROS production due to Nox4, which is responsible for cardiac dysfunction and RV failure. Our findings show that PAB, within the first 6 h, induces a primary increase in Nox4-derived ROS, and an upregulation of mitochondria-localized Nox4.
MATERIAL AND METHODS
Pulmonary artery banding.
Male C57BL6/J wild-type, Nox2 null, and p47phox null mice, 6–8 wk old, were obtained from Jackson Laboratory (Bar Harbor, ME). PAB was performed as previously described (20). Briefly, mice were anesthetized with an intraperitoneal injection of etomidate (5–10 mg/kg-1), intubated, and placed on a ventilator (Type845; Harvard Apparatus, Holliston, MA) using a tidal volume of ∼225 μl and a respiratory rate of ∼200 breaths/min. Mice were then placed supine on a heated pad to maintain a body temperature of 37°-39°C. Body temperature was monitored via rectal probe thermometer (THM100; Indus Instruments, Houston, TX). A left thoracotomy was performed and the PA carefully dissected free from the aorta. A surgical clip (Weck 005200; Research Triangle Park, North Carolina, NC) that had been calibrated to a 27-gauge diameter was placed around the PA to create pressure afterload on the RV. After banding, the thoracic cavity was closed in layers, and the mice were placed on a heating pad until they regained their righting reflex and became ambulatory. This banding produces a pressure overload of the RV with an RV end-systolic pressure of ∼35 mmHg (data not shown). Sham-operated animals underwent the same procedure except for the banding of the PA. Animals were euthanized at the indicated times and hearts collected. The RV was carefully separated from the LV and septum and Fulton Index (RV wet weight/LV + septum wet weight) determined. For all assays the tissues were homogenized using a scinted glass mortar on glass pestle connected to an electric motor. An appropriate buffer of homogenization for the assay employed was used and maintained at 4°C during homogenization. All animal studies were performed under a protocol approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
H2O2 production (Amplex Red assay).
H2O2 production was measured in tissue homogenates using Amplex Red. Frozen RV or LV from sham or mice subjected to PAB were homogenized in PBS containing protease inhibitors cocktail (Roche Diagnostics GmbH, Mannheim, Germany), Phospho-Stop cocktail (Roche Diagnostics GmbH, Mannheim, Germany), and 0.1 mM PMSF. The homogenates were centrifuged at 1,000 g for 10 min at 4°C to eliminate unbroken tissues, and an appropriate amount of supernatant (equivalent to 50 μg of protein) was loaded into black 96-well plates in assay buffer (25 mM Hepes, pH 7.4, containing 0.12 M NaCl, 3 mM KCl, 1 mM MgCl2, 0.1 mM Amplex Red, and 0.32 U/ml horseradish peroxidase) in the presence or absence of different inhibitors: 3,000 U/ml catalase, 20 μM diphenylene iodonium (DPI), 200 U/ml SOD, 10 nM febuxostat, 10 μM indomethacin, and 100 μM Nω-nitro-l-arginin methyl ester (l-NAME). Fluorescence was measured using a Biotek Synergy 4 hybrid multimode microplate reader with a 530/25-excitation and a 590/35-emission filter, for 1 h and 30 min at 37°C.
Superoxide production (cytochrome c assay).
Frozen RV or LV from sham mice and mice subjected to PAB were homogenized in lysis buffer composed of 8 mM potassium, sodium phosphate buffer (pH 7.0), 131 mM NaCl, 340 mM sucrose, 2 mM NaN3, 5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, protease inhibitor cocktail, and 1 mM dithiothreitol and then subjected to 5 freeze/thaw cycles and passed five times through a 30-gauge needle to lyse the tissues. Homogenates were centrifuged at 1,000 g for 10 min at 4°C, and the supernatants were centrifuged at 28,000 g for 15 min at 4°C to separate the membrane fraction from the cytosol. Finally, pellets were resuspended in 50 μl (for the RV) or 100 μl (for the LV) of lysis buffer. Membrane fractions (5 μg) in oxidase assay buffer consisting of 65 mM sodium phosphate buffer (pH 7.0), 1 mM EGTA, 10 μM FAD, 1 mM MgCl2, 2 mM NaN3, and 0.2 mM cytochrome c (3, 26, 35, 37, 38) were loaded into a clear 96-well plate. O2·− production was initiated by the addition of 180 μM NADPH, and absorbance measured at 550 nm in a Biotek Synergy 4 hybrid multimode microplate reader. The initial linear rate of the curves with or without SOD was measured and O2·− production was expressed as SOD-inhibitable cytochrome c reduction. The rate of O2·− generated was tabulated using extinction coefficient 21.1 mM−1cm−1.
Western blot of SOD1, catalase, glutathione peroxidase (GPx), and Nox4.
Frozen RV or LV from sham-operated mice and mice subjected to PAB were homogenized in radioimmunoprecipitation assay (RIPA) buffer composed of 150 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton, 1% deoxycolate, and 0.1% SDS (pH 7.4) with the addition of protease inhibitor and Phospho-Stop cocktails. The homogenates were centrifuged at 13,000 rpm for 15 min at 4°C. Tissue supernatants (50 μg of protein) were subjected to SDS-PAGE. Blots were probed with goat polyclonal anti-SOD1 (1:1,000; Everest Biotech), rabbit polyclonal anti-catalase (1:1,000), rabbit polyclonal anti-GPx (1:10,000), and rabbit polyclonal anti-Nox4 (1:1,000 Catalog No. NB110-58849, Novus Biologicals, Littleton, CO) antibodies and then incubated with their respective secondary antibodies (1:10,000, IRDye antibodies; Li-Cor Biotechnology). For mitochondrial Nox4 evaluation, tissues were homogenized in sucrose-Tris-EGTA (STE) mitochondrial extraction buffer composed of (in mM) 300 sucrose, 50 KCL, 2 EGTA, and 20 Tris (pH 7.35) in the presence of protease inhibitor and Phospho-Stop cocktails, centrifuged at 1,300 g for 5 min and the supernatant centrifuged at 10,000 g for 10 min to pellet mitochondria. The pellet was then resuspended in RIPA buffer, and Western blot was carried out as described above. All membranes were reprobed with a mouse monoclonal β-actin antibody (1:500; Santa Cruz Biotechnology) as loading controls. Membranes from isolated mitochondria blots were reprobed with cyochrome c oxidase I (COX I) antibody (1:1,000, Mitosciences, Eugene, OR) as loading controls. Blots were scanned using the Odyssey infrared imaging system (Li-Cor Biotechnology) and densitometric analysis performed using the program ImageJ.
Catalase activity.
Catalase activity was determined by spectrophotometry according to the standard protocol following the method by Aebi (1). Briefly, 50 μg of RV homogenates was added to phosphate buffer solution (pH = 7) containing H2O2 (10 mM). H2O2 absorbance was measured at 240 nm to obtain the rate of H2O2 decomposition as absorbance per minute. Catalase activity was calculated in terms of units per milligram of protein where 1 unit is defined as the quantity of catalase that scavenges 1.0 μmol of H2O2 per min at pH = 7.0 and 25°C.
Real-time quantitative PCR.
Total RNA was extracted from tissues using RNeasy Plus Kit (Qiagen) as per the manufacturer's protocol. All RNA samples were quantified by spectrophotometry, and 1 μg of total RNA was used in reverse transcription to obtain cDNA. Commercially available primers used in quantitative PCR were Nox2 Catalog No. 4331182 Mm01287742_m1 TaqMan Gene Expression assay; Nox4 Catalog No. 4331182 Mm00479246_m1 TaqMan Gene Expression assay; and HPRT1 Catalog No. 4331182 Mm03024075_m1 TaqMan Gene Expression assay (Applied Biosystems). HPRT1 was used as a housekeeping gene. Real-time quantitative PCR was performed with the ABI 7700 detection system (Applied Biosystems). The reaction was carried out using TaqMan Gene expression Assays (TaqMan MGB probes, FAM dye-labeled): 5 μl of TaqMan Universal PCR Master Mix, No Amperase UNG (2×) fast reaction, 0.5 μl 20× TaqMan Gene expression Assay Mix (primers), and 4.5 μl of the proper dilution of cDNA for a final volume of 10 μl per reaction (assays were performed in 384-well plates). The thermal cycling program consisted of an initial denaturation for 20 s at 95°C, followed by 40 cycles of 1 s at 95°C and 20 s at 60°C. All analyses were done in triplicate. All results are normalized for the housekeeping gene HPRT1, and relative quantification was obtained using the ΔCt (threshold cycle) method; relative expression was calculated as 2−ΔΔCt.
Statistical analysis.
All results are expressed as means ± SE. Paired Student's t-test for single-paired point comparisons and a two-way ANOVA with replication followed by a Tukey's post hoc modified t-test for group comparisons were used. P < 0.05 was considered statistically significant. The number of animals used in each experiment is represented by n values. For each animal, experiments were performed in triplicate with the exception of Western blots, which were performed in singlicate.
RESULTS
Increased RV and LV ROS production after PAB.
For a comprehensive overview of the production of ROS in RV and LV, we began by measuring H2O2 production, a more general indicator of oxidase activity, at different time points after PAB. In the RV of mice subjected to PAB, an increase in fluorescence, expressed as relative fluorescence units (RFU), compared with its respective sham time control (assigned a value of 100%) is shown. There was an 18.00 ± 12.00%, 39.17 ± 19.86%, and 49.85 ± 10.96% increase over sham at 1, 3, and 6 h after PAB, respectively (n = 3–9; P < 0.05 vs. sham at 6 h). No increase was observed at 1 day after PAB (96.87 ± 16.39% of sham; n = 3; Fig. 1A). In contrast, there was no change in H2O2 production in the LV of PAB mice at any time point (87.84 ± 11.27%, 86.56 ± 7.47%, 86.55 ± 11.36%, and 95.02 ± 3.25% vs. sham at 1, 3, and 6 h and 1 day after PAB, respectively; Fig. 1B).
Fig. 1.

H2O2 levels increase in early points of pulmonary artery banding (PAB) in right ventricle (RV) but not in left ventricle (LV). Homogenates of RV and LV were incubated with Amplex Red solution, and fluorescence was measured for 1 h and 30 min at 37°C. H2O2 level is expressed as percentage of relative fluorescence units (RFU; arbitrary fluorescence units) of the respective sham for each time point. The RFU refers to the fluorescence measured in the linear part of the curve of H2O2 production. Results are expressed as means ± SE; n = 3–11 animals for the RV (A) and n = 3 animals for the LV (B). *P < 0.05 indicates significant difference vs. sham-operated animals.
RV and LV O2·− production after PAB.
O2·− production was measured by SOD-inhibitable cytochrome c reduction in RV (Fig. 2A) and LV (Fig. 2B). In the RV, O2·− production expressed as picomole O2·− per minute per milligram protein was 1.51 ± 0.19 vs. 1.27 ± 0.18 at 1 h, 1.79 ± 0.28 vs. 1.06 ± 0.10 at 3 h, 1.58 ± 0.17 vs. 1.65 ± 0.32 at 6 h, and 1.48 ± 0.27 vs. 1.69 ± 0.21 at 1 day (24 h) for sham versus PAB, respectively (n = 3–5). These differences were not significant at 1, 6, and 24 h. However, PAB caused a paradoxical significant decrease at 3 h (Fig. 2A). There was no difference between sham and PAB mice LV O2·− production at any time point (1, 3, and 6 h and 1 day; Fig. 2B).
Fig. 2.

O2·− production does not change after PAB in RV and LV. To membrane fractions of RV and LV, 180 μM of NADPH were added to initiate the reaction of O2·− production. O2·− production was measured by the linear rate of SOD-inhibitable cytochrome c reduction in RV (A) and LV (B). Results are expressed as picomoles O2·− per minute per milligram protein (prot) as means ± SE; n = 3–5 animals. *P < 0.05 indicates significant difference vs. sham.
Effect of ROS inhibitors on H2O2 production in RV after PAB.
Pharmacological inhibitors were used to explore whether RV Nox could be involved in whether RV Nox could be involved in H2O2 production, to verify that H2O2 measured was not due to the dismutation of O2·−, and to verify the identity of H2O2. RV homogenates from 1-, 3-, and 6 h and 1 day PAB or their respective sham samples were treated with DPI (20 μM), l-Name (100 μM), febuxostat (10 nM), indomethacin (10 μM), SOD (200 U/ml), and catalase (3,000 U/ml). DPI significantly decreased activity in sham and PAB at all time points (1, 3, and 6 h and 1 day), suggesting that a flavoprotein such as Nox was involved in the RV H2O2 generation (Fig. 3, A–D). Suppression of fluorescence for sham-operated and PAB mice at all time points by catalase corroborated that the fluorescence was specific for H2O2. There was no effect of SOD in any group, suggesting that O2·− did not contribute to the signal; that is, the data indicated that H2O2 production is primary and immediate and not due to dismutation of O2·−. l-NAME, febuxostat, and indomethacin inhibited H2O2 production at 1 and 3 h, suggesting a contribution of nitric oxide synthase, xanthine oxidase, and cyclooxygenase, respectively, on H2O2 generation at these time points. Importantly, there was no significant inhibition by l-NAME, febuxostat, and indomethacin at 6 h of PAB, supporting a primarily Nox-derived H2O2 production at this time point.
Fig. 3.

Effect of reactive oxygen species (ROS) scavengers and inhibitors on H2O2 levels after PAB. RV homogenates of mice subjected to sham vs. PAB for 1 h (A), 3 h (B) 6 h (C), or 1 day (D) were preincubated with catalase (Cat; 3,000 U/ml), SOD (200 U/ml), diphenylene iodonium (DPI; 20 μM), Nω-nitro-l-arginin methyl ester (l-NAME; 100 μM), febuxostat (Feb; 10 nM), or indomethacin (Ind; 10 μM), and H2O2 production was assessed by Amplex Red fluorescence measured for 1 h and 30 min at 37°C. H2O2 production is expressed as percentage of RFU of the respective sham for each time point. Results are expressed as means ± SE; n = 3–6 animals. *P < 0.05 indicates significant difference vs. sham without inhibitors (w/o inhib); #P < 0.05 indicates significant difference vs. PAB without inhibitors.
mRNA expression of Nox4 and Nox2 at early time points after PAB in the RV.
To confirm the role of Nox in PAB, we analyzed RV tissue for mRNA expression of the two most prevalent Nox isoforms reportedly expressed in mouse hearts, Nox2 and Nox4. We observed an upregulation of Nox4 mRNA expression in the early phase of PAB (significant after 1 h vs. sham, n = 3; Fig. 4) and a later more modest upregulation of Nox2 mRNA expression (statistically significant after 1 day, n = 5; Fig. 4). These findings suggested an early activation of Nox4 in PAB followed by induction of Nox2 expression.
Fig. 4.

Increase of Nox2 and Nox4 mRNA expression in RV after PAB. Nox2 and Nox4 mRNA expression was measured in RV homogenates normalized to HRPT1 as a housekeeping gene. Results are expressed as fold change of respective sham for each time point. Data represent means ± SE; n = 3–6 animals. *P < 0.05 indicates significant difference vs. sham.
Nox4 protein expression in mitochondria and in total homogenates from RV.
Nox4 has been reported to be expressed in the mitochondria of various cell types (9, 24). Inasmuch as the mitochondria are expected to be an abundant and significant source of ROS in the RV, we isolated RV mitochondria from sham- and PAB-operated mice at 6 h and measured Nox4 protein levels. We found a nearly twofold increase in Nox4 protein expression in mitochondria from PAB versus sham mice (Fig. 5, A and B). To test whether the increase in mitochondrial Nox4 protein expression in response to PAB also coincided with global increase in Nox4 in RV cardiomyocytes, we analyzed Nox4 expression in RV homogenates in wild-type and Nox2 null mice subjected to PAB. Our data show that Nox4 in total homogenates did not change in response to PAB compared with sham-operated controls (Fig. 6). Moreover, Nox4 expression was similar in PAB Nox2 null mice compared with PAB wild-type mice, suggesting that the absence of Nox2 did not affect Nox4 protein expression under PAB (Fig. 6).
Fig. 5.

Nox4 protein expression increases in mitochondria after 6 h of PAB. Nox4 protein expression was measured by Western blot in isolated mitochondria from RV and normalized (Norm) to protein levels of mitochondria-specific cytochrome c oxidase I (COXI). A: representative blot. B: densitometry quantification of the blots. Data are expressed as means ± SE of fold change from sham; n = 4–8 animals. *P < 0.05 indicates significant difference vs. sham.
Fig. 6.
Nox4 expression in RV total homogenates. Nox4 protein expression was measured in total RV homogenates of wild-type (WT) and Nox2 null mice after 6 h PAB and normalized to β-actin. Representative blot. n = 2 to 3 animals.
Increased RV H2O2 production at early time points of PAB in Nox2 null mice.
We measured H2O2 production in Nox2 null mice to test whether the absence of Nox2 negates RV H2O2 production after PAB as a result of Nox2 activity or Nox2-induced Nox4 activity (15, 19). Nox2-derived O2·− is typically dismuted to H2O2, and Nox4 appears to directly generate H2O2. Thus Amplex Red fluorescence is a measure of ROS production from both Nox isoforms. We detected an increase in signal comparable with the increase shown in wild-type mice (see Fig. 3C) after 6 h PAB (n = 3, P < 0.05; Fig. 7), an effect abolished by catalase and DPI but unaltered by SOD. These findings suggest that Nox4 rather than Nox2 is responsible for ROS after 6 h PAB and that Nox2 deletion is inconsequential at early time points of pressure overload-induced ROS production.
Fig. 7.

H2O2 levels in RV of Nox2 null mice after 6 h of PAB. Homogenates of RV from Nox2 null mice were preincubated with Cat (3,000 U/ml), SOD (200 U/ml), or DPI (20 μM), and H2O2 production was measured by Amplex Red fluorescence for 1 h and 30 min at 37°C. H2O2 production is expressed as percentage of RFU of the respective sham for each time point. Results are expressed as means ± SE; n = 3 animals. *P < 0.05 indicates significant difference vs. sham without inhibitors; #P < 0.05 indicates significant difference vs. PAB without inhibitors.
Protein expression of RV antioxidant enzymes (catalase, SOD, and GPx) does not change in response to PAB.
An increase of oxidative stress is usually followed by an increase in antioxidant enzymes expression and/or activity. Therefore, we analyzed the protein expression of the major antioxidant systems present in the heart, catalase, SOD, and GPx (Fig. 8). We did not find a difference in catalase (Fig. 8B), SOD (Fig. 8C), or GPx (Fig. 8D) expression between sham-operated and PAB mice at any time point. This suggested that alterations in ROS levels were not a consequence of changes in the expression of major antioxidant systems controlling ROS.
Fig. 8.
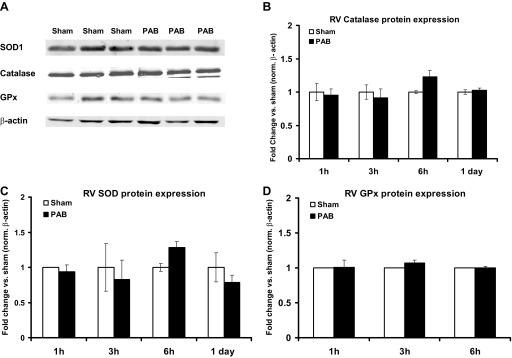
Catalase, SOD, and glutathione peroxidase (GPx) protein expression do not change after PAB. Catalase, SOD, and GPx protein expression were measured in RV after PAB by Western blot. β-Actin was used as a loading control. A: representative blots of protein expression in sham and PAB at 3 h. B–D: quantification of catalase (B), SOD (C), and Gpx (D) protein expression in response to PAB for the indicated times. Results are expressed as means ± SE of fold change from the respective sham for each time point; n = 3–6 animals. Norm, normalized.
Catalase activity increase after 6 h of PAB.
Since expression of antioxidants was not altered in response to PAB, we tested whether activity of antioxidants increased. Because H2O2 was the major ROS observed to increase in response to PAB, we measured catalase activity. There was a twofold increase in catalase activity after 6 h of PAB compared with sham-operated mice (Fig. 9). In contrast, catalase activity was unaltered between PAB and sham at 1 and 3 h.
Fig. 9.

Catalase activity increases after 6 h of PAB. Catalase activity was measured 1, 3, and 6 h after PAB in RV. Results are expressed as means ± SE of fold changes from sham; n = 3 animals. *P < 0.05 indicates significant difference between PAB and sham.
DISCUSSION
In this study, we show for the first time a rapid induction in RV Nox expression and H2O2 levels within the first 6 h (acute phase) of pressure overload in PA-banded mice, a surgical model capable of inducing pure hemodynamic stress in the RV and maladaptive remodeling and failure within 2 to 3 wk (chronic phase) of sustained pressure overload (20). This effect was specific for the RV, because no increase of H2O2 was shown in LV, suggesting that this model does not affect generalized heart ROS production. In contrast, at all time points there was no difference in RV O2·− levels between sham and PAB mice, suggesting that O2·− is not the major ROS produced following acute PAB. In contrast, pharmacological experiments supported that H2O2 is the major ROS in the RV post-PAB and suggested that NADPH oxidase is the likely source of the H2O2 at 6 h of PAB. Nox isoform expression profile revealed an early fourfold increase in Nox4 expression 1 h post-PAB followed by a twofold increase in Nox2 expression at 1 day post-PAB. Western blot analysis demonstrated an early approximately twofold increase in mitochondrial Nox4 expression in response to 6 h PAB, supporting a role for mitochondrial Nox4 in this process. This finding was supported by data in which PAB-induced ROS was not attenuated in RV from Nox2 null mice. Finally, our data demonstrated that changes in H2O2 production in response to PAB coincided with an increase in catalase activity at 6 h of PAB but not with changes in expression of catalase or the other ROS scavenging enzymes SOD, and GPx.
Of the major findings of this study are data generated from two independent gold-standard techniques of ROS detection demonstrating that the primary ROS produced as an immediate result of pressure overload was H2O2 and not O2·−. Incubation with catalase abolished PAB-induced ROS elevation in RV at every time point, supporting that the observed Amplex Red fluorescence was H2O2 specific and not due to other sources, such as autofluorescence of the tissue homogenates for example. The lack of effect of added SOD confirmed that the observed H2O2 is not a consequence of O2·− production and subsequent dismutation at the time points analyzed. In fact, O2·− levels were significantly reduced 3 h postbanding. These data are different from previous findings which implicated mainly O2·− in the dysfunctional heart (8, 24). Inasmuch as the current study explored the effects of pressure overload on the right ventricle per se, this discrepancy may be due to multiple factors including 1) differences in the cellular Nox composition in those studies which were performed in the LV (vs. the RV), 2) the particular model used and differences in the level and type of hemodynamic stress, and 3) unique cellular signaling within the two tissue types. Assessing distinctions in signaling upstream and downstream of Nox between the right and left ventricle is an area of current investigation.
The Nox family is a major source of ROS and consists of seven members, Nox1, Nox2, Nox3, Nox4, Nox5, and dual oxidase 1 and 2, that display distinct subcellular localizations, tissue distributions, mechanisms of regulation and activities, and (patho)physiological functions (11). In general, the literature reports that Nox2 and Nox4 are major cardiomyocyte Nox isoforms. However, most, if not all, of these data are derived from studies of the LV. Thus, little to no information is available on the role of Nox in the RV. Our data suggest that Nox4 is the source of the increase in H2O2 observed following PAB. That is, PAB-induced H2O2 increase 1) was attenuated by incubation of RV homogenates with the iodonium compound DPI, an inhibitor of flavin-containing enzymes including Nox at all time points but not by incubation with l-NAME, indomethacin, or febuxostat; 2) exhibited an early increase in RV Nox4 and not Nox2 mRNA as well as an increase in mitochondrial Nox4 protein; and 3) was unaffected by deletion of Nox2. The data are in agreement with mounting evidence that the primary product of Nox4 is H2O2 (4, 42) and is supported by findings of attenuated H2O2 generation in heart tissue from Nox4 null mice (42). Our data are also in agreement with studies by Byrne et al. (12) who demonstrated that the rise in ROS levels in the LV in response to pressure overload was not attenuated in Nox2 null hearts. Moreover, Looi et al. (31) observed a similar lack of effect of Nox2 deletion on ROS in a model of myocardial infarction. Our data are unique in that they are, to our knowledge, the first to implicate Nox4-derived H2O2 in RV pressure overload and are supported by studies showing that Nox4 is involved in LV pressure overload-induced heart failure (12, 24) and that Nox4 worsens heart function (24).
mRNA expression profiles demonstrated an increase in Nox4 mRNA 1 h post-PAB followed by an increase in Nox2 mRNA 1 day post-PAB. Thus the data appear to suggest a positive feed-forward mechanism whereby Nox4-derived H2O2 during the early phase of pressure overload leads to subsequent upregulation of Nox2-derived ROS in response to sustained chronic pressure overload and a chain reaction, which ultimately leads to RV dysfunction. Nox2 activation (i.e., as a result of post-translational modification and/or recruitment of cytosolic activating subunits) did not likely play a role in the early phase as evidenced by sustained (<1 day) PAB-induced ROS in Nox2 null mice. Indeed, pressure overload leads to RV dysfunction in this model, and deletion of Nox2 oxidase essential cytosolic subunit p47phox ameliorates the dysfunction (Fig. 10). Thus both Nox4 and Nox2 play critical roles in pressure overload-induced RV dysfunction. This is in close agreement with reports suggesting that Nox enzymes can feed-forward activate other Nox proteins and oxidases via ROS production (4, 15, 19). The factors and mechanisms mediating this feed-forward chain of Nox activation leading to RV dysfunction require deeper investigation and are currently part of a much larger study.
Fig. 10.

RV hypertrophy is attenuated in p47phox null mice after PAB. Fulton Index [RV wet weight/LV + septum (S) wet weight] was determined in hearts from sham or 3-wk PAB wild-type mice (WT) vs. 3-wk PAB p47phox nulls. n = 4–6 animals. *P < 0.005 indicates significance between PAB and sham in wild type mice; ***P < 0.005 indicates significance between PAB in wild-type (WT) and p47phox null mice.
Our data demonstrate that mitochondrial protein expression of Nox4 was increased approximately twofold 6 h post-PAB compared with sham-operated mice. To date, Nox4 has been localized to multiple subcellular organelles, and in cardiomyocytes, it appears to be expressed primarily in the mitochondria (9, 24), although this remains controversial (33). Our data showing the presence of Nox4 in isolated mitochondria appear to go some way to solve this controversy. The mitochondria are an important source of ROS in any cell, but in cardiomyocytes evidence suggests that they are the major source of ROS involved in heart failure (2, 4, 24). Thus the upregulation of Nox4 we observe herein in the mitochondria is likely to play a key role in the dysfunction following PAB. Indeed, previous studies demonstrate that Nox4 mediates mitochondrial and cardiac dysfunction in a model of LV pressure overload (24). It is also supported by data in other cells in which mitochondrial Nox4 is a major source of oxidative stress (23). Nox4 has also been found in the endoplasmic reticulum (ER) in vascular smooth muscle endothelium (5, 14) and in focal adhesions, plasma membrane and nucleus in different cell types (6, 22, 27). Mitochondrial preparations used for this set of studies did not contain ER-specific protein disulfide isomerase activity (data not shown). Thus the possibility of contamination of mitochondrial preparations by ER membranes (and thus ER-localized Nox4) was ruled out. Taken together, our data suggest the potential for a feed-forward mechanism in which an increase in mitochondrial Nox4-derived ROS leads to mitochondrial dysfunction and subsequent electron leak from the mitochondrial respiratory chain. This would undoubtedly result in further ROS production, exacerbating cardiomyocyte dysfunction and ultimately RV failure.
Finally, we assessed the potential regulation of ROS scavenging enzymes SOD, catalase, and GPx in response to pressure overload in this model to determine whether they could be playing a role in an elevated ROS steady state. Our data reveal that there is no difference in SOD, catalase, or GPx protein expression in RV between PAB- and sham-operated animals at any of the time points in this study. We did observe, however, a twofold increase in catalase activity at 6 h of PAB, which may explain the observation that H2O2 levels reached after 3 h of PAB are similar to those at 6 h. That is, the increase in catalase activity at 6 h PAB may suggest a steady state between H2O2 production and H2O2 scavenging at this time point. Our findings with respect to SOD expression are in contrast with studies demonstrating an amplification of left heart dysfunction in response to pressure overload in mice deficient in extracellular SOD (32). Consistent with our findings, however, other studies showed a failure to activate ROS, scavenging enzymes in response to pressure overload in the RV (16). These discrepancies in the literature and from our study are likely to be the result of differential responses in antioxidant activation mechanisms between the RV and LV.
Limitations of the study.
A number of limitations in the current study warrant discussion. First, pharmacological inhibitors under some conditions display off-target effects and thus remain limited in their ability to identify the enzymatic source of ROS in tissues. This limitation was addressed by the use of a spectrum of ROS source inhibitors and a process of elimination to implicate a Nox source of PAB-induced ROS in the RV. Second, specificity of Nox antibodies remains a challenge for the field. To address this issue, we tested the specificity of the Nox antibodies in heterologous COS-7 cell systems expressing one particular Nox isoform at a time (data not shown) and confirmed that the immunoblot bands being detected were indeed Nox isoform specific. Our findings using Western blot were also supported by mRNA expression profiles using quantitative PCR. Third, our current study investigated ROS production largely in an acute model of pressure overload, thus limiting the detection of functional effects that do not typically appear until 2 to 3 wk of PAB. Nonetheless, Fig. 10 does support a Nox-dependent mechanism of RV hypertrophy in response to prolonged PAB. Finally, although our data do not currently show direct causality of Nox4 in the acute response to pressure overload, a number of experiments evaluated herein strongly predict its seminal role in early and pivotal ROS production. Future acute and chronic studies will require the use of global or conditional Nox4 nulls, small interfering RNA, or yet unavailable specific inhibitors to demonstrate such causality.
Conclusion
This study sheds important light on the early cellular mechanisms of pressure overload injury in the RV. Our data suggest that mitochondrial Nox4-derived ROS are involved in propagating cardiomyocyte phenotypic changes in PAH that ultimately lead to RV failure. The use of the PAB model afforded us the opportunity to separate RV responses from changes that occur in the pulmonary circulation and thus focus on the cardiomyocyte. There are only a handful of studies investigating the RV in pressure overload and ROS production, and no studies are available on the role of Nox enzymes in this model. Thus the current findings identify an important gap in knowledge and spotlight a novel role for Nox4 in RV dysfunction. Future studies are planned interrogating the mechanisms by which PAB leads, respectively, to Nox4- and Nox2-mediated hypertrophy and RV dysfunction.
GRANTS
For this work, G. Frazziano received support from Fondazione Ri.MED and P. J. Pagano received support from the National Heart, Lung, and Blood Institute (R01HL-079207 and P01HL-103455-01) and is an Established Investigator of the American Heart Association. All Vascular Medicine Institute investigators received support from the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania, PA.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: G.F., I.A.G., H.C.C., and P.J.P. conception and design of research; G.F., I.A.G., J.B., and S.S. performed experiments; G.F., I.A.G., and S.S. analyzed data; G.F., I.A.G., H.C.C., and P.J.P. interpreted results of experiments; G.F., I.A.G., and S.S. prepared figures; G.F., I.A.G., and P.J.P. drafted manuscript; G.F., I.A.G., S.S., H.C.C., and P.J.P. edited and revised manuscript; G.F., I.A.G., J.B., S.S., H.C.C., and P.J.P. approved final version of manuscript.
REFERENCES
- 1.Aebi H. Catalase in vitro. Methods Enzymol 105: 121–126, 1984 [DOI] [PubMed] [Google Scholar]
- 2.Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, Sadoshima J. The NADPH oxidase Nox4 and aging in the heart. Aging (Albany NY) 2: 1012–1016, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al Ghouleh I, Frazziano G, Rodriguez AI, Csanyi G, Maniar S, St Croix CM, Kelley EE, Egana LA, Song GJ, Bisello A, Lee YJ, Pagano PJ. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res 97: 134–142, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med 51: 1271–1288, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 279: 45935–45941, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Anilkumar N, Jose GS, Sawyer I, Santos CX, Sand C, Brewer AC, Warren D, Shah AM. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler Thromb Vasc Biol 33: e104–e112, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105: 293–296, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA 106: 14385–14390, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med 49: 687–706, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 47: 1239–1253, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93: 802–805, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 120: 992–1007, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., JrRegulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol 181: 1129–1139, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datla SR, Griendling KK. Reactive oxygen species, NADPH oxidases, and hypertension. Hypertension 56: 325–330, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ecarnot-Laubriet A, Rochette L, Vergely C, Sicard P, Teyssier JR. The activation pattern of the antioxidant enzymes in the right ventricle of rat in response to pressure overload is of heart failure type. Heart Dis 5: 308–312, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, Chamera E, Corretti MC, Champion HC, Abraham TP, Girgis RE, Hassoun PM. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 174: 1034–1041, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, Slingerland J, Arnold RS, Lambeth JD, Cohen C, Hilenski L, Griendling K, Martinez-Diez M, Cuezva JM, Arbiser JL. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest 117: 719–729, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A–11A, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Hemnes AR, Maynard KB, Champion HC, Gleaves L, Penner N, West J, Newman JH. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ 2: 352–358, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41: 2164–2171, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 677–683, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Kim SM, Kim YG, Jeong KH, Lee SH, Lee TW, Ihm CG, Moon JY. Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PLos One 7: e39739, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565–15570, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Laufs U, Wassmann S, Czech T, Munzel T, Eisenhauer M, Bohm M, Nickenig G. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol 25: 809–814, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Lee YM, Kim BJ, Chun YS, So I, Choi H, Kim MS, Park JW. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell Signal 18: 499–507, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, Steger K, Krause KH, Jaconi ME. The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell 17: 3978–3988, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40: 477–484, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Li S, Tabar SS, Malec V, Eul BG, Klepetko W, Weissmann N, Grimminger F, Seeger W, Rose F, Hanze J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid Redox Signal 10: 1687–1698, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, Marber M, Monaghan MJ, Shah AM. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51: 319–325, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Lu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ, Chen Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 51: 19–25, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol 50: 408–416, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Molshanski-Mor S, Mizrahi A, Ugolev Y, Dahan I, Berdichevsky Y, Pick E. Cell-free assays: the reductionist approach to the study of NADPH oxidase assembly, or “all you wanted to know about cell-free assays but did not dare to ask”. Methods Mol Biol 412: 385–428, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev 16: 5–12, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci USA 94: 14483–14488, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peluffo G, Calcerrada P, Piacenza L, Pizzano N, Radi R. Superoxide-mediated inactivation of nitric oxide and peroxynitrite formation by tobacco smoke in vascular endothelium: studies in cultured cells and smokers. Am J Physiol Heart Circ Physiol 296: H1781–H1792, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med 50: 777–793, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wosniak J, Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 11: 1265–1278, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA 107: 18121–18126, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]