

Abstract
Creatine and phosphocreatine levels are decreased in heart failure, and reductions in myocellular phosphocreatine levels predict the severity of the disease and portend adverse outcomes. Previous studies of transgenic mouse models with increased creatine content higher than two times baseline showed the development of heart failure and shortened lifespan. Given phosphocreatine's role in buffering ATP content, we tested the hypothesis whether elevated cardiac creatine content would alter cardiac function under normal physiological conditions. Here, we report the creation of transgenic mice that overexpress the human creatine transporter (CrT) in cardiac muscle under the control of the α-myosin heavy chain promoter. Cardiac transgene expression was quantified by qRT-PCR, and human CrT protein expression was documented on Western blots and immunohistochemistry using a specific anti-CrT antibody. High-energy phosphate metabolites and cardiac function were measured in transgenic animals and compared with age-matched, wild-type controls. Adult transgenic animals showed increases of 5.7- and 4.7-fold in the content of creatine and free ADP, respectively. Phosphocreatine and ATP levels were two times as high in young transgenic animals but declined to control levels by the time the animals reached 8 wk of age. Transgenic mice appeared to be healthy and had normal life spans. Cardiac morphometry, conscious echocardiography, and pressure-volume loop studies demonstrated mild hypertrophy but normal function. Based on our characterization of the human CrT protein expression, creatine and phosphocreatine content, and cardiac morphometry and function, these transgenic mice provide an in vivo model for examining the therapeutic value of elevated creatine content for cardiac pathologies.
Keywords: cardiac failure, creatine transporter, energy metabolism, phosphocreatins
a hallmark of cardiac failure is a marked disturbance of energy metabolism. In this condition, the manner in which ATP is buffered and transferred, via the creatine kinase (CK) system, from the mitochondria to utilization sites, is profoundly altered (26). In the early stages of heart failure, myocardial creatine (Cr) and phosphocreatine (PCr) are reduced by as much as 70%, whereas a slow and progressive ATP loss of up to 30–40% is observed at the latter stages of the disease (4, 28, 39, 45). Moreover, the PCr-to-ATP ratio decreases, and this decrease is a better predictor of overall and cardiovascular-related mortality than traditional indexes such as ejection fraction or New York Heart Association symptomatology (27). These deficits in high-energy metabolites are accompanied by decreases in the activity and expression levels of the CKs in the myocardium (22, 38). Recent reports elegantly illustrated that increased CK function has a protective effect in murine models of global cardiac ischemia and hypertension-induced heart failure (1, 14). Cardiomyocytes are not capable of Cr synthesis and depend on transport from the extracellular environment by the creatine transporter (CrT) to maintain an adequate intracellular Cr pool. CrT function is reduced in the failing heart (29, 43), and, despite recent progress in our understanding of cellular energetics and the relevance of Cr and PCr in cardiac muscle, a clear understanding of how CrT is regulated in normal and failing hearts is lacking. We have recently shown that, in cardiomyocytes in culture, its function is regulated by substrate availability, AMP-activated protein kinase (AMPK) (8), and protein kinase C (9).
Given the pivotal role of Cr as a component of a spatial/temporal energy shuttle that sustains ATP levels at sites of energy consumption, it has been hypothesized that an increase in myocellular Cr content would be therapeutically valuable in the setting of acute or chronic cardiac injury. Attempts to increase cardiac Cr content and ameliorate cardiac and energetic disarray by oral supplementation in a rat heart failure model have proven unsuccessful (16). On the other hand, depletion of Cr by chronic pharmacological Cr transport or synthesis inhibition had an adverse effect on cardiac function (19, 44). Recent characterization of physiologically relevant regulation of cardiac Cr transport explains these earlier findings; in cardiomyocytes, Cr transport decreases quickly and very significantly in response to increases in extracellular Cr within the time frame and concentrations observed during oral supplementation with Cr (8). To circumvent these limitations and to determine if cardiac Cr content elevations would be beneficial to stressed out cardiac muscle, transgenic mice that overexpress the rabbit CrT were prepared. In these animals, myocardial Cr content was augmented, but these transgenic animals also developed heart failure spontaneously (46). It was proposed that the increase in intracellular Cr led to increased free ADP concentrations that in turn lowered the free energy change of ATP hydrolysis (ΔGATP) required to drive ATP production. However, whether these changes represented an adaptive or causal event of cardiac failure is unknown (34, 46). Further analysis showed that CrT overexpression, resulting in a Cr increase below twofold that of the baseline, was well tolerated. Moreover, such an increase was clearly demonstrated to be protective in the setting of acute myocardial infarction (21).
Recently, the importance of Cr as a spatial and temporal energy buffer in cardiomyocytes has been questioned. Knockout mice lacking the enzyme (guanidine acetate N-methyltransferase) required for the Cr biosynthesis had normal cardiac function and, furthermore, did not appear to have worsened outcomes following myocardial infraction (20). It is not clear how these findings can be reconciled with reports demonstrating the deleterious effects of Cr depletion to cardiac muscle (44).
Here, we report the generation of a transgenic mouse line where cardiac specific overexpression of the human CrT protein resulted in a 5.7-fold increase in Cr content but with normal ventricular function and lifespan.
METHODS
Construction of human CrT transgenic mice.
Human CrT-cDNA was a kind gift from Dr. Marc Caron (Department of Cell Biology, Duke University). The Flag epitope was introduced to the 5′-end of the cDNA. The cDNA encoding the tagged CrT protein was subcloned into pCDNA3.0, yielding a construct where expression was under the control of the α-myosin heavy chain (α-MHC) promoter (10). This promoter is active in both atrial and ventricular cardiomyocytes (35, 40). The resulting construct integrity was verified by expression and function studies in Griptite cells (data not shown; Life Technologies, Carlsbad CA). The Flag-CrT protein functional properties were similar to those obtained from the untagged CrT. Transgenic animals were generated at Duke University's Transgenic Mouse Facility. The above-mentioned construct was microinjected in the pronucleus of fertilized FVB oocytes. The oocytes were implanted in the oviducts of pseudopregnant Swiss-Webster female mice. Pups were screened for transgene insertion by polymerase chain reactions from DNA isolated from their tails and toes, using the 5′-aagaggcagggaagtggtgg-3′ as the forward primer and the 5′-ccccagacagcctcaagact-3′ as the reverse primer. Adult male mice bearing the transgene were bred with FVB mice (NCI-Frederick, Frederick, MD). Animals were handled according to the approved protocols and animal welfare regulations of the Institutional Review Board at Duke University Medical Center.
Transthoracic echocardiography.
Echocardiography was performed on conscious mice using either an HDI 5000 echocardiograph (Phillips) or a Vevo 770 high-resolution imaging system (VisualSonics), as previously described by Tanaka et al. (42).
In vivo pressure-volume analysis in anesthetized mice.
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (2.5 mg/kg), and the left ventricle was cannulated with a 1.4-French conductance catheter (Millar, Houston, TX) as previously described (32). Data were analyzed with PVAN software (versions 3.3 and 3.6; Millar).
Immunoblotting.
Mice were sedated with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (2.5 mg/kg). The thoracic cavity was opened, and the heart was removed, rinsed in saline, separated into the four chambers, weighed, and then flash-frozen in liquid nitrogen in ∼20 s. The heart was subsequently stored at −80°C. The tissue was homogenized over ice in 150 mM NaCl, 50 mM Tris, pH 8.0, 5 mM EDTA, 1% Nonidet P-40, and 0.5% deoxycholate supplemented with protease inhibitors (Complete Mini protease inhibitors; Roche, Indianapolis, IN) and phosphatase inhibitors (in mM: 5 NaF, 1 phenylmethylsulfonyl fluoride, 2.5 Na2P2O7, 50 β-glycerol, and 1 Na3VO3; Sigma, St. Louis, MO) using an electrical tissue homogenizer. Homogenates were centrifuged at 4°C and 100,000 g for 45 min. The protein concentration in the supernatant was determined using the bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL), and equal amounts of protein (5.5 μg/lane) isolated from wild-type (WT) or creatine transporter transgenic (CrT-Tg) hearts were subjected to electrophoresis on a 4–12% Novex Tris-Bis precast gradient gel using MES buffer (Invitrogen, Carlsbad, CA) and Western blot analysis, as described (7).
Immunohistochemistry.
Formalin-fixed ventricles were embedded in paraffin. Four-micrometer sections were cut from the tissue blocks, placed on positive charged slides, allowed to air dry, and then heated in a 65°C oven for 30 min. After removing the paraffin with xylene and clearing with alcohol, the slides were placed in hydrogen peroxide and methanol to quench endogenous peroxidase activity. Sections were hydrated and washed in deionized water. It was determined during antibody optimization that proteinase K (concentrate diluted 0.05 ml in 1.0 ml of 0.05 M Tris, pH 7.5; Dako, Carpinteria, CA) was the proteolytic enzyme of choice. Comparative tissue pretreatment studies were performed using heat-induced epitope retrieval. The tissue sections were digested for 5 min in the working proteinase K solution and then rinsed in deionized water and placed in Tris-buffered saline (TBS), pH 7.5. Primary anti-CrT rat monoclonal antibody 4B9, (7) at a 1:500 dilution, was applied and incubated for 1 h at room temperature. Following a rinse and wash with TBS, the bound primary antibody was linked with biotinylated goat anti-rat IgG (H&L specific, 5 μg/ml; Vector Laboratories, Burlingame, CA). The formed immune complex was further amplified and labeled with horseradish peroxidase conjugated to the avidin biotin complex (ABC Elite; Vector Laboratories). Diaminobenzidine was used to visualize the bound 4B9 antibody. The slides were then washed with tap water. Hematoxylin counterstain was applied, followed by dehydration with absolute alcohol. Finally, the slides were cleared with xylene and cover slipped with a permanent mounting media. For quantification of immunostained cardiomyocytes, sections were analyzed using darkfield and phase-contrast imaging modes. Images were captured and postprocessed using a modified version of ImageJ (National Institutes of Health, Bethesda, MD). Sample edges were discarded to avoid counting errors resulting from mechanical deformation of the sectioned tissue. Segmentation histograms were computed, and all segments within two SDs (total area) of the training set (hand selected) were included in the analysis. A total of 1,935 valid segments were selected in this manner, of which 82 corresponded to fully stained segments (as computed from the average pixel color value). Segmentation across unstained sections presented a challenge in some areas of the sample; thus, an average area calculation was performed as well. The ratio of stained/unstained area (0.047) was very similar to the result previously obtained counting single segments (0.042). The slight increase obtained using the area comparison can be explained because of the presence of very small segments not accounted for in the segmentation procedure.
Real-time RT-PCR.
Total RNA was extracted from left ventricle tissue using an Aurum Total RNA Fatty and Fibrous Kit (Bio-Rad, Hercules, CA), and genomic DNA was eliminated by DNase I digestion. Equal RNA concentrations were used to perform a two-step RT-PCR using oligo(dT), random hexamer primers, and the IScript cDNA Synthesis Kit (Bio-Rad). After cDNA was obtained, qRT-PCR was performed to determine the abundance of mRNA encoding the CrT transgene or the native CrT relative to β-actin mRNA. For the quantification of the CrT transgene, the following primers were used: forward primer: 5′-ctgttgctgcttggtctc-3′ and reverse primer: 5′-ttggaaacggaagtagtagg-3′. For β-actin mRNA, the primers used were: forward primer: 5′-gacaggatgcagaaggagattact-3′ and reverse primer: 5′-tgatccacatctgctggaaggt-3′, as previously described (22, 33). qRT-PCR was done using iQ SYBR Green Supermix (Bio-Rad) and an MX 3005P thermal cycler (Strategene, Santa Clara, CA). Reactions were performed in triplicate and cycle threshold values were analyzed as reported by Pfaffl (33). Efficiencies were calculated using serial dilutions generated from CrT transgene plasmid and WT mice RNA, respectively.
Quantification of ATP, ADP, Cr, and PCr.
Beating hearts were clamped while still in the chest using liquid N2 precooled clamps. The frozen heart “wafers” were stored at −80°C until extract preparation. Samples were submerged in liquid N2, weighed, and ground to a fine powder using disposable individual tissue grinders. Tissue extracts were prepared and subjected to HPLC analysis following the method described by Wiseman et al. (47), assuming an intracellular volume of 0.5 ml/g wet wt. ATP, Cr, and PCr were measured by HPLC, and free ADP levels were calculated based on the CK equilibrium equation: [ADP] = [ATP][Cr]/[PCr] × where square brackets denote concentration. The value for the creatine kinase equilibrium constant depends on the pH and free Mg2+ measurements. Thus, we used the values determined by phosphorus nuclear magnetic resonance spectroscopy in murine hearts as reported by Himmelreich and Dobson (free Mg2+ = 0.4 mM, pH = 7.32) (15). The at 38°C was as reported by Golding et al. (13). Metabolite ratios were calculated using only data from heart extracts where values for both metabolites (e.g., PCr and ATP) were obtained.
Statistical analyses.
Data are reported as means ± SE and were analyzed using STATISTICA 6.1 (StatSoft, 1984–2003) with one- or two-way ANOVA for multiple groups of independent samples, followed by post hoc, pairwise comparisons. A probability value of ≤0.05 was considered significant. Most group comparisons had a 1:1 proportion of male to female animals. Kaplan-Meyer survival analysis was performed using Graphpad Prism (GraphPad, La Jolla, CA).
RESULTS
Phenotype and transgene expression.
Expression of the transgene in CrT-Tg animals was characterized by qRT-PCR (Table 1), Western blotting (Fig. 1A), and immunohistochemical analysis (Fig. 1B). The mRNA encoding the human CrT and native CrT protein was quantified in 2-, 4-, and 8-wk-old animals. The mRNA encoding transgene (human CrT) was ∼60 times more abundant than that encoding the native (mouse) CrT protein (Table 1). We observed a decrease in the mRNA encoding for both the native and human CrT in 4-wk-old CrT-Tg, but both rebounded in 8-wk-old Cr-Tg animals, resulting in 8-wk-old mice having the same expression level for the transgene message as 2-wk-old animals. In WT animals, the level of mRNA for native CrT increased as the animals matured, a trend also observed in transgenic animals (Table 1).
Table 1.
Transcription of CrT encoding mRNA in CrT-Tg and wild-type mice
| 2 Weeks (n = 6) |
4 Weeks (n = 5) |
8 Weeks (n = 6) |
||||
|---|---|---|---|---|---|---|
| CrT-Tg | WT | CrT-Tg | WT | CrT-Tg | WT | |
| Native CrT mRNA | 0.0073 ± 0.001 | 0.0050 ± 0.0003 | 0.0037 ± 0.0008 | 0.0108 ± 0.0013 | 0.014 ± 0.001 | 0.0162 ± 0.0017 |
| Human CrT mRNA | 0.56 ± 0.17 | NA | 0.22 ± 0.06 | NA | 0.63 ± 0.11 | NA |
Values are means ± SE; n, no. of animals. Real-time quantification of mRNA encoding the native (mouse) and transgene (human) forms of the creatine transporter (CrT) protein in both creatine transporter transgenic (CrT-Tg) and wild-type (WT) age-matched control animals. The abundance of the respective mRNAs was determined in 2-, 4-, and 8-wk-old animals and was normalized to β-actin and expressed in arbitrary units based on the cycle threshold (Ct) values, as described in methods. NA, not applicable.
Fig. 1.

Human creatine transporter (hCrT) is expressed in creatine transporter transgenic (CrT-Tg) cardiac muscle. A: Western blot of cardiac tissue homogenates from adult transgenic or wild-type (WT) mice. In CrT-Tg homogenates, a band of ∼55 kDa corresponding to the hCrT protein was detected using a specific rat monoclonal antibody against the human CrT protein isoform. B: cardiac ventricle tissue sections from 8-wk-old CrT-Tg animals or WT age-matched controls were prepared for histological analysis as described in methods. Staining was observed only in CrT-Tg animals (bottom), and localized to the cell surface in a striated pattern.
The presence of the human CrT protein was also verified by probing Western blots of cardiac tissue homogenates from 6-mo-old CrT-Tg and WT animals with an anti-CrT antibody that preferentially recognized the human isoform of the protein (7). A band of ∼55 kDa was observed only in the solubilized homogenates from CrT-Tg hearts (Fig. 1A). Immunohistochemistry was used to further document the expression of the protein encoded by the transgene in cardiac muscle sections. Interestingly, not every cardiomyocyte was stained, and quantification of these cells in the ventricle sections used for immunohistochemistry indicated that 4% of the cardiomyocytes were immunopositive. This “mottled” pattern was observed in hearts from both male and female transgenic animals, thereby excluding integration of the transgene in the X-chromosome. A similar nonhomogenous expression pattern was previously reported for transgenes under the control of the α-MHC promoter (31). In the cells that were stained by the anti-CrT antibody, the signal concentrated at the cell membrane and had a striated pattern. This staining was not observed in WT animals (Fig. 1B).
Cardiac energy metabolite measurements.
Cr, PCr, ATP, and ADP were quantified in flash-frozen cardiac tissue extracts from 2-, 4-, and 8-wk-old CrT-Tg animals and age-matched WT controls by HPLC, and free ADP was calculated as described in methods. CrT-Tg animals had significantly higher myocellular Cr levels (Fig. 2A). By the time animals reached adulthood (8 wk), cardiac Cr was 5.7-fold higher in 8-wk-old CrT-Tg mice than in WT animals. PCr content was also significantly elevated in 2- and 4-wk-old CrT-Tg mice, but, by adulthood, had decreased to levels observed in WT age-matched animals (Fig. 2B). Similarly, ATP concentrations were also significantly higher in 2- and 4-wk-old CrT-Tg mice. However, ATP levels were lower than those of age-matched controls by week 8 (Fig. 2C). Total ADP (tADP) levels were significantly reduced in 8-wk-old CrT-Tg animals compared with WT animals. There were no significant differences in tADP content in younger animals (Fig. 2D). Free ADP content was determined as described in methods and found to be significantly elevated at all ages in CrT-Tg hearts compared with WT hearts (Fig. 2E). The resulting PCr-to-ATP ratio was significantly elevated in 4- and 8-wk-old CrT-Tg mice compared with WT age-matched controls (Fig. 2F), whereas PCr-to-Cr ratios were reduced in CrT-Tg hearts at all ages (Fig. 2G), likely reflecting the increased size of the Cr pool secondary to augmented Cr transport. The ATP-to-tADP ratio (Fig. 2H) was significantly elevated in CrT-Tg 2- and 4-wk-old animals compared with the respective age-matched WT animals. This difference was not observed in 8-wk-old animals.
Fig. 2.
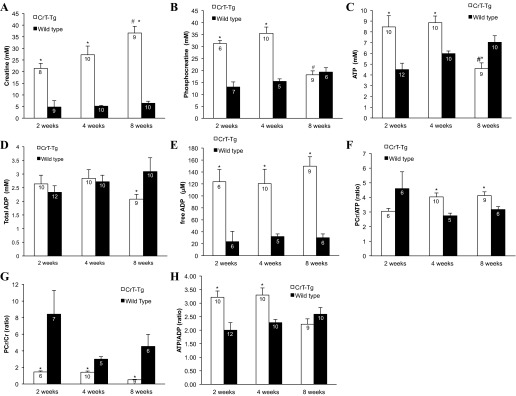
High-energy metabolite analysis. Hearts from control or CrT-Tg mice were harvested, and the content of creatine (Cr), phosphocreatine (PCr), ATP, and free ADP was determined by HPLC as described in methods. The number within each bar indicates the number of hearts analyzed. A: CrT-Tg mice hearts had significantly higher Cr content compared with WT mice. B: PCr levels were significantly increased in CrT-Tg levels in 2- and 4-wk-old animals (2.3- to 2.5-fold vs. WT controls). By 8 wk of age, the content of PCr in CrT-Tg mice was the same as WT controls. C: ATP levels were significantly elevated in CrT-Tg at 2 and 4 wk of age but fell below those measured in WT animals in 8-wk-old CrT-Tg hearts. D: total ADP content was significantly elevated in 8-wk-old WT animals. Two- and 4-wk-old animals had similar ADP content, irrespective of transgene expression. E: free ADP content (calculated as described in methods) was elevated in CrT-Tg animals at all time points. F: the PCr-to-ATP ratio was significantly elevated in CrT-Tg animals at 4 and 8 wk of age. G: CrT-Tg animals had significantly greater PCr-to-Cr ratios than age-matched WT controls. H: the ATP-to-ADP ratio (calculated using the measured total ADP content) was significantly higher in 2- and 4-wk-old CrT-Tg animals compared with WT animals. This difference was not observed in 8-wk-old animals. *P < 0.05 vs. WT, age-matched control, 2-way ANOVA, Fisher's least significant difference (LSD). #P < 0.05 vs. same line at 2 wk, 2-way ANOVA, Fisher's LSD.
Cardiac morphometry and function.
The morphometry and function of CrT-Tg hearts was analyzed throughout development and into maturity and compared with WT age-matched animals (Tables 2 and 3). Significant increases were observed in the right atrium-to-tibial length ratio in 2-, 4-, and 8-wk- and 9-mo-old CrT-Tg animals and the left atrium-to-tibial length ratio of 4- and 8-wk- and 9-mo-old CrT-Tg animals. Although the LV/body weight ratios were not significantly different, the LV/TL ratios were slightly but significantly greater at all ages among CrT-Tg compared with WT animals. Lung weight was significantly less among CrT-Tg at 4 and 8 wk. Echocardiography showed no differences in fractional shortening or left ventricular chamber diameters at any time point and significantly increased posterior wall thickness at 4 wk and 9 mo (Table 2). Taken together, these data indicate mild left ventricular hypertrophy and atrial enlargement without echocardiographic evidence of chamber enlargement of contractile dysfunction among CrT-Tg animals. Left ventricular function was further evaluated in vivo by pressure-volume loop analysis. At both 8 wk and 9 mo (Table 3), there were no significant differences in systolic or diastolic function between CrT-Tg and WT mice. Kaplan-Mayer plots of WT and CrT-Tg animals overlapped, indicating that lifespans and mortality rates were not different from those of controls animals (data not shown).
Table 2.
Cardiac morphometry and function of CrT-Tg mice
| 2 Weeks |
4 Weeks |
8 Weeks |
9 Months |
|||||
|---|---|---|---|---|---|---|---|---|
| CrT-Tg (n = 10) | WT (n = 10) | CrT-Tg (n = 10) | WT (n = 10) | CrT-Tg (n = 8) | WT (n = 10) | CrT-Tg (n = 24) | WT (n = 26) | |
| Body wt, g | 7.20 ± 0.27 | 7.05 ± 0.24 | 16.95 ± 0.61 | 17.63 ± 0.50 | 24.16 ± 1.34 | 24.09 ± 1.06 | 40.5 ± 1.82 | 36.07 ± 1.36 |
| TL, mm | 10.70 ± 0.20 | 10.63 ± 0.12 | 15.37 ± 0.22 | 15.32 ± 0.09 | 17.54 ± 0.22 | 17.56 ± 0.12 | 18.84 ± 0.09 | 18.61 ± 0.06 |
| Lungs, mg | 97.37 ± 2.50 | 98.17 ± 2.34 | 118.56 ± 3.42* | 125.00 ± 3.32 | 125.51 ± 4.40* | 137.59 ± 3.76 | 157.66 ± 4.30 | 162.12 ± 4.14 |
| RV wt, mg | 8.47 ± 0.38 | 8.23 ± 0.52 | 8.47 ± 0.38 | 8.23 ± 0.52 | 22.31 ± 1.49 | 20.45 ± 1.48 | 28.86 ± 1.55 | 24.71 ± 0.93 |
| LV wt, mg | 26.16 ± 0.66* | 22.41 ± 0.55 | 63.28 ± 1.64* | 55.04 ± 1.14 | 79.53 ± 4.33* | 73.97 ± 1.86 | 114.11 ± 4.13* | 104.90 ± 3.79 |
| LV/BW, mg/g | 3.66 ± 0.09 | 3.74 ± 0.12 | 3.76 ± 0.10 | 3.13 ± 0.05 | 3.30 ± 0.09 | 3.10 ± 0.08 | 2.86 ± 0.08 | 2.95 ± 0.10 |
| LV/TL, mg/mm | 2.45 ± 0.03* | 2.11 ± 0.06 | 4.11 ± 0.06* | 3.59 ± 0.06 | 4.53 ± 0.24* | 4.21 ± 0.09 | 6.05 ± 0.20* | 5.63 ± 0.20 |
| RV/TL, mg/mm | 0.77 ± 0.05 | 0.80 ± 0.04 | 1.14 ± 0.05 | 1.08 ± 0.08 | 1.27 ± 0.08 | 1.16 ± 0.08 | 1.53 ± 0.08* | 1.33 ± 0.05 |
| LA/TL, mg/mm | 0.14 ± 0.01 | 0.13 ± 0.01 | 0.27 ± 0.02* | 0.21 ± 0.01 | 0.30 ± 0.02* | 0.24 ± 0.02 | 0.49 ± 0.03* | 0.31 ± 0.02 |
| RA/TL, mg/mm | 0.22 ± 0.01* | 0.15 ± 0.01 | 0.41 ± 0.06* | 0.21 ± 0.01 | 0.59 ± 0.06* | 0.25 ± 0.01 | 0.46 ± 0.04* | 0.27 ± 0.02 |
| HR, beats/min | 547 ± 21 | 535 ± 14 | 696 ± 7.2 | 691 ± 6.5 | 619 ± 10.2 | 673 ± 8.3 | 645 ± 7.1 | 653 ± 10.1 |
| SW, mm | 0.48 ± 0.04 | 0.54 ± 0.05 | 0.76 ± 0.05 | 0.66 ± 0.06 | 0.95 ± 0.03 | 0.89 ± 0.04 | 1.07 ± 0.03 | 1.10 ± 0.04 |
| PW, mm | 0.53 ± 0.04 | 0.51 ± 0.04 | 0.87 ± 0.06* | 0.69 ± 0.03 | 0.85 ± 0.05 | 0.84 ± 0.02 | 1.15 ± 0.04* | 0.99 ± 0.04 |
| FS, % | 65.82 ± 3.48 | 67.58 ± 3.11 | 65.57 ± 3.39 | 64.23 ± 1.36 | 45.33 ± 1.33 | 48.12 ± 1.59 | 47.37 ± 1.39 | 51.03 ± 1.50 |
| LVDd, mm | 2.41 ± 0.06 | 2.48 ± 0.07 | 2.93 ± 0.06 | 3.03 ± 0.10 | 3.13 ± 0.10 | 3.06 ± 0.05 | 3.67 ± 0.07 | 3.60 ± 0.06 |
| LVDs, mm | 0.82 ± 0.08 | 0.81 ± 0.09 | 1.02 ± 0.11 | 1.09 ± 0.05 | 1.71 ± 0.08 | 1.59 ± 0.06 | 1.93 ± 0.06 | 1.77 ± 0.07 |
Values are means ± SE; n, no. of animals. BW, body wt; HR, heart rate; SW, septal wall thickness; PW, posterior wall thickness; LVDd, left ventricle dimension (diastole); LVDs, left ventricle dimension (systole). The mass of the left ventricle (LV), left atrium (LA), right ventricle (RV), or right atrium (RA) was normalized to the corresponding animal's tibial length (TL) and compared between age-matched CrT-Tg and WT control animals. The LV-to-TL and RA-to-TL ratios of CrT-Tg animals were significantly greater at all ages compared with WT controls, as were the LA-to-TL ratios of 4- and 8-wk- and 9-mo-old CrT-Tg animals. The percent fractional shortening (%FS), left ventricular chamber dimensions, and septal/posterior wall thicknesses were determined in conscious 2-, 4-, and 8-wk-old and 9-mo-old mice as described in methods. Posterior wall thickness was significantly greater at 4-wk and 9-mo among CrT-Tg. No other echocardiographic differences were detected between CrT-Tg mice and age-matched WT control animals.
Significant difference with age-matched WT controls, P < 0.05 (ANOVA, Tukey's honest significant difference).
Table 3.
Cardiac function of adult and aged CrT-Tg animals: pressure-volume loop measurement
| 8 Weeks |
9 Months |
|||
|---|---|---|---|---|
| WT (n = 10) | CrT-Tg (n = 7) | WT (n = 8) | CrT-Tg (n = 9) | |
| Systolic function | ||||
| Heart rate, beats/min | 399 ± 15 | 397 ± 17 | 455.44 ± 15.05 | 399.14 ± 24.37 |
| End-systolic volume, μl | 17.17 ± 1.48 | 17.13 ± 3.63 | 7.15 ± 0.96 | 7.74 ± 0.97Ŧ |
| End-diastolic volume, μl | 35.94 ± 2.34 | 32.36 ± 2.78 | 17.04 ± 1.81* | 17.91 ± 1.80Ŧ |
| Maximum SBP, mmHg | 91.35 ± 3.67 | 85.34 ± 1.46 | 86.46 ± 3.45 | 81.04 ± 3.09 |
| End-systolic pressure, mmHg | 83.68 ± 4.76 | 77.05 ± 3.18 | 75.01 ± 4.62 | 71.13 ± 3.09 |
| dP/dtmax, mmHg/s | 8,302 ± 518 | 9,176 ± 604 | 8,099 ± 460 | 8,535 ± 710 |
| Cardiac output, μl/min | 8.70 ± 0.40 | 7.51 ± 0.92 | 5.05 ± 0.50* | 4.54 ± 0.21Ŧ |
| Ejection fraction | 58.75 ± 2.47 | 56.47 ± 6.96 | 62.69 ± 2.84 | 62.63 ± 1.66 |
| End-systolic elastance | 5.78 ± 0.56 | 5.23 ± 0.56 | 7.41 ± 1.07* | 6.32 ± 0.50Ŧ |
| Diastolic Function | ||||
| End-diastolic pressure, mmHg | 7.98 ± 1.20 | 5.64 ± 0.61 | 3.74 ± 0.40* | 4.08 ± 0.56Ŧ |
| dP/dtmin, mmHg/s | −6,773 ± 503 | −6,116 ± 310 | −6,996 ± 719 | −6,000 ± 319 |
| Tau (Glatz), ms | 10.87 ± 0.41 | 11.57 ± 1.07 | 10.88 ± 0.79 | 12.01 ± 0.61 |
| EDPVR | 0.62 ± 0.09 | 0.80 ± 0.18 | 0.97 ± 0.15 | 0.84 ± 0.14 |
Values are means ± SE; n, no. of animals. SBP, systolic blood pressure; EDPVR, end-diastolic pressure volume relationship. Cardiac function was evaluated by pressure volume measurements as described in methods in 8-wk- and 9-mo-old animals. Significant differences between the 8-wk-old animals compared with 9-mo-old animals of the same genotype are demoted by for WT (*) or CrT-Tg (Ŧ) (P < 0.05, 2-way ANOVA, nonrepeated measures and Bonferroni post hoc test).
DISCUSSION
We report the creation of a transgenic mouse that expresses the human Cr transporter in the heart with normal cardiac function and survival. Cardiac Cr content in CrT-Tg animals was nearly six times higher than that of WT mice as demonstrated by HPLC analysis. In addition, CrT protein expression was documented by Western blotting and immunohistochemical analysis of cardiac muscle sections (Fig. 1B), demonstrating for the first time that functional CrT protein is found at the cell membrane. Approximately 4% of the cardiomyocytes in the sections subjected to immunohistochemical analysis had robust staining with the anti-CrT antibody. Nonhomogenous cardiac expression of a transgene driven by the α-MHC promoter has been previously reported (31).
No functional ventricular cardiac abnormalities were observed on echocardiographic or pressure-volume studies, and the structural differences observed in morphometric studies indicate a mild left ventricular hypertrophy with no deleterious effects on cardiac function (Tables 2 and 3). Our results differ significantly from those initially described by Wallis et al. where mice with two- to fourfold increases in intracellular Cr concentrations developed cardiac dysfunction spontaneously (21, 34, 46). Moreover, in these animals, the severity of dysfunction was positively correlated with intracellular Cr concentrations. The authors hypothesized that the increase in total Cr led to increased free ADP concentration, which in turn caused the free energy change of ATP hydrolysis (ΔGATP) to decrease. In this scenario, and in conjunction with a CK system that remained at the same functional level as in WT animals, the Cr shuttle's main function (keeping ADP levels low and ΔGATP high) would be impaired. The authors also reported diminished α-enolase activity and reduced flux through the glycolytic pathway. However, whether these changes represented an adaptive or a causal event of cardiac failure was unknown (34). Upon further screening of this transgenic animal model, it was determined that very moderate increases in Cr transport capacity, up to a twofold increase in cardiac Cr content, were well tolerated. More importantly, the authors demonstrated that these increases afforded protection from ischemic insults to the cardiac muscle (21).
There are several possible explanations for the discrepancies noted between the transgenic line studied here and those reported in the studies by Wallis and Phillips. First, mice of different genetic backgrounds may react differently to the same cardiac stressor (3, 6, 36, 41). In the studies by Wallis and Phillips, C57BL/6 mice genetic backgrounds were used, whereas FVB mice were used in our study. C57BL/6 mice appear to be more susceptible to developing cardiomyopathy and have increased mortality rates following transverse aortic constriction compared with other strains (3). FVB mice tend to develop myocardial hypertrophy without severe contractile alterations after the same intervention (10, 12). Thus it is plausible that differences in animal strains may be involved in the adaptation of the heart to mechanical as well as to biochemical stress factors. In an effort to address this specific issue, we have begun backcrossing the CrT-Tg mice into the C57/Bl6 background. At this time, we have bred the sixth generation, and these animals do not display any sign of cardiac malfunction. Second, the site of transgene insertion may disrupt sensitive coding or regulatory gene(s) sequences with the end result being the development of cardiac dysfunction phenotype. Notably, transgenic animals expressing green fluorescent protein in cardiac muscle may spontaneously develop heart failure (17), raising the possibility that the site or copy number of transgene insertion(s) may be responsible for adverse cardiac effects unrelated to the function of the transgene itself. During the initial stages of our study, we isolated several founders that died as pups, in addition to a transgenic lineage that did develop heart failure. This lineage also had higher expression of the human CrT, and the Cr content was increased 8- to 10-fold than that measured in WT littermates (data not shown). We did not determine the copy number or site of insertion of the transgene for any of our lines, and thus we cannot rule out that in these transgenic animals the deleterious effects were the result of genetic disruptions or cellular toxicity because of the very elevated Cr content. Taken together with the findings from other CrT overexpression models, our results suggest that the heart can tolerate very significant (6-fold) and chronic increases in cardiac Cr content.
Third, although both mouse models used cardiac specific promoters (MLC2v, Wallis et al., and α-MHC in our study), they differ in their spatial-temporal expression activation profile during gestation and after birth, which could influence how the transgene is regulated (23, 30). Last, the two transgenic models differ in the species of the transgene inserted. In our model we used the human isoform, whereas Wallis and colleagues used a rabbit CrT. Protein sequence analysis (ClustalO Uniprot) shows that the human CrT (Uniprot identifier P48029) is 98.12% identical to the mouse CrT (Uniprot identifier Q8VBW1), whereas the rabbit CrT (Uniprot Identifier P31661) amino acid sequence differs slightly more (96.87% identity). These differences are very small and consist mostly of conserved amino acid changes. Thus, it seems unlikely that sequence differences could underlie the markedly different phenotypes of the two transgenic lines.
CK, a key element in the “creatine energy shuttle,” is the conduit by which high-energy phosphates are transferred from the sites of generation (ATP in the mitochondria) to sites of consumption (contractile machinery and ion pumps) using PCr as a carrier. CK function and distribution analysis indicated that the transgenic animals generated by Wallis and colleagues had no changes in the activity or distribution of CK isoforms (46). Interestingly, the high-energy phosphate metabolite profile observed in our CrT-Tg mice was similar to that reported by Wallis and Phillips. Namely, PCr and ADP levels were elevated in adult animals, whereas ATP levels in the same age group were below those observed in control animals. In our transgenic model, we observed that 2- and 4-wk-old CrT-Tg animals had elevated ATP content, which decreased to below the levels measured in WT littermates (controls) by the time the animals were 8 wk old. Although we did not measure the activity or distribution of CK isotypes in CrT-Tg animals, the similarities in the profiles of ATP, ADP, and PCr suggest that Cr kinase enzymatic capabilities of CrT-Tg were not altered.
Previous reports had hypothesized that ADP removal from areas surrounding cellular sites with elevated ATPase activity (i.e., myosin ATPase and sarcoplasmic reticulum ATPase) helps regulate energy regeneration locally, e.g., where ATPase activity is lowered by product accumulation (18, 24). Moreover, it was proposed that direct ATP channeling between the mitochondria, the myofibrils, and the sarcoplasmic reticulum might increase when the CK system becomes insufficient (18, 37). Thus, whereas both our models demonstrate an increase in free ADP concentration, which would lower the energy available for ATPase activity required for excitation-contraction coupling, the results of our functional studies suggest the existence of an alternative pathway that allows the cell to maintain and buffer ATP within normal ranges at the sites of consumption (contractile machinery and ion pumps). This mechanism may also be responsible for the capacity of CrT-Tg juvenile mice capacity to maintain an increased ATP and ATP-to-tADP ratios (at 2 and 4 wk of age) despite elevated free ADP concentrations during maturation to adulthood, which could explain why these mice did not develop heart failure. Understanding how these developing mice were able to maintain high ATP levels despite high free ADP concentrations could shed light on the mechanisms cardiomyocytes use to buffer high-energy metabolites and respond to functional and biochemical demands and potential therapeutic interventions to ameliorate the energy derangement observed in failing hearts.
Previously we showed in HL-1 cells and rat neonatal cardiomyocytes that Cr transport decreases rapidly in response to increased Cr in the extracellular environment. Similar observations were made in isolated heart preparations (5). The molecular mechanism by which the cell reduces Cr transport in response to increased Cr is largely unknown. However, we have shown that, in cardiomyocytes in culture, the decrease in Cr transport is the result of a reduction in the cell surface content of CrT transporter protein (8). The expression of CrT protein detected on Western blots from CrT-Tg animals appeared robust (Fig. 1, A and B). The mRNA encoding the native transporter increased as the animals matured in both WT and CrT-Tg mice, just as it occurs in developing rat hearts (11). These observations suggest that the increased intracellular Cr present in CrT-Tg animals did not exert a significant repressive effect at the transcriptional level (Table 1) and that the chronically increased cardiac Cr content did not cause a decrease in the content of human CrT mRNA. This observation would also be consistent with what is known about substrate regulation of Cr transport, i.e., increased extracellular Cr content decreases Cr transport capacity.
A significant question remains surrounding the impact of sustained Cr, PCr, and free ADP elevations on intracellular pH and key energy homeostasis enzymatic complexes, such as AMPK, acetyl-CoA carboxylase, or lactate dehydrogenase, all of which are perturbed in heart failure (2, 11, 25). In addition of answering these questions, the fact that not every cardiomyocyte in the CrT-Tg heart appears to express the human CrT protein may be particularly useful in answering how elevated Cr content is cardioprotective, because within the same organ (heart) the effects of Cr elevations can be studied and contrasted in vivo to control cells. Another interesting question is how the additional Cr that is brought into the cells expressing the transgene diffuses into neighboring cells, increasing Cr content “by proxy” and what would be the effect on the overall metabolic state of these cells. In particular, given our observation that a small number of modified cardiomyocytes (4%) can cause a significant elevation of cardiac Cr content, the exploration of therapeutic gene therapy approaches that would increase Cr transport capacity in a reduced number of cardiomyocytes warrants further investigation.
In conclusion, we report that transgenic overexpression of the human CrT protein in murine cardiac muscle can result in a significant elevation in cardiac Cr content and, despite a mild hypertrophic response, does not result in deleterious effects on cardiac function under normal conditions. These transgenic hearts provide a unique system where a combination of advanced imaging platforms and experimental models can be applied to study in situ the mechanisms by which Cr exerts its protective effect in the heart. Future studies focused on understanding how the cell surface CrT content is modulated will clarify how the cell controls its Cr transport capacity and thus its Cr content.
GRANTS
This work was funded by the Department of Surgery at Duke University Medical Center and National Heart, Lung, and Blood Institute Grants HL-056687 and T32-HL-07101 to H. A. Rockman, which provided funding to J. Nienaber.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.S., A.H., and J.N. conception and design of research; L.S., A.H., J.N., R.M., M.P., J.B., and L.M. performed experiments; L.S., A.H., J.N., M.P., and L.M. analyzed data; L.S., A.H., J.N., M.P., and L.M. interpreted results of experiments; L.S. prepared figures; L.S. drafted manuscript; L.S., A.H., J.N., M.P., L.M., H.A.R., and D.O.J. edited and revised manuscript; L.S., A.H., J.N., R.M., M.P., J.B., L.M., H.A.R., and D.O.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Lauren Goers for technical assistance, Quique Toloza for assistance in the preparation of this manuscript, Eric Toloza and Marcus Darrabie for assistance with statistical analysis, and Dawn Bowles, Kumar Pandya, and Bryan Feger for critical reading and insightful comments.
Current addresses: L. Santacruz, Dept. of Biochemistry and Molecular Biology, University of Texas, Medical Branch, Galveston, TX 77555; A. Hernandez, Dept. of Anesthesiology, Yale University School of Medicine, New Haven, CT; J. Nienaber, Charles George VA Medical Center, 1100 Tunnel Rd., Asheville, NC 28805; M. Pinilla, UPMC Montefiore Hospital, N-715, 200 Lothrop St., Pittsburgh, PA 15213; and D. O. Jacobs, School of Medicine, University of Texas Medical Branch, 301 University Blvd., Galveston, TX 77555-0133.
REFERENCES
- 1.Akki A, Su J, Yano T, Gupta A, Wang Y, Leppo MK, Chacko VP, Steenbergen C, Weiss RG. Creatine kinase overexpression improves ATP kinetics and contractile function in postischemic myocardium. Am J Physiol Heart Circ Physiol 303: H844–H852, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res 100: 474–488, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am J Physiol Heart Circ Physiol 292: H2119–H2130, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 40: 1267–1274, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Boehm E, Chan S, Monfared M, Wallimann T, Clarke K, Neubauer S. Creatine transporter activity and content in the rat heart supplemented by and depleted of creatine. Am J Physiol Endocrinol Metab 284: E399–E406, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Cook SA, Clerk A, Sugden PH. Are transgenic mice the ‘alkahest’ to understanding myocardial hypertrophy and failure? J Mol Cell Cardiol 46: 118–129, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Darrabie MD, Arciniegas AJ, Mantilla JG, Mishra R, Vera MP, Santacruz L, Jacobs DO. Exposing cardiomyocytes to subclinical concentrations of doxorubicin rapidly reduces their creatine transport. Am J Physiol Heart Circ Physiol 303: H539–H548, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Darrabie MD, Arciniegas AJ, Mishra R, Bowles DE, Jacobs DO, Santacruz L. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am J Physiol Endocrinol Metab 300: E870–E876, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Darrabie MD, Zhao ZF, Goers L, Santacruz-Toloza L, Toloza EM, Jacobs DO. Creatine transport is modulated by PKC and PPI/PP2A. Biophys J 63a–63a, 2007. 17827222 [Google Scholar]
- 10.Dorn GW, 2nd, Robbins J, Ball N, Walsh RA. Myosin heavy chain regulation and myocyte contractile depression after LV hypertrophy in aortic-banded mice. Am J Physiol Heart Circ Physiol 267: H400–H405, 1994 [DOI] [PubMed] [Google Scholar]
- 11.Fischer A, Ten Hove M, Sebag-Montefiore L, Wagner H, Clarke K, Watkins H, Lygate CA, Neubauer S. Changes in creatine transporter function during cardiac maturation in the rat (Abstract). BMC Dev Biol 10: 70, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giampietri C, Petrungaro S, Musumeci M, Coluccia P, Antonangeli F, De Cesaris P, Filippini A, Marano G, Ziparo E. c-Flip overexpression reduces cardiac hypertrophy in response to pressure overload. J Hypertens 26: 1008–1016, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Golding EM, Teague WE, Jr, Dobson GP. Adjustment of K′ to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment. J Exp Biol 198: 1775–1782, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Gupta A, Akki A, Wang Y, Leppo MK, Chacko VP, Foster DB, Caceres V, Shi S, Kirk JA, Su J, Lai S, Paolocci N, Steenbergen C, Gerstenblith G, Weiss RG. Creatine kinase-mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J Clin Invest 122: 291–302, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himmelreich U, Dobson GP. Detection and quantification of free cytosolic inorganic phosphate and other phosphorus metabolites in the beating mouse heart muscle in situ. NMR Biomed 13: 467–473, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Horn M, Remkes H, Dienesch C, Hu K, Ertl G, Neubauer S. Chronic high-dose creatine feeding does not attenuate left ventricular remodeling in rat hearts post-myocardial infarction. Cardiovasc Res 43: 117–124, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med 6: 482–483, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res 89: 153–159, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Kapelko VI, Kupriyanov VV, Novikova NA, Lakomkin VL, Steinschneider A, Severina M, Veksler VI, Saks VA. The cardiac contractile failure induced by chronic creatine and phosphocreatine deficiency. J Mol Cell Cardiol 20: 465–479, 1988 [DOI] [PubMed] [Google Scholar]
- 20.Lygate CA, Aksentijevic D, Dawson D, ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag-Montefiore L, Hunyor I, Channon KM, Clarke K, Zervou S, Watkins H, Balaban RS, Neubauer S. Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ Res 112: 945–955, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lygate CA, Bohl S, ten Hove M, Faller KM, Ostrowski PJ, Zervou S, Medway DJ, Aksentijevic D, Sebag-Montefiore L, Wallis J, Clarke K, Watkins H, Schneider JE, Neubauer S. Moderate elevation of intracellular creatine by targeting the creatine transporter protects mice from acute myocardial infarction. Cardiovasc Res 96: 466–475, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lygate CA, Fischer A, Sebag-Montefiore L, Wallis J, ten Hove M, Neubauer S. The creatine kinase energy transport system in the failing mouse heart. J Mol Cell Cardiol 42: 1129–1136, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Lyons GE, Schiaffino S, Sassoon D, Barton P, Buckingham M. Developmental regulation of myosin gene expression in mouse cardiac muscle. J Cell Biol 111: 2427–2436, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minajeva A, Ventura-Clapier R, Veksler V. Ca2+ uptake by cardiac sarcoplasmic reticulum ATPase in situ strongly depends on bound creatine kinase. Pflugers Arch 432: 904–912, 1996 [DOI] [PubMed] [Google Scholar]
- 25.Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, Mochizuki S. Optimization of cardiac metabolism in heart failure. Curr Pharm Des 17: 3846–3853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 96: 2190–2196, 1997 [DOI] [PubMed] [Google Scholar]
- 28.Neubauer S, Krahe T, Schindler R, Horn M, Hillenbrand H, Entzeroth C, Mader H, Kromer EP, Riegger GA, Lackner K. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease Altered cardiac high-energy phosphate metabolism in heart failure. Circulation 86: 1810–1818, 1992 [DOI] [PubMed] [Google Scholar]
- 29.Neubauer S, Remkes H, Spindler M, Horn M, Wiesmann F, Prestle J, Walzel B, Ertl G, Hasenfuss G, Wallimann T. Downregulation of the Na(+)-creatine cotransporter in failing human myocardium and in experimental heart failure. Circulation 100: 1847–1850, 1999 [DOI] [PubMed] [Google Scholar]
- 30.O'Brien TX, Lee KJ, Chien KR. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc Natl Acad Sci USA 90: 5157–5161, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandya K, Cowhig J, Brackhan J, Kim HS, Hagaman J, Rojas M, Carter CW, Jr, Mao L, Rockman HA, Maeda N, Smithies O. Discordant on/off switching of gene expression in myocytes during cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 105: 13063–13068, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest 116: 1547–1560, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR (Abstract). Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips D, Ten Hove M, Schneider JE, Wu CO, Sebag-Montefiore L, Aponte AM, Lygate CA, Wallis J, Clarke K, Watkins H, Balaban RS, Neubauer S. Mice over-expressing the myocardial creatine transporter develop progressive heart failure and show decreased glycolytic capacity. J Mol Cell Cardiol 48: 582–590, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rindt H, Subramaniam A, Robbins J. An in vivo analysis of transcriptional elements in the mouse alpha-myosin heavy chain gene promoter. Transgenic Res 4: 397–405, 1995 [DOI] [PubMed] [Google Scholar]
- 36.Sato Y, Schmidt AG, Kiriazis H, Hoit BD, Kranias EG. Compensated hypertrophy of cardiac ventricles in aged transgenic FVB/N mice overexpressing calsequestrin. Mol Cell Biochem 242: 19–25, 2003 [PubMed] [Google Scholar]
- 37.Shannon TR, Chu G, Kranias EG, Bers DM. Phospholamban decreases the energetic efficiency of the sarcoplasmic reticulum Ca pump. J Biol Chem 276: 7195–7201, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation 114: 1151–1158, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem 180: 171–177, 1998 [PubMed] [Google Scholar]
- 40.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 266: 24613–24620, 1991 [PubMed] [Google Scholar]
- 41.Suzuki M, Carlson KM, Marchuk DA, Rockman HA. Genetic modifier loci affecting survival and cardiac function in murine dilated cardiomyopathy. Circulation 105: 1824–1829, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J., Jr Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation 94: 1109–1117, 1996 [DOI] [PubMed] [Google Scholar]
- 43.Ten Hove M, Chan S, Lygate C, Monfared M, Boehm E, Hulbert K, Watkins H, Clarke K, Neubauer S. Mechanisms of creatine depletion in chronically failing rat heart. J Mol Cell Cardiol 38: 309–313, 2005 [DOI] [PubMed] [Google Scholar]
- 44.ten Hove M, Lygate CA, Fischer A, Schneider JE, Sang AE, Hulbert K, Sebag-Montefiore L, Watkins H, Clarke K, Isbrandt D, Wallis J, Neubauer S. Reduced inotropic reserve and increased susceptibility to cardiac ischemia/reperfusion injury in phosphocreatine-deficient guanidinoacetate-N-methyltransferase-knockout mice. Circulation 111: 2477–2485, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 104: 1664–1669, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Wallis J, Lygate CA, Fischer A, ten Hove M, Schneider JE, Sebag-Montefiore L, Dawson D, Hulbert K, Zhang W, Zhang MH, Watkins H, Clarke K, Neubauer S. Supranormal myocardial creatine and phosphocreatine concentrations lead to cardiac hypertrophy and heart failure: insights from creatine transporter-overexpressing transgenic mice. Circulation 112: 3131–3139, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Wiseman RW, Moerland TS, Chase PB, Stuppard R, Kushmerick MJ. High-performance liquid chromatographic assays for free and phosphorylated derivatives of the creatine analogues beta-guanidopropionic acid and 1-carboxy-methyl-2-iminoimidazolidine (cyclocreatine). Anal Biochem 204: 383–389, 1992 [DOI] [PubMed] [Google Scholar]