

Abstract
Slow moving blood flow and changes in flow direction, e.g., negative wall shear stress, can cause increased superoxide (O2·−) production in vascular endothelial cells. The mechanism by which shear stress increases O2·− production, however, is not well established. We tested the hypothesis that actin depolymerization, which occurs during flow reversal, mediates O2·− production in vascular endothelial cells via NADPH oxidase, and more specifically, the subunit p47phox. Using a swine model, we created complete blood flow reversal in one carotid artery, while the contralateral vessel maintained forward blood flow as control. We measured actin depolymerization, NADPH oxidase activity, and reactive oxygen species (ROS) production in the presence of various inhibitors. Flow reversal was found to induce actin depolymerization and a 3.9 ± 1.0-fold increase in ROS production as compared with forward flow. NADPH oxidase activity was 1.4 ± 0.2 times higher in vessel segments subjected to reversed blood flow when measured by a direct enzyme assay. The NADPH oxidase subunits gp91phox (Nox2) and p47phox content in the vessels remained unchanged after 4 h of flow reversal. In contrast, p47phox phosphorylation was increased in vessels with reversed flow. The response caused by reversed flow was reduced by in vivo treatment with jasplakinolide, an actin stabilizer (only a 1.7 ± 0.3-fold increase). Apocynin (an antioxidant) prevented reversed flow-induced ROS production when the animals were treated in vivo. Cytochalasin D mimicked actin depolymerization in vitro and caused a 5.2 ± 3.0-fold increase in ROS production. These findings suggest that actin filaments play an important role in negative shear stress-induced ROS production by potentiating NADPH oxidase activity, and more specifically, the p47phox subunit in vascular endothelium.
Keywords: endothelial cells
the mechanotransduction of wall shear stress (WSS) has been a subject of intense investigation (11). Transient near-wall retrograde flow (negative WSS) occurs during diastole with each heart beat in peripheral vessels (38). More prominent retrograde arterial blood flows have been reported in diseased conditions, especially in heart failure (12). In general, low blood flow and changes in flow direction cause endothelial dysfunction, whereas vessel regions that are exposed to steady blood flow are not as prone to atherosclerosis (14). The mechanisms that dictate the interplay between WSS and biochemical responses of vessel endothelium are of major interest in health and atherogenesis. Although the cause of endothelial cell dysfunction due to change in WSS may be complex, increased evidence suggests that excessive production of reactive oxygen species (ROS) may play an important role in this process. We have previously shown that NADPH oxidase has a directional response to shear stress (15).
The cytoskeleton plays an important role in maintaining cellular structural integrity through the three-dimensional actin filament network. It may also mediate the signal transduction between surface membrane and intracellular target. Actin network disruption has been shown to reduce angiotensin II-induced Ca2+ release in smooth muscle cells (37) or angiotensin II-induced ERK1/2 activity (29). It has been confirmed that fluid shear stress exerts significant influence on the endothelial cells in cell orientation and morphology, cytoskeleton organization, signal transduction, growth factor mediation, and gene expression (7, 11). Endothelial cells can sense differences in the temporal and/or spatial characteristics of flow and translate these biochemical stimuli into biological responses (13).
It has been suggested that the endothelial cell membrane integrin may serve as sensor for WSS (25). Several actin binding proteins serve as candidates for integrin-mediated signal transduction (2). In conjunction with external ligands, integrins activate signaling cascades that regulate the formation, turnover, and linkage of actin filaments. One of the outcomes of this pathway may be the change in NADPH oxidase activity and O2·− production. Actin may transmit surface mechanostimulation to regulate NADPH oxidase.
Currently, there is no in vivo evidence of NADPH oxidase regulation by actin in the endothelial cells during reversed flow. Here, we tested the hypothesis that actin depolymerization during blood flow reversal induces ROS production in vascular endothelial cells mediated by NADPH oxidase, presumably by translocation of p47phox. We provide evidence that reversed blood flow induces ROS production in endothelial cells via actin microfilament depolymerization, which potentiates NADPH oxidase activity both in vitro and in vivo.
MATERIAL AND METHODS
Animal preparation.
All animal experiments were performed in accordance with national and local ethical guidelines, including the Principles of Laboratory Animal Care, the Guide for the Care and Use of Laboratory Animals (34a), and the National Association for Biomedical Research (4), and an approved Indiana University Purdue University Indianapolis IACUC protocol regarding the use of animals in research.
Twenty-four Yorkshire pigs (40–55 kg body wt) of either sex were used in this study. The pigs were fasted overnight, and surgical anesthesia was induced with TKX (telazol, 10 mg/kg; ketamine, 5 mg/kg; xylazine, 5 mg/kg) and maintained with 1% to 2% isoflurane-balance O2 during acute, nonaseptic surgery. ECG leads were attached to the swine limbs. Body temperature was maintained at 37.5 ± 0.5°C and pH at 7.4 ± 0.1. At the end of the experiments, the animals were euthanized under deep anesthesia.
A midincision was made on the neck to expose both carotid arteries while maintaining an intact adventitia. To create a complete reversed blood flow in one carotid, two pieces of silicon tubes of the same length obtained from a blood set (V2410; B. Braun Medical) were inserted into the proximal (close to the heart) and distal (toward the head) portions of the vessel and secured with 2-0 silk suture (Fig. 1A). Two additional silicon tubes were inserted into the contralateral artery in such way that blood forward flow was maintained (control; Fig. 1B). An ultrasonic perivascular flow probe (Transonic Systems, Ithaca, NY) was placed around the middle portion of each carotid artery to monitor flow rate (Fig. 1).
Fig. 1.

Schematic representation of the carotid arteries in vivo setup showing the connections made with the silicon tubes at the proximal (toward the heart) and distal (toward the head) portions of the artery to create complete blood flow reversal (A) and blood forward flow (B). The total length of the arterial segment was ∼15 cm.
The animals were divided into four groups of six animals each. Group I was subjected to forward (right carotid) and reversed (left carotid) flows for 4 h. Group II was subjected to forward and reversed flows after the endothelium had been denuded in both arteries. Group III tested the effect of jasplakinolide during reversed flow where the left carotid was incubated for 10 min with jasplakinolide before the establishment of flow reversal, while the right carotid was incubated with saline before forward flow. Group IV tested the effect of apocynin on both forward (right carotid) and reversed (left carotid) flows.
The carotid vessels were prepared as shown in Fig. 1A (flow reversal) and Fig. 1B (forward flow) for 4-h duration. In Group II a 6-mm peripheral dilatation catheter (Guidant, Santa Clara, CA) was inserted and inflated into the carotid arteries before flow reversal or forward flow was established. The inflated catheter was pulled and pushed across the intimal surface 3 times, deflated, and withdrawn to ensure endothelial denudation. The loss of endothelium-dependent vasodilation was subsequently confirmed.
In vivo pharmacological treatment.
In Group III the left carotid artery was treated with 0.1 μM of jasplakinolide, an actin-stabilizing agent, and incubated for 10 min before flow reversal was established. A low concentration of jasplakinolide was used to stabilize the actin as previously reported (35) to avoid nonspecific effects of higher concentrations (46). The contralateral vessel was prefilled with saline before forward flow was established.
In Group IV, a bolus injection of apocynin (4 mg/kg iv) was given to the animals before the establishment of reversed and forward flows, followed by continuous infusion (3.5 μg·kg−1·min−1 iv) of apocynin during the entire experimental procedure. Apocynin is not a specific NADPH oxidase inhibitor but affects reactive oxygen species (ROS) production by acting as an antioxidant (19, 44). Apocynin inhibits the release of O2·− by NADPH oxidase by blocking migration of p47phox to the membrane, critically involved in initiating assembly of the functional NADPH oxidase complex (40).
After the in vivo experiments, the carotid arteries from all four groups were quickly removed and placed into a heated (37°C) chamber filled with HEPES buffered PSS containing (in mM) 5.5 D-glucose, 4.7 KCl, 142 NaCl, 2.7 HEPES sodium salt, 1.17 MgSO4, 2.79 CaCl2, at pH 7.4, for ROS/O2·− quantification, NADPH oxidase activity analysis, and protein expression measurements by Western blotting.
ROS/O2·− measurements.
The harvested vessels were cut into ∼3-mm rings and placed into a 96-well plate with HEPES-PSS buffer. Various compounds [cytochalasin D, latrunculin B, jasplakinolide, and phorbol 12-myristate 13-acetate (PMA)] were then added to the corresponding wells. Cytochalasins, a class of fungal metabolites, inhibit the rate of actin filament polymerization due to inhibition of actin subunit addition to the fast-growing end of actin filaments, and are used as actin depolymerization agents (5). Latrunculin B is commonly used to disrupt the actin cytoskeleton of cells and to block actin polymerization (32) both in vitro and in vivo. Jasplakinolide is a potent inducer of actin polymerization and stabilization at low concentrations (3). PMA is a strong O2·− stimulator (activator of protein kinase C) commonly used as a positive control to stimulate ROS/O2·− production in different types of cells (36). After these compounds were added, the plate was incubated at 37°C for 30 min. Finally, L012 (100 μM) was added to all wells and the luminescence was recorded for 60 min at 1-min interval using a multilabel plate reader (En vision Xcite; Perkin Elmer, Shelton, CT) at ultra-luminescent mode. The total reaction volume was 200 μl. After ROS/O2·− quantification, the rings were air dried and weighed. Each condition was tested in triplicates. ROS levels were measured as relative light units after subtracting the background luminescence and the number was divided by the dry tissue weight (mg) of the arterial rings (27). The values were then normalized to ratio relative to forward flow condition as the control.
NADPH oxidase activity assay.
The arterial samples were placed in chilled lysis buffer containing 20 mM of KH2PO4, 1 mM of EGTA, and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO), and homogenized with an electric homogenizer (Power Gen 35; Fisher Scientific) for 20 s. The homogenates were centrifuged at 1,000 g for 10 min at 4°C to remove the unbroken cells and large tissue debris. The supernatant was collected and kept on ice. The enzyme activity was determined in 200 μl of assay buffer at pH 7.4 containing the following (in mM): 25 HEPES, 120 NaCl, 5.9 KCl, 1.2 MgSO4, 1.75 CaCl2, 11 glucose; 0.1 β-NADPH, and 0.1 L012. The reaction started by adding 10 μl of homogenate. The luminescence was measured every 15 s for 5 min using a plate reader at ultra-sensitive mode. The protein concentration was determined with a BCA kit (Bio-Rad) and the results of the enzyme activity were standardized to the protein level of each sample.
Immunoprecipitation and Western blotting for NADPH oxidase expression and phosporylation.
The arterial vessels were homogenized in the lysis buffer containing 50 mM glycerophosphate, 100 μM sodium orthovanadate, 2 mM magnesium chloride, 1 mM of EGTA, 0.5% Triton X-100, 1 mM DL-dithiothreitol, 20 μM pepstatin, 20 μM leupeptin, 0.1 units/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride, and then incubated on ice for 1 h. The sample was centrifuged at 12,500 rpm for 15 min at 4°C, and the supernatant collected. The total protein was measured with a BCA kit (Bio-Rad). To determine the NADPH oxidase expression, equal amounts of protein (50 μg) from each sample were loaded on each lane and electrophoresed in 4–20% Tris-Glycine gel (Invitrogen) and then transferred onto a polyvinylidene difluoride membrane (Millipore). After being blocked for 1 h in 6% dried milk in TBS-Tween buffer, the membrane was incubated overnight at 4°C with a specific primary antibody (anti-p47phox and anti-gp91phox; Santa Cruz Biotechnology) at 1:200 dilution in blocking buffer. The membrane was then rinsed and incubated with a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) for 2 h at 1:5,000 dilutions in blocking buffer. All samples from each group were also probed with an anti-β-actin antibody (1:1,000 dilution in blocking buffer; Santa Cruz Biotechnology) to correct for sample loading.
To quantify p47phox phosphorylation, the total protein with p47phox was first immunoprecipitated according to an established method (28). Briefly, the samples were incubated with anti-p47phox at 4°C. They were then mixed with Protein G PLUS-Agarose (Santa Cruz Biotechnology) and incubated for 3 h at 4°C. The samples were then centrifuged and washed three times with a lysis buffer, and the pallet was suspended in loading buffer and denatured. The phosphorylated p47phox was then semiquantified by a rabbit polyclonal antibody to phosphoserine (Abcam, Cambridge, MA) by Western blotting. For quantification of phos-p47phox, the levels of total p47phox immunoprecipitated were first checked by prerun, and the final gel loading was calculated and confirmed to achieve equal amount of total p47phox between samples (43).
Actin immunostaining.
The carotid vessels were cut into ∼3-mm rings and immediately fixed for 2 h in 4% methanol-free formaldehyde-PBS solution (Thermo Scientific, Rockford, IL) for actin staining. Fixed samples were permeabilized with 0.1% Triton X-PBS solution. After nonspecific sites were blocked with 1% BSA in PBS, the actin was labeled by incubating the samples with Alexa Fluor 488-phalloidin (Invitrogen, Carlsbad, CA) for 10 min at room temperature. Each sample was then cut into ∼3 × 3 mm pieces, mounted on glass slides, covered with glass cover slips in the presence of 70% glyceral-PBS, and sealed with glue. The endothelial cytoskeleton was examined using a confocal microscope (Olympus FV1000-MPE) with excitation wavelength of 488 nm and emission of 500–530 nm for detecting actin.
4-Hydroxynonenal immunohistochemistry.
Fixed samples were embedded in paraffin, cut into 3-μm thick sections, mounted on glass slides and processed for immunohistology. Heat-mediated antigen retrieval was performed using citrate buffer. The samples were blocked with 5% serum for 30 min at 20°C and then incubated with a primary antibody against mouse monoclonal 4-Hydroxynonenal (1:50; Abcam) at 4°C overnight. A goat anti-mouse IgG-FITC (1:100; Santa Cruz Biotechnology) was used as the secondary antibody. Sections were counterstained with DAPI while a slide without primary antibody was used as a negative control. Fluorescence images were acquired with a ZEISS observer Z1 microscope.
Data analysis.
All data were expressed as means ± SD. Comparisons of results between two groups were performed by using unpaired two-tailed t-tests assuming unequal population variance. Multiple group comparisons were done with ANOVA, and statistical significance among multiple groups was determined by Tukey tests. P < 0.05 was considered statistically significant.
RESULTS
Endothelial ROS/O2·− production in response to blood flow.
We examined the role of actin in flow-induced ROS/O2·− generation where ROS was measured in the carotid arteries subjected to normal blood forward flow in the absence and presence of cytochalasin D, latrunculin B, and jasplakinolide. Basal ROS (control) was detected in the vessels with forward flow in the absence of treatment (Fig. 2), whereas a fivefold and twofold increase was found with 10 μM cytochalasin D (p = 0.004) and 1 μM latrunculin B (p = 0.044) treatments, respectively (Fig. 2) as compared with the control. ROS levels remained unchanged with 0.1 μM jasplakinolide (Fig. 2).
Fig. 2.

Effect of in vitro treatment with cytochalasin D (Cyto-D), latrunculin B (Latr), and jasplakinolide (Jasp) on reactive oxygen species (ROS) production of carotid arteries subjected to forward blood flow. Measurements were determined using chemiluminescence and reported as ratio relative to control (untreated sample). *P < 0.05, experimental groups vs. control.
Similar experiments were also conducted in endothelium denuded vessels subjected to forward (Fig. 3A) and reversed blood (Fig. 3B) flows. ROS production was significantly (p < 0.05) reduced in vessels with denuded endothelium subjected to forward flow (Fig. 3A) and reversed flow (Fig. 3B) with and without treatment with PMA and cytochalasin D. Treatment with PMA dramatically reduced ROS generation in endothelial denuded vessels subjected to both forward (Fig. 3A; p = 0.001) and reversed blood (Fig. 3B; p = 0.003) flows, as compared with vessels with intact endothelium. ROS production was also reduced in forward (Fig. 3A; p = 0.04) and reversed (Fig. 3B; p = 0.007) flow-endothelium denuded vessels treated with cytochalasin D. The results suggest that the major source of ROS/O2·− is the endothelial cell.
Fig. 3.

Effect of endothelial denudation on ROS production of carotid arteries subjected to forward blood flow (A) and reversed blood flow (B). The vessels were stimulated in vitro with phorbol 12-myristate 13-acetate (PMA) and Cyto-D. *P < 0.05, denuded vs. intact endothelium.
ROS production was significantly larger (p = 0.004) in reversed blood flow vessels, as compared with forward flow (Fig. 4). Treatment with jasplakinolide, however, significantly decreased ROS production in reversed flow vessels (p = 0.007; Fig. 4). Cytochalasin D, which has a general stimulatory effect, increased ROS production in reversed blood flow, as compared with forward flow (p = 0.04; Fig. 4). Pretreatment with jasplakinolide largely prevented the effects of cytochalasin D significantly reducing ROS production in reversed flow vessels (p = 0.003; Fig. 4).
Fig. 4.

Effect of reversed blood flow on ROS production in carotid vessels treated in vivo with Jasp and then stimulated in vitro with cytochalasin D. *P < 0.05, reversed vs. forward; **P < 0.05, reversed + Jasp vs. reversed; *^P < 0.05, reversed vs. forward in CytoD group; **^P < 0.05, reversed + Jasp vs. reversed in CytoD group.
Endothelial actin polymerization during flow reversal.
To confirm that flow reversal inhibits actin polymerization in endothelial cells, the pattern of actin (labeled with fluorescence conjugated phalloidin) in endothelial cells was examined using confocal microscopy. Figure 5A corresponds to a carotid segment with denuded endothelium (negative control). Figure 5B corresponds to actin endothelial cells during forward flow. Disruption and clumping was observed in vessels exposed to reversed flow (Fig. 5C) but to a lesser degree as compared with vessels treated with cytochalasin D. Cytochalasin D caused significant disruption of actin filaments during forward blood flow. Actin filaments appeared disarranged and clumped (Fig. 5D). The changes in actin cytoskeleton were visible when the concentration of cytochalasin D reached 20 nM. At a concentration >2 μM, all the long actin filament bundles were disrupted and replaced by large focal aggregates of F-actin. Treatment with jasplakinolide before and during blood flow reversal prevented the increase in monomeric actin due to the disruption of actin cytoskeleton (Fig. 5E), which was found to be similar to vessels with forward flow.
Fig. 5.
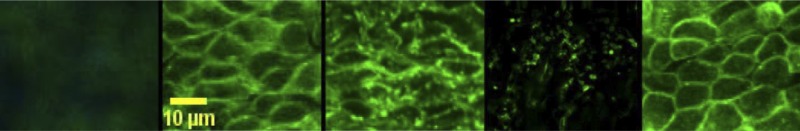
Immunostaining of carotid artery endothelial cells under different conditions. Denuded endothelium (negative control), forward blood flow, reversed blood flow, forward blood flow + cytochalasin D treatment, and reversed blood flow + jasplakinolide treatment, respectively, are shown. Scale bar = 10 μm.
Flow reversal increased NADPH oxidase activity.
Superoxide from reversed flow vessels was significantly higher than the controls, which suggests higher NADPH oxidase activity during blood flow reversal (Fig. 6A, left). NADPH oxidase activity was decreased in reversed flow vessels after treatment with jasplakinolide as compared with the control group, whereas reversed flow-induced ROS production was abolished in vessels treated with apocynin (Fig. 6A, left). The results of these experiments were validated by immunostaining of the arterial samples assessed with 4-hydroxynonenal-protein adducts, a biomarker of lipid peroxidation (Fig. 6A, right).
Fig. 6.

A: effect of reversed blood flow on NADPH oxidase activity. Data are expressed as the ratio between reversed flow and forward flow (RF/FF), reversed flow + Jasp and forward flow (RF + Jasp/FF), and reversed flow and forward flow + apocynin (RF/FF + Apo) (left). Carotid arterial sections were immunostained with anti-4-hydroxynonenal (HNE) protein (green). Scale bar = 20 μm (right). B: effect of reversed blood flow vs. forward blood flow on NADPH oxidase protein expression (gp91phox and p47phox) determined by Western blot in samples untreated and treated with jasplakinolide. C: protein expression of gp91phox and p47phox in forward and reversed flow samples, untreated (left) and treated (right) with jasplakinolide. D: Western blot bands of gp91phox and p47phox from carotid vessels and white blood cells (WBC). DAPI, 4, 6-diamidino-2-phenylindole. *P < 0.05, RF/FF vs. FF/FF; **P < 0.05, RF + Jasp/FF vs. RF/FF; **^P < 0.05, RF/FF + Apo vs. RF/FF.
NADPH oxidase protein levels were determined by Western blot. The levels of two major subunits, gp91phox (Nox2) and p47phox, are shown in Fig. 6B. The ratio of protein band density between reversed flow and forward flow vessels was close to unity, which suggests equal levels of protein expression under these conditions. The expressions of these subunits did not change after treatment with jasplakinolide. The protein expression of gp91phox and p47phox in forward and reversed flow samples, untreated and treated with jasplakinolide, are shown in Fig. 6C. Western blot bands of gp91phox and p47phox from carotid vessels and white blood cells (WBC) with their corresponding molecular weights are shown in Fig. 6D.
Blood flow reversal and actin depolymerization increased p47phox phosphorylation.
We examined p47phox phosphorylation to understand the molecular basis for increased ROS/O2·− production subsequent to actin depolymerization and blood flow reversal. As shown in Fig. 7A, treatment with PMA and cytochalasin D induced increased p47phox phosphorylation as compared with control (p < 0.05 in both cases). Under in vivo conditions, p47phox phosphorylation was also increased in vessels exposed to reversed blood flow as compared with those with forward flow (Fig. 7B; p < 0.05). Figure 7C shows the p47phox protein expression before and after immunoprecipitation in carotid vessels subjected to forward and reversed flow.
Fig. 7.

A: in vitro p47phox phosphorylation in untreated and treated samples with PMA and cytochalasin D with their corresponding protein expression. *P < 0.05, PMA or CytoD vs. control. B: in vivo p47phox phosphorylation during forward flow and reversed flow experiments with their corresponding protein expression. The loading controls were from different blots. *P < 0.05, reversed vs. forward. C: p47phox protein expression before (pre) and after (post) immunoprecipitation (IP) of carotid vessels subjected to forward and reversed flow. Post-IP samples were double loaded.
DISCUSSION
The major finding of this study was that the directionality of the mechanical loading (positive vs. negative WSS) on actin filaments plays an important role in the activity of the NADPH oxidase system in vascular endothelial cells. The findings suggest that actin depolymerization, which occurs during reversed blood flow, activates the NADPH oxidase system via p47phox phosphorylation, which induces ROS (mainly O2·− production). These novel observations may reveal the mechanisms of this mechanical-biochemical interaction, which has pathological implications for atherogenesis.
The regulation of NADPH oxidase has been mostly studied in phagocytes where disruption of actin polymerization potentiates O2·− production (22). Activation of phagocytic NADPH oxidase requires serine phosphorylation of the cytosolic p47phox, p67phox, and p40phox components, assembly of the phosphorylated subunits with Rac2, and translocation to the phagosomes for association with cytochrome b558 reductase, which consists of p22phox and gp91phox (Nox2) (45). The endothelial NADPH oxidase (Nox) shares many similarities with that of phagocytes. All the classical NADPH oxidase subunits are expressed in endothelial cells (17), and recent studies have indicated that several novel Nox family isoforms, in addition to Nox2 (formerly known as gp91phox), are highly expressed in vascular endothelial and smooth muscle cells (45). The mRNA and protein of p22phox can be detected in both endothelial and smooth muscle cells. In contrast, the expression of gp91phox (Nox2) is confined to endothelial cells (17). In nonstimulated endothelial cells, a proportion of the NADPH oxidase enzyme exists as a preassembled intracellular complex associated with the cytoskeleton in mainly perinuclear distribution (23). Endothelial oxidase constitutively generates small amounts of O2·− in nonstimulated cells, which is in agreement with our findings of basal constitutive production of ROS in the intact vessel (Figs. 2–4).
The interaction of actin and NADPH oxidase in endothelial cells is not well understood (1). Although the regulation of NADPH oxidase has been widely investigated in phagocytes, the data cannot be directly translated to endothelial cells. Protein kinase C has been shown to play a role in NADPH oxidase regulation in both phagocytes and nonphagocytes (8, 20). Cytochalasin D-induced O2·− production, however, is protein kinase C independent in phagocytes (22). Our findings in endothelial cells suggest that cytochalasin D does induce ROS/O2·− production (Figs. 2–4).
NADPH oxidase regulation may vary depending on cell type. Many previous studies have focused on NADPH oxidase and actin interaction in nonphagocytes under culture conditions. In these studies, increased depolymerization enhanced ROS production (26, 30, 35, 45), which is in agreement with our findings. The actin network in cultured cells, however, differs from primary cells, where cells in different stages of culture conditions migrate and maintain different surface tensions, which require variable actin polymerization and depolymerization rates. Therefore, the findings from culture cells should be tested under in vivo conditions.
A novel finding of this study is that actin depolymerization-mediated reversed blood flow induces ROS production in endothelial cells in vivo by increasing p47phox phosphorylation. Nam et al. (34) have also demonstrated ROS production in a p47phox-dependent manner in a mouse model of partial carotid ligation and disturbed flow. DHE staining intensity in the vessel walls of carotid arteries subjected to disturbed flow was significantly higher than that of vessels with normal flow, which suggests that ROS was produced in response to disturbed flow in a p47phox NADPH oxidase-dependent manner (34). Similarly, Castier et al. (6) also found that ROS production is enhanced in mice carotid arteries exposed to chronic high flow. They also showed that the p47phox subunit was the major generator of shear stress-induced ROS in the vascular wall. In this study, under blood flow reversal, ROS production was increased (Fig. 4) and actin depolymerization was observed in endothelial cells (Fig. 5C). The increase in ROS production was prevented by stabilizing the actin filaments with an actin stabilizer, jasplakinolide (Figs. 2 and 5E). Jasplakinolide is a cyclo-depsipeptide commonly used as an actin filament polymerizing and stabilizing drug (3). It is membrane permeable and can easily diffuse into the cells and selectively binds to F-actin. A low concentration (nanomolar range) of jasplakinolide was used in this study, which has been shown to be an effective dose (35). Jasplakinolide did not change basal ROS production (Fig. 2), suggesting that the change that occurred after treatment with jasplakinolide was not due to cell damage. To support this finding, we also showed that ROS production was increased in intact vessels under in vitro conditions when actin filaments were depolymerized by cytochalasin D (Fig. 2). In phagocytes, cytochalasins have been shown to induce or potentiate ROS/O2·− production via NADPH oxidase (33). To confirm that this was not due to nonspecific effects, we showed that latrunculin B, another actin polymerization inhibitor, activated ROS/O2·− production (Fig. 2).
Several cytoplasmic components of the NADPH oxidase are associated with the actin cytoskeleton (40, 47). This association is not permanent but inducible, as suggested by the fact that VEGF stimulation of endothelial cells triggers translocation of p47phox to the membrane ruffles, where the most active actin reorganization takes place (47). We speculate that actin depolymerization due to flow reversal regulated p47phox serine phosphorylation and facilitated p47phox translocation, i.e., increased NADPH oxidase activity, as seen in Figs. 6, B and C, and 7, C and D.
The NADPH oxidase enzyme assay supports the hypothesis that flow reversal increases the formation of the NADPH oxidase complex (Fig. 6A), and jasplakinolide prevented this elevation. The NADPH oxidase enzyme assay was performed in tissue homogenate after removal of intact cells and mitochondria by centrifugation. The ROS produced by this assay was derived from preformed and activated enzyme complex. As shown by Western blot, the protein levels were unchanged (Fig. 6B), which suggests that the increased in enzyme activity was not due to increased oxidase expression. Apocynin blocked the translocation of NADPH oxidase subunits. The enzyme activity was reduced in vessels from animals treated with apocynin (Fig. 6A). Various forms of actin polymer may have different binding affinity and effects on NADPH oxidase. For example, in a cell-free system, the addition of G-actin activated human neutrophil NADPH oxidase (31), whereas the addition of F-actin did not enhance oxidase activation. An actin binding site was identified in p47phox (41). The present study showed that increased depolymerization of actin facilitated ROS production (Figs. 2 and 4). This may be partially due to increased G-actin around NADPH oxidase, which presumably facilitated p47phox phosphorylation and translocation (i.e., upregulated enzyme activity). Although this study only focused on two subunits of the NADPH oxidase complex (Nox2 and p47phox), future studies should consider the role of other subunits in flow reversal, like Nox4, which has been shown to be regulated by shear stress (16, 21).
Study limitations.
First, the origin of ROS (mainly O2·−) was not directly determined. There are several enzymatic sources of ROS in mammalian cells, including NADPH oxidase system, mitochondrial electron transport chain, xanthine oxidase, and cytochrome p450 (23). Among these, the major source of ROS/O2·− in endothelial cells is the NADPH oxidase system (1). In our vessel preparation, ROS/O2·− was primarily derived from the endothelial cells. Removal of the endothelial layer decreased ROS/O2·− production by nearly 80% in all vessels (under various conditions). In support of our findings, others have shown that PMA stimulates ROS production in aortic segments only when the endothelial layer is intact (17). In the same study, PMA stimulated ROS production in cultured endothelial cells, but not in cultured smooth muscle cells. We have previously shown (15) in an in vitro blood vessel preparation that the major source of ROS/O2·− during complete reversed flow is the NADPH oxidase.
Second, an accurate method for real time direct measurement of ROS in vivo does not exist. We used a chemiluminescence probe, L-012, for ROS detection. Previous studies (9, 39) have shown that L-012 is a very sensitive probe for ROS measurement in cell free assay, isolated cells, or whole tissue. This probe has also been validated for ROS measurements in vascular tissues (9, 39). L-012 has also been shown to detect peroxynitrite in cell free solutions in the presence of nitric oxide (NO) (10) and hence raises the concern that NO might interfere with ROS measurement. L-012 also detects peroxynitrite in isolated mitochondria, but requires much higher concentration of NO. L-012, when used in intact tissue, predominantly detects extracellular O2·−. The contribution of peroxynitrite in L-012 signal in our study is likely to be less than that from ROS. In future studies, other methods to confirm ROS measurements as well as specific scavengers may be used concomitantly with L-012.
Third, jasplakinolide paradoxically stabilizes actin filaments in vitro, but it can disrupt actin filaments and induce polymerization of monomeric actin into amorphous masses in vivo. Our results show that the activation of NADPH oxidase is regulated by actin polymerization/depolymerization and that jasplakinolide attenuates ROS production from NADPH oxidase. The mechanistic regulation of actin polymerization/depolymerization on NADPH oxidase is unclear. We speculate that the concentration of monomeric actin may regulate the activation of NADPH oxidase through the translocation of cytoplasmic subunits of NADPH oxidase, e.g., p47phox. Therefore, administration of jasplakinolide decreases the concentration of monomeric actin by the polymerization of monomeric actin into an amorphous mass, even if jasplakinolide does not stabilize actin filaments in endothelial cells. Clearly, the mechanism of actin-NADPH oxidase interaction needs to be further studied.
Finally, there is a possible contribution of infiltrating leukocytes to the ROS/O2·− production in the carotid artery preparations; however, vessels were harvested fresh from the animals, and hence, unlikely to have significant infiltration of leukocytes. Furthermore, vessels subjected to reversed and forward flows were obtained under identical conditions. In addition, when the vessel was denuded, the O2·− production was dramatically reduced, which suggests that the endothelium was a predominant source of ROS/O2·−. We quantified IL-8 in the carotid vessels using ELISA. The presence of leukocytes in these specimens was not significantly different in reversed flow versus forward flow with intact (p = 0.98) and denuded (p = 0.92) endothelium, reversed flow with and without jasplakinolide treatment (p = 0.60), and reversed flow versus forward flow in the presence of apocynin (p = 0.98). Hence, the contribution of leukocytes to the relative ROS/O2·− production was either small or similar in control and treated vessels.
Conclusions
This study demonstrated that actin cytoskeleton plays an important regulatory role in reversed blood flow-induced ROS/O2·− production from NADPH oxidase in vascular endothelial cells. Negative shear stress leads to actin network reorganization, which induced p47phox phosphorylation and NADPH oxidase complex formation and ROS/O2·− production. Our results suggest that actin stabilization may be protective in vascular regions of blood flow reversal prone to atherogenesis.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute Grant HL-084529.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.S.C. and X.L. conception and design of research; J.S.C., X.L., and Z.-D.Z. performed experiments; J.S.C. and X.L. analyzed data; J.S.C., Z.-D.Z., and G.S.K. interpreted results of experiments; J.S.C. prepared figures; J.S.C. drafted manuscript; J.S.C. and G.S.K. edited and revised manuscript; J.S.C., X.L., Z.-D.Z., and G.S.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Benjamin Jansen, Charles Butterfield, and Margaret Brass for excellent technical assistance.
REFERENCES
- 1.Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life 50: 267–269, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Brakebusch C, Fassler R. The integrin-actin connection, an eternal love affair. EMBO J 22: 2324–2333, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem 269: 14869–14871, 1994 [PubMed] [Google Scholar]
- 4.Cardon AD, Bailey MR, Bennett BT. The Animal Welfare Act: from enactment to enforcement. J Am Assoc Lab Anim Sci 51: 301–305, 2012 [PMC free article] [PubMed] [Google Scholar]
- 5.Casella JF, Flanagan MD, Lin S. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 293: 302–305, 1981 [DOI] [PubMed] [Google Scholar]
- 6.Castier Y, Brandes RP, Leseche G, Tedgui A, Lehoux S. p47Phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res 97: 533–540, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curnutte JT, Erickson RW, Ding J, Badwey JA. Reciprocal interactions between protein kinase C and components of the NADPH oxidase complex may regulate superoxide production by neutrophils stimulated with a phorbol ester. J Biol Chem 269: 10813–10819, 1994 [PubMed] [Google Scholar]
- 9.Daiber A, August M, Baldus S, Wendt M, Oelze M, Sydow K, Kleschyov AL, Munzel T. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic Biol Med 36: 101–111, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Daiber A, Oelze M, August M, Wendt M, Sydow K, Wieboldt H, Kleschyov AL, Munzel T. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radic Res 38: 259–269, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev 75: 519–560, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gharib M, Beizaie M. Correlation between negative near-wall shear stress in human aorta and various stages of congestive heart failure. Ann Biomed Eng 31: 678–685, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 902: 230–239, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Gnasso A, Irace C, Carallo C, De Franceschi MS, Motti C, Mattioli PL, Pujia A. In vivo association between low wall shear stress and plaque in subjects with asymmetrical carotid atherosclerosis. Stroke 28: 993–998, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Godbole AS, Lu X, Guo X, Kassab GS. NADPH oxidase has a directional response to shear stress. Am J Physiol Heart Circ Physiol 296: H152–H158, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goettsch C, Goettsch W, Brux M, Haschke C, Brunssen C, Muller G, Bornstein SR, Duerrschmidt N, Wagner AH, Morawietz H. Arterial flow reduces oxidative stress via an antioxidant response element and Oct-1 binding site within the NADPH oxidase 4 promoter in endothelial cells. Basic Res Cardiol 106: 551–561, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res 87: 26–32, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Hong NJ, Silva GB, Garvin JL. PKC-α mediates flow-stimulated superoxide production in thick ascending limbs. Am J Physiol Renal Physiol 298: F885–F891, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res 93: 1225–1232, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imagawa N, Nagasawa K, Nagai K, Kawakami-Honda N, Fujimoto S. Protein kinase C-independent pathway for NADPH oxidase activation in guinea pig peritoneal polymorphonuclear leukocytes by Cytochalasin D. Arch Biochem Biophys 438: 119–124, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287: R1014–R1030, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277: 19952–19960, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech 38: 1949–1971, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Li H, Bubolz AH, Zhang DX, Gutterman DD. Endothelial cytoskeletal elements are critical for flow-mediated dilation in human coronary arterioles. Med Biol Eng Comput 46: 469–478, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu X, Dang CQ, Guo X, Molloi S, Wassall CD, Kemple MD, Kassab GS. Elevated oxidative stress and endothelial dysfunction in right coronary artery of right ventricular hypertrophy. J Appl Physiol 110: 1674–1681, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, Bernstein KE. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 375: 247–250, 1995 [DOI] [PubMed] [Google Scholar]
- 29.Matrougui K, Tanko LB, Loufrani L, Gorny D, Levy BI, Tedgui A, Henrion D. Involvement of Rho-kinase and the actin filament network in angiotensin II-induced contraction and extracellular signal-regulated kinase activity in intact rat mesenteric resistance arteries. Arterioscler Thromb Vasc Biol 21: 1288–1293, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal 1: 29–43, 1999 [DOI] [PubMed] [Google Scholar]
- 31.Morimatsu T, Kawagoshi A, Yoshida K, Tamura M. Actin enhances the activation of human neutrophil NADPH oxidase in a cell-free system. Biochem Biophys Res Commun 230: 206–210, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol 2: 376–378, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Nakagawara A, Minakami S. Role of cytoskeletal elements in cytochalasin E-induced superoxide production by human polymorphonuclear leukocytes. Biochim Biophys Acta 584: 143–148, 1979 [DOI] [PubMed] [Google Scholar]
- 34.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 297: H1535–H1543, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals Guide for the Care and Use of Laboratory Animals (8th ed.). Washington, DC: National Academies Press, 2011 [PubMed] [Google Scholar]
- 35.Qian Y, Liu KJ, Chen Y, Flynn DC, Castranova V, Shi X. Cdc42 regulates arsenic-induced NADPH oxidase activation and cell migration through actin filament reorganization. J Biol Chem 280: 3875–3884, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Saitoh T, Dobkins KR. Protein kinase C in human brain and its inhibition by calmodulin. Brain Research 379: 196–199, 1986 [DOI] [PubMed] [Google Scholar]
- 37.Samain E, Bouillier H, Perret C, Safar M, Dagher G. ANG II-induced Ca2+ increase in smooth muscle cells from SHR is regulated by actin and microtubule networks. Am J Physiol Heart Circ Physiol 277: H834–H841, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Snow HM, Markos F, O'Regan D, Pollock K. Characteristics of arterial wall shear stress which cause endothelium-dependent vasodilatation in the anaesthetized dog. J Physiol 531: 843–848, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohn HY, Gloe T, Keller M, Schoenafinger K, Pohl U. Sensitive superoxide detection in vascular cells by the new chemiluminescence dye L-012. J Vasc Res 36: 456–464, 1999 [DOI] [PubMed] [Google Scholar]
- 40.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11: 95–102, 1994 [DOI] [PubMed] [Google Scholar]
- 41.Tamura M, Itoh K, Akita H, Takano K, Oku S. Identification of an actin-binding site in p47phox an organizer protein of NADPH oxidase. FEBS Lett 580: 261–267, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Tamura M, Kai T, Tsunawaki S, Lambeth JD, Kameda K. Direct interaction of actin with p47(phox) of neutrophil NADPH oxidase. Biochem Biophys Res Commun 276: 1186–1190, 2000 [DOI] [PubMed] [Google Scholar]
- 43.Teng L, Fan LM, Meijles D, Li JM. Divergent effects of p47(phox) phosphorylation at S303–4 or S379 on tumor necrosis factor-α signaling via TRAF4 and MAPK in endothelial cells. Arterioscler Thromb Vasc Biol 32: 1488–1496, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Touyz RM. Apocynin, NADPH oxidase, and vascular cells: a complex matter. Hypertension 51: 172–174, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Usatyuk PV, Romer LH, He D, Parinandi NL, Kleinberg ME, Zhan S, Jacobson JR, Dudek SM, Pendyala S, Garcia JG, Natarajan V. Regulation of hyperoxia-induced NADPH oxidase activation in human lung endothelial cells by the actin cytoskeleton and cortactin. J Biol Chem 282: 23284–23295, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol 288: H1296–H1305, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Wu RF, Gu Y, Xu YC, Nwariaku FE, Terada LS. Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem 278: 36830–36840, 2003 [DOI] [PubMed] [Google Scholar]