

Abstract
A hallmark of cigarette smoking is a shift in the protease/antiprotease balance, in favor of protease activity. However, it has recently been shown that smokers have increased expression of a key antiprotease, secretory leukoprotease inhibitor (SLPI), yet the mechanisms involved in SLPI transcriptional regulation and functional activity of SLPI remain unclear. We examined SLPI mRNA and protein secretion in differentiated nasal epithelial cells (NECs) and nasal lavage fluid (NLF) from nonsmokers and smokers and demonstrated that SLPI expression is increased in NECs and NLF from smokers. Transcriptional regulation of SLPI expression was confirmed using SLPI promoter reporter assays followed by chromatin immunoprecipitation. The role of STAT1 in regulating SLPI expression was further elucidated using WT and stat1−/− mice. Our data demonstrate that STAT1 regulates SLPI transcription in epithelial cells and slpi protein in the lungs of mice. Additionally, we reveal that NECs from smokers have increased STAT1 mRNA/protein expression. Finally, we demonstrate that SLPI contained in the nasal mucosa of smokers is proteolytically cleaved but retains functional activity against neutrophil elastase. These results demonstrate that smoking enhances expression of SLPI in NECs in vitro and in vivo, and that this response is regulated by STAT1. In addition, despite posttranslational cleavage of SLPI, antiprotease activity against neutrophil elastase is enhanced in smokers. Together, our findings show that SLPI regulation and activity is altered in the nasal mucosa of smokers, which could have broad implications in the context of respiratory inflammation and infection.
Keywords: cigarette smoking, epithelial cells, antiprotease
cigarette smoking is a significant public health burden and has been linked with various cancers, heart disease, infection, and respiratory pathologies (15, 32). Each year in the United States, cigarette smoking results in more than $100 billion lost to cover healthcare and indirect costs (11). In the context of the respiratory mucosa, a hallmark of cigarette smoking is a shift in protease/antiprotease balance, in favor of protease expression and activity, resulting in increased inflammation and pathology (1, 14, 28, 43, 47, 53). As a potential balance to the increased protease expression and activity, recent studies indicate that a key antiprotease secretory leukoprotease inhibitor (SLPI) is elevated in smokers compared with nonsmokers (8). Moreover, in patients with chronic obstructive pulmonary disease (COPD) and in patients with COPD and secondary bacterial infection, SLPI levels are elevated in the respiratory tract (23, 29). However, the mechanisms mediating this induction of SLPI in the respiratory tract of smokers and patients with COPD are not known.
The SLPI promoter contains many regulatory sites, including interferon-sensitive response element (ISRE) binding sites (49). Thus transcription factors activated by interferon signaling pathways, such as signal transducers and activators of transcription 1 (STAT1), could be potential regulators of SLPI transcription by binding to ISRE and ISRE-like sites present in the SLPI promoter region. Although STAT1 has not been examined in the context of cigarette smoking, recent studies demonstrate a positive correlation between STAT1 induction and COPD status, as well as elevation of downstream STAT1-dependent genes such as NOS2, suggesting enhanced STAT1 activation and STAT1-dependent regulation of SLPI in smokers (4, 10).
In addition to transcriptional regulation, extracellular SLPI can be posttranslationally cleaved by respiratory proteases such as cathepsins, matrix metalloproteinases, chymase, and neutrophil elastase (NE), which can dramatically reduce SLPI activity (6, 30, 40, 46). Because increased protease levels are associated with smoking, this suggests that SLPI is cleaved and less active in smokers (1, 28, 53). Because cleaved SLPI can be proinflammatory, we believe that examining extracellular SLPI cleavage and activity is important in understanding the pathophysiology associated with smoking (29, 34).
Investigating differences in innate immune mechanisms in potentially susceptible subpopulations is important in understanding the underlying mechanisms for enhanced pathology and disease. SLPI is a key antiprotease involved in respiratory homeostasis and antimicrobial responses. Understanding the mechanisms that regulate SLPI transcriptional regulation in smokers is important because SLPI is a potent antiprotease in the lung and possesses anti-inflammatory and antimicrobial qualities. On the basis of the presence of ISRE sites in the promoter region of SLPI, we hypothesized that STAT1 may regulate SLPI expression in smokers. Using in vitro and in vivo approaches, we demonstrate that SLPI expression is enhanced in nasal epithelial cells (NECs) from smokers, that the expression of SLPI is regulated by STAT1, and that the proteolytic cleavage of SLPI in smokers does not affect the anti-NE activity.
MATERIALS AND METHODS
Study subjects and nasal lavage fluid collection.
Fourteen healthy young adults (9 men, 5 women); 7 nonsmokers (29.2 ± 7.2 yr old) and 7 smokers (27.1 ± 3.3 yr old), as characterized by our previous studies, were recruited to participate in this study (20). Informed consent was obtained from all subjects and the protocol was approved by the University of North Carolina Biomedical Institutional Review Board. Table 1 describes the demographic and cigarette smoking status of the participants. Nasal lavage was performed as previously described (33). Nasal lavage fluid (NLF) was filtered, centrifuged, and cell-free NLF supernatants were stored at −80°C.
Table 1.
Subject characteristics and smoking status
| Sex | Race | Age, yr | Body Mass Index | Packs per Day | |
|---|---|---|---|---|---|
| Nonsmokers | 3 male, 4 female | 5 white, 2 black | 29.2 ± 7.2 | 26.6 ± 4.3 | 0 |
| Smoker | 6 male, 1 female | 2 white, 4 black, 1 Asian | 27.1 ± 3.3 | 22.8 ± 4.1 | 0.75 ± 0.25 |
| Statistical relationship | P = 0.125 | P = 0.93 | P = 0.14 | P = 0.0156 |
Data are means ± SE. There were no significant differences between sex, age, and body mass index.
Differentiated human NECs and bronchial epithelial cell line.
NECs from nonsmoker and smoker volunteers were obtained, expanded, and cultured as described by us previously (33). Briefly, NECs were obtained from nonsmokers (n = 5) and smokers (n = 5) by sampling the inferior surface of the turbinate with a Rhino-Probe curette (Arlington Scientific, Arlington, TX), which was inserted through a nasoscope. This protocol was approved by the University of North Carolina School of Medicine Institutional Review Board for Biomedical Research. Primary NECs were expanded to passage 2, then plated on collagen-coated filter supports with a 0.4-μm pore size (Trans-CLR; Costar, Cambridge, MA). Upon confluency, air liquid interface culture conditions (removal of the apical medium) to promote differentiation was established as described by us previously (34). We obtained and grew bronchial epithelial cell line, BEAS-2B, as previously described (33).
Quantitative RT-PCR.
Total RNA was extracted using TRIzol. First-strand cDNA synthesis and quantitative RT-PCR (qRT-PCR) were performed using commercially available primers and probes (Applied Biosystems, Life Technologies, Carlsbad, CA) for SLPI, STAT1, and β-actin. Gene-specific mRNA levels were normalized to β-actin mRNA levels.
Western blotting.
Cell lysates and apical washes from NECs and lung homogenates from wild-type (WT) and stat1−/− mice were harvested. In some experiments, 7.5 μg of recombinant human SLPI (rhSLPI; R&D, Minneapolis, MN) was incubated with 0.15 U of NE (Enzo Life Sciences, Farmingdale, NY) at 37°C for 30 min prior to analysis by Western blotting. All samples were separated by 15% SDS-PAGE and transferred to nitrocellulose. Proteins were detected using specific antibodies (Santa Cruz, Dallas, TX) to SLPI and STAT1 (1:1,000) or β-actin (1:2,000), which served as a loading control. Antigen-antibody complexes were incubated with horseradish peroxidase-conjugated secondary antibody and were detected using chemiluminescence.
SLPI ELISA.
NLF was collected as described above and analyzed for SLPI protein using a commercially available ELISA (R&D).
SLPI transcriptional activation.
BEAS-2B cells were seeded overnight and transfected using Lipofectamine (Life Technologies) following the manufacturer's suggestions, as previously reported (33). To measure SLPI transcriptional activation, a plasmid containing luciferase E under the control of the SLPI promoter, containing 1,385 bp of the 5′ regulatory region on the porcine SLPI gene, was constructed (41). Cells were transfected with 1 μg of SLPI-Luc and 500 ng of TK-Renilla for 24 h. Cells were treated with 25 μM AG490 (Sigma Aldrich, St. Louis, MO), a JAK/STAT inhibitor, for 4 h and/or 1 ng/ml of recombinant human interferon-γ (IFN-γ; Calbiochem, Billerica, MA) for 2 h. Cell lysates were harvested and subjected to dual luciferase assay (Promega, Madison, WI).
Chromatin immunoprecipitation.
BEAS-2B cells were seeded on 10-cm dishes overnight and processed for Chromatin immunoprecipitation (ChIP) using a ChIP-IT Express Kit (Active Motif, Carlsbad, CA). Chromatin was immunoprecipitated with mouse anti-human phospho-STAT1 monoclonal antibody (Cell Signaling, Beverly, MA) or mouse anti-human RNA polymerase II IgG (Active Motif). Antibody-bound protein-DNA complexes were recovered using protein G-coated magnetic beads, and the DNA was analyzed by polymerase chain reaction (PCR). The oligonucleotide primers were designed to amplify the −500 to −700 region of the SLPI promoter, which contained a STAT1 binding site and were as follows: 5-CCTGAACCCTACTCCAAGCA-3 and 5-AGAAAGACACTTGCCCAGGA-3 (forward and reverse, respectively; 179 bp).
WT and stat1−/− mice.
Male WT and stat1−/− knockout (KO) mice bred on a 129S6/Sv/Ev background were purchased (Taconic Laboratories, Germantown, NY). Mice were housed in a climate-controlled animal care facility and given food and water ad libitum. All aspects of animal care and experimentation described in this study were conducted according to National Institutes of Health guidelines and approved by the North Carolina State University Institutional Animal Care and Use Committee. Mice were euthanized, lungs were harvested, and either flash frozen in liquid nitrogen or fixed with formalin for histological evaluation.
Immunohistochemistry.
Formalin-fixed lungs were embedded in paraffin, cut into sections, and prepared as previously described (20). Slides were incubated with SLPI antibody (1:200; Santa Cruz) followed by incubation with a biotin-labeled anti-rabbit antibody, washed, incubated with avidin-biotin complex, and washed. The signal was then detected with DAB (3,3′-diaminobenzidine), washed, and evaluated under light microscopy.
Anti-NE assay using NLF from nonsmokers and smokers.
SLPI activity was measured by examining the ability of SLPI to halt cleavage of NE-specific chromogenic substrate (52). Briefly, rhNE (3.5 μM) was incubated with NLF samples from nonsmokers and smokers for 20 min. After incubation, NE-specific chromogenic substrate (MeOSuc-AAPV-pNA; 20 mM, Sigma) was mixed into each sample and absorbance at 405 nm was measured. No inhibitor was used as a positive control. Anti-NE activity was determined by comparing anti-NE activity of samples to the anti-NE activity of no inhibitor and was expressed as percent activity.
Statistical analysis.
Data are presented as means ± SE for normally distributed data. Densitometric quantification was performed using Multi Gauge analysis software (Fuji Film, Tokyo, Japan). Mann-Whitney tests were used to compare SLPI/STAT1 mRNA, secreted SLPI levels, and SLPI activity in samples from nonsmokers and smokers. Paired Student's t-tests were used to analyze the effects of AG490 and/or IFN-γ on SLPI promoter reporter activity. An unpaired Student's t-test was used to assess differences in slpi levels in WT and stat1−/− mice. We performed the Mann-Whitney test to compare age and body mass index, column statistics to compare packs per day, and a two-tailed χ2 test to compare gender in nonsmokers or gender in smokers.
RESULTS
NECs from smokers have increased SLPI expression in vitro and in vivo.
Recent data demonstrate that the airways of smokers have increased SLPI expression and secretion, yet the mechanisms mediating this phenomenon remain unclear (23, 29). To examine SLPI levels in respiratory epithelial cells from nonsmokers and smokers, we used an in vitro model of differentiated primary NECs, as previously described (33). Intracellular and extracellular secreted SLPI levels were measured in NECs from nonsmokers and smokers. Figure 1, A–C, reveals that NECs from smokers have increased SLPI mRNA and secreted SLPI protein compared with nonsmokers. These data reveal that at baseline, NECs from smokers express significantly higher levels of SLPI compared with cells from nonsmokers. To confirm and expand our in vitro findings, we collected NLF from nonsmokers and smokers and analyzed it for SLPI proteins levels by ELISA. Figure 1D indicates that SLPI levels are significantly higher in NLF from smokers compared with nonsmokers. These data demonstrate that SLPI mRNA expression and SLPI secretion is increased in the nasal mucosa of smokers in vitro and in vivo.
Fig. 1.

SLPI mRNA and protein secretion from in vitro and in vivo samples. A: SLPI mRNA expression was examined by qRT-PCR and normalized to β-actin mRNA levels in nasal epithelial cells (NECs) from nonsmokers (NS) and smokers (SM). B and C: apical washes from NEC from NS and SM were normalized to total protein, examined for SLPI protein levels by Western blot, and analyzed by densitometry. In B, samples were run on the same blot but were nonadjacent. The 2 images were spliced together to form the figure as denoted by the black separating line. D: nasal lavage fluid from NS and SM was used to measure secreted SLPI protein by ELISA. n = 5–7 NS and SM. *P ≤ 0.05, **P ≤ 0.01.
STAT1 regulates SLPI transcriptional activation and binds to the SLPI promoter.
Analyses using the Basic Local Alignment Search Tool (BLAST) revealed that the SLPI promoter contains several ISRE-like and ISRE sites and specifically contains a STAT1 (AGGGCC)-specific binding site, starting at position −601 of the SLPI promoter. We hypothesized that STAT1 could activate SLPI expression because the SLPI promoter contains ISRE and ISRE-like sites (49). Exposure of BEAS-2B cells to IFN-γ increased SLPI transcription, whereas treatment with AG490, a JAK/STAT signaling inhibitor, decreased SLPI transcriptional activation (Fig. 2, A and B). Transfected cells pretreated with AG490 and exposed to IFN-γ resulted in significantly lower SLPI promoter activation, despite IFN-γ exposure (Fig. 2C). Using ChIP assays, we confirmed phospho-STAT1 binding to the SLPI promoter utilizing oligonucleotide primers that amplified the −500 to −700 region of the SLPI promoter, which contained a STAT1 binding site (Fig. 2D). These data mechanistically confirm that STAT1 regulates SLPI transcriptional activation in bronchial epithelial cells.
Fig. 2.

Transcriptional regulation of SLPI in bronchial epithelial cells. A: BEAS-2B cells were transfected with an SLPI promoter reporter construct, treated with vehicle or IFN-γ for 4 h and assayed for SLPI transcriptional activation by luciferase assay. Data were normalized to Renilla levels and expressed as relative luciferase units (RLU). After transfection with the SLPI promoter reporter construct, cells were treated with vehicle or AG490 for 4 h (B), or vehicle or AG490 for 4 h (C) prior to stimulation with IFN for 2 h, and assayed as in (A). D: BEAS-2B cells were processed for chromatin immunoprecipitation using a phospho-STAT1 antibody. PCR was performed using SLPI primers designed to include the STAT1 binding site located on the SLPI promoter. Data are representative of 3 independent experiments. *P ≤ 0.05.
Stat1−/− mice have decreased SLPI protein expression.
Because we show that STAT1 binds to the SLPI promoter, we wanted to further elucidate the significance of this association in vivo. To do so, lungs from 129S6/SvEv (WT) and stat1−/− mice were analyzed by immunohistochemisitry for SLPI expression. As expected, slpi expression was robust in epithelial cells lining the airways of WT mice, and slpi expression was lower in the lungs from stat1−/− mice than those of WT mice (Fig. 3A). Additionally, lung tissue from WT and stat1−/− mice was homogenized and assayed for slpi protein levels using Western blotting. Lung homogenate from stat1−/− mice had significantly less slpi protein compared with WT mice (Fig. 3, B and C). These data suggest that STAT1 regulates SLPI levels in the respiratory epithelium in vivo.
Fig. 3.
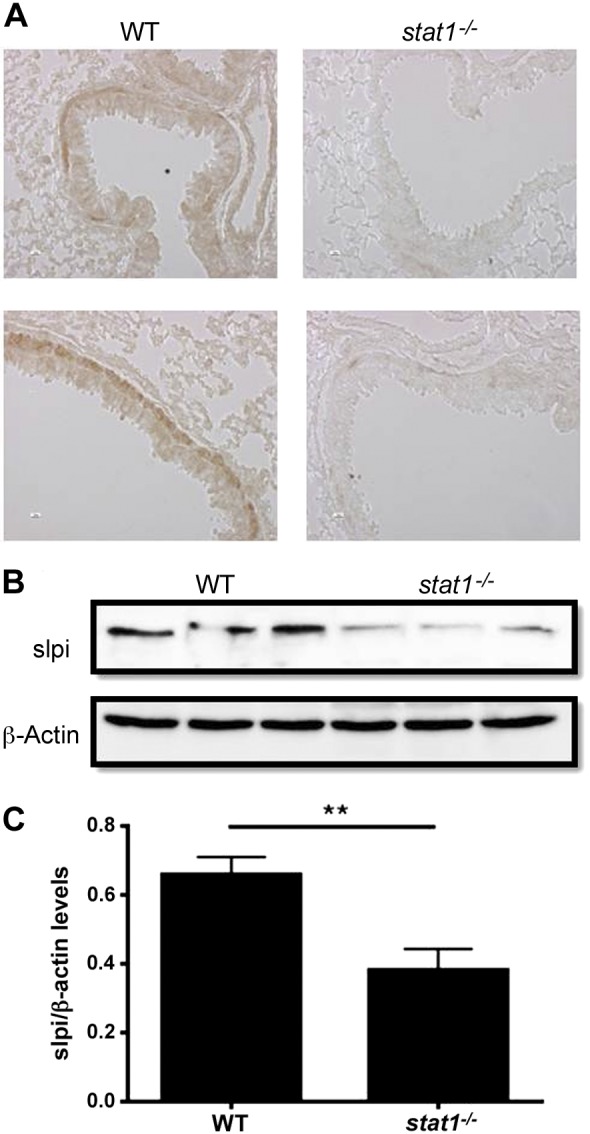
Expression of slpi in wild-type (WT) and stat1−/− mice. A: lungs from 129S6/SvEv (WT) and stat1−/− mice were harvested, fixed, and analyzed for slpi protein by immunohistochemistry. B and C: lungs from WT and stat1−/− mice were harvested, flash-frozen, and assayed for SLPI protein expression by Western blot and analyzed by densitometry. Data are representative of 2 independent experiments. n = 6 WT and stat1−/− mice. **P ≤ 0.01.
NECs from smokers have increased STAT1 mRNA and protein levels.
Although elevated STAT1 has not been reported in smokers, downstream STAT1-dependent genes such NOS2 are induced in smokers (10). Additionally, there is a positive correlation between STAT1 levels and patients with COPD, indicating that STAT1 expression and activation may be elevated in smokers (4). To examine smoking-induced changes in STAT1 expression, mRNA and protein were harvested and analyzed for STAT1 mRNA and protein expression in NECs from nonsmokers and smokers (Fig. 4, A–C). Our data reveal that at baseline, NECs from smokers had significantly increased STAT1 mRNA and a trend for increased STAT1 protein expression compared with nonsmokers.
Fig. 4.

STAT1 mRNA and protein levels in NECs from NS and SM. A: STAT1 mRNA expression was examined by qRT-PCR and normalized to β-actin levels in NECs from NS and SM. B and C: intracellular STAT1 protein levels were examined by Western blot and analyzed by densitometry in NECs from NS and SM. In B, samples were run on the same blot but were nonadjacent. The 2 images were spliced together to form the figure as denoted by the black separating line. n = 5 NS and SM. *P ≤ 0.05.
Extracellular SLPI protein is cleaved in smokers in vitro and in vivo.
It has previously been shown that SLPI can be cleaved by serine and cysteine protease such as cathepsins, matrix metalloproteinase, chymase, and NE (30, 40, 46, 52). To confirm these studies, we incubated rhSLPI with and without NE and examined SLPI cleavage by Western blotting. We chose NE because it is one of the major proteases elevated in the airways of smokers, and is the main target for the antiprotease activity of SLPI. Figure 5A demonstrates the cleavage pattern of rhSLPI induced by NE. Because smoking is characterized by increased serine and cysteine protease activity, we hypothesized that smokers would have increased cleavage of SLPI (1, 28, 53). To test this hypothesis, NLF from nonsmokers and smokers was collected, examined for SLPI cleavage by Western blotting, and analyzed using densitometry. Figure 5, B and C, demonstrates a trend for increased baseline SLPI cleavage in smokers. These data imply that although SLPI expression and secretion is higher in smokers, SLPI activity may be altered due to proteolytic cleavage.
Fig. 5.

Extracellular posttranslational modification of SLPI. A: rhSLPI was incubated with NE for 30 min and examined for SLPI cleavage by Western blot. B and C: NLF from NS and SM were analyzed for SLPI cleavage products by Western blot and analyzed by densitometry. In B, samples were run on the same blot but were nonadjacent. The 2 images were spliced together to form the figure as denoted by the black separating line. n = 6–7 NS and SM.
Anti-NE activity of SLPI is maintained despite posttranslational cleavage of SLPI in smokers.
On the basis of our data showing that SLPI is cleaved in smokers, and previous studies demonstrating that SLPI cleavage alters SLPI functional activity, we hypothesized that the functional activity of SLPI may be altered in smokers. We chose to examine SLPI function by evaluating the ability of SLPI to inhibit NE, its main target (26, 48). To assess total anti-NE capacity, equal NLF volumes from nonsmokers and smokers were incubated with rhNE and an NE-specific chromogenic substrate (MeOSuc-AAPV-pNA; 20 mM). Figure 6 indicates that the increased levels of SLPI observed in smokers were functionally active against NE, despite proteolytic cleavage and processing that was observed in Fig. 5. Additionally, when the percent activity is normalized to respective SLPI levels, the anti-NE activity remains greater in NLF from smokers (data not shown). We believe it is not possible to conclude that the anti-NE activity in smokers is due to higher SLPI levels or more functionally active SLPI in the NLF. However, we are able to conclude that the SLPI cleavage observed in NLF from smokers does not affect anti-NE activity.
Fig. 6.

SLPI activity against NE in nasal lavage fluid (NLF). NE was incubated with NLF from NS and SM for 20 min. After incubation, NE-specific chromogenic substrate was added and absorbance was measured. Data were normalized to the activity of no inhibitor and expressed as percent activity. n = 7 NS and SM. *P ≤ 0.05.
DISCUSSION
Using in vitro and in vivo models, we compared the expression of SLPI in the nasal mucosa of nonsmokers and smokers, examined the transcriptional regulation of SLPI by STAT1, and evaluated the extracellular processing and activity of SLPI in nonsmokers and smokers. Several reports have detailed increased SLPI secretion in smoking-related lung diseases such as COPD, yet the mechanisms mediating this effect remain unclear (8, 23, 29). We confirm these findings by showing increased SLPI mRNA and protein secretion in NEC and NLF from smokers (Fig. 1). We and others had previously reported that SLPI expression is regulated by a variety of transcription factors such as activator protein-1, nuclear factor-κB (NF-κB), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (33, 44). Previous reports and BLAST analysis of the SLPI promoter indicate that there are multiple ISRE and ISRE-like sites located on SLPI promoter, suggesting STAT1-dependent regulation of SLPI expression (35, 49). Our data indicate that in the nasal mucosa of smokers, enhanced baseline expression of STAT1 mediates increased SLPI expression.
In our study, we observed elevated STAT1 mRNA and protein expression at baseline in NECs from smokers (Fig. 4). Although we could not examine phosphorylated (activated) STAT1 in NECs from nonsmokers or smokers at baseline (data not shown), we believe that that downstream SLPI activation is indicative of STAT1 activity. We speculate that increased baseline STAT1 expression is independent of external type I or type II IFN stimuli, the primary inducers for STAT1, because it has been shown that smokers have decreased levels of type I and type II IFN, and epithelial cells are not a major source for type II IFN (12). As such, we hypothesize that STAT1 may be regulated by cigarette smoke-induced modifications to the epithelium. We have previously shown that epigenetic modifications regulate expression of genes in NECs from smokers. More specifically, we demonstrated that STAT6, a mediator involved in STAT signaling, is hypomethylated and expressed at higher levels in NECs from smokers (22, 39).
It is also possible that increased STAT1 may act as a compensatory protective mechanism in the epithelium. It has been previously reported that STAT1 acts as a tumor suppressor in a variety of capacities (21). The antitumor role of STAT1 is further supported by the report that STAT1 activation is repressed by micro-RNAs in cancer cells (17). Finally, it has been previously shown that STAT1 is protective in pulmonary fibrogenesis after bleomycin treatment in mice (51). Thus by upregulating SLPI expression, STAT1 may serve as a compensatory mediator to ameliorate the damaging effects of cigarette smoke. Together, these studies may suggest that there are alternative mechanisms and functions that regulate STAT1 activation.
The link between cigarette smoke and STAT1 activation is unclear, and various studies have provided conflicting data as to the mechanisms for STAT1 activation in the context of cigarette smoke exposure. Although elevated STAT1 has not been reported in NECs from smokers, STAT1 levels are correlated with COPD diagnosis, and downstream STAT1-dependent genes such as NOS2 are induced in smokers (4, 10). Additionally, studies performed in bronchial epithelial cell lines have shown that pretreatment with cigarette smoke extract (CSE) reduces STAT1 activation, whereas other studies report that CSE treatment increased STAT1 activation (13, 37). It should be noted that for these studies, the acute effects of CSE rather than the chronic effects of cigarette smoking on the respiratory epithelium after multiple years, or possible decades, of smoking, were examined. Moreover, various groups have reported that nicotine enhances STAT1 activation, and because nicotine is a main component of cigarettes, this may be an alternative explanation for increased STAT1 levels in smokers (3, 27). Additionally, it has been shown that STAT1 is strongly induced by reactive oxygen species (ROS) such as H2O2. This could alternatively explain a mechanism for ROS-dependent STAT1 induction because H2O is generated after cigarette smoke exposure (2). Taken together, these data may indicate that elevated baseline STAT1 may serve as a response to chronic oxidative stress (Figs. 2 and 3).
Although we detail the intracellular mechanism for SLPI regulation, we maintain that testing the extracellular functional activity of SLPI is important because patients with COPD with secondary bacterial infection or cystic fibrosis have increased levels of cleaved SLPI, which has been speculated as a main mechanism for inactivation of SLPI (1, 6, 29, 52). Extracellular SLPI can be cleaved by respiratory proteases such as cathepsin L, matrix metalloproteinase 12 (MMP12), and NE, and can serve as a biomarker for airway disease (5, 6, 30, 40, 46). It has been shown that cathepsins and MMPs are induced in smokers as well as in chronic cigarette smoke exposure models (9, 28). Additionally, alternatively processed SLPI has been shown to induce interleukin-8 and to recruit neutrophils, leading to enhanced pathophysiology associated with smoking (34). Because increased protease levels and activity are associated with smoking, this could explain the phenomenon of decreased SLPI activity in smokers (1, 28, 53). However, we show that although extracellular SLPI is cleaved, the functional activity against NE of SLPI is maintained in smokers (Figs. 5 and 6).
Although the major role of SLPI is to inhibit the activity of NE, additional functions of SLPI are being discovered. For example, SLPI exerts cellular anti-inflammatory effects by preventing the degradation of regulatory components of the NF-κB pathway, such as IκBα and IκBβ (16). SLPI further suppresses inflammation by directly competing for NF-κB binding sites in the promoter regions of proinflammatory cytokines such as interleukin-8 and tumor necrosis factor-α (44, 45). SLPI is also protective in the context of fungal, bacterial, and viral infections (24, 31, 38, 42, 50). In the context of the respiratory mucosa, SLPI has been shown to prevent the proposed proteases in influenza A virus (IAV) activation and infection, demonstrating another alternative action of SLPI (7, 25). Thus although the anti-NE activity of SLPI is elevated in smokers, secreted SLPI levels in smokers may be altered and nonfunctional in the context of inflammation or infection. This is an important area of study considering that cigarette smoking results in proinflammatory events and impairs anti-IAV response both in vitro and in vivo (20, 22, 36).
Each year, cigarette smoking results in significant loss due to healthcare and associated costs (11). We and others have shown that cigarette smoking modifies mucosal host defense, yet the specific mechanisms mediating this effect remain unclear (19, 20, 22, 36). A hallmark of cigarette smoking is a shift in the protease/antiprotease balance in favor of protease expression and activity (1, 28, 53). Although it has been previously reported that smokers and patients with COPD have increased SLPI secreted levels, we believe this study is the first to detail the mechanisms regulating SLPI transcription and to characterize the extracellular activity of SLPI in respiratory epithelial cells from smokers (23, 29). We demonstrate that STAT1 regulates SLPI transcriptional activation, and that although extracellular SLPI is cleaved in smokers, the functional activity of SLPI against NE is maintained in smokers. Because the canonical SLPI cleavage site occurs within an anti-NE active site and anti-NE activity was maintained in smokers, we predict that an alternative cleavage site exists within the SLPI protein. On the basis of our data and previous reports, these alternative cleavage products could dramatically affect the alternative functions of SLPI (18, 46). Detailing alternative extracellular processing and alternative activity of SLPI could be of extreme importance, because SLPI possesses anti-inflammatory, antibacterial, antifungal, and antiviral properties. Finally, because the protease/antiprotease balance is found in a variety of organs, understanding the regulation and activity of SLPI could have significant implications beyond the respiratory mucosa.
GRANTS
This study was supported by National Institutes of Health Grants R01 HL-095163, R01 CA-122906, and R01 ES-020897, and by the Flight Attendant Medical Research Institute.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.M., L.E.B., E.T., R.C.S., J.B., and I.J. conception and design of research; M.M., R.N.B., B.D.L., L.E.B., and E.T. performed experiments; M.M., R.N.B., B.D.L., L.E.B., and I.J. analyzed data; M.M., R.N.B., B.D.L., J.B., and I.J. interpreted results of experiments; M.M. prepared figures; M.M. and I.J. drafted manuscript; M.M., R.N.B., B.D.L., E.T., R.C.S., J.B., and I.J. edited and revised manuscript; M.M., R.N.B., B.D.L., L.E.B., E.T., R.C.S., J.B., and I.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Martha Almond, Carole Robinette, and Margaret Herbst for subject recruitment.
REFERENCES
- 1.Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis 12: 361–367, 2008 [PubMed] [Google Scholar]
- 2.Antao-Menezes A, Turpin EA, Bost PC, Ryman-Rasmussen JP, Bonner JC. STAT-1 signaling in human lung fibroblasts is induced by vanadium pentoxide through an IFN-beta autocrine loop. J Immunol 180: 4200–4207, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Arredondo J, Chernyavsky AI, Grando SA. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol Ther 5: 511–517, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Bakke PS, Zhu G, Gulsvik A, Kong X, Agusti AG, Calverley PM, Donner CF, Levy RD, Make BJ, Pare PD, Rennard SI, Vestbo J, Wouters EF, Anderson W, Lomas DA, Silverman EK, Pillai SG. Candidate genes for COPD in two large data sets. Eur Respir J 37: 255–263, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Belkowski SM, Boot JD, Mascelli MA, Diamant Z, de Garavilla L, Hertzog B, Polkovitch D, Towers M, Batheja A, D'Andrea MR. Cleaved secretory leucocyte protease inhibitor as a biomarker of chymase activity in allergic airway disease. Clin Exp Allergy 39: 1179–1186, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Belkowski SM, Masucci J, Mahan A, Kervinen J, Olson M, de Garavilla L, D'Andrea MR. Cleaved SLPI, a novel biomarker of chymase activity. Biol Chem 389: 1219–1224, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Beppu Y, Imamura Y, Tashiro M, Towatari T, Ariga H, Kido H. Human mucus protease inhibitor in airway fluids is a potential defensive compound against infection with influenza A and Sendai viruses. J Biochem 121: 309–316, 1997 [DOI] [PubMed] [Google Scholar]
- 8.Bouloukaki I, Tsiligianni IG, Tsoumakidou M, Mitrouska I, Prokopakis EP, Mavroudi I, Siafakas NM, Tzanakis N. Sputum and nasal lavage lung-specific biomarkers before and after smoking cessation. BMC Pulm Med 11: 35, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bracke K, Cataldo D, Maes T, Gueders M, Noel A, Foidart JM, Brusselle G, Pauwels RA. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol 138: 169–179, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Brindicci C, Kharitonov SA, Ito M, Elliott MW, Hogg JC, Barnes PJ, Ito K. Nitric oxide synthase isoenzyme expression and activity in peripheral lung tissue of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 181: 21–30, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention Ten great public health achievements—United States, 1900–1999. MMWR Morb Mortal Wkly Rep 48: 241–243, 1999 [PubMed] [Google Scholar]
- 12.Dhillon NK, Murphy WJ, Filla MB, Crespo AJ, Latham HA, O'Brien-Ladner A. Down modulation of IFN-gamma signaling in alveolar macrophages isolated from smokers. Toxicol Appl Pharmacol 237: 22–28, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol 44: 118–126, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gompertz S, Bayley DL, Hill SL, Stockley RA. Relationship between airway inflammation and the frequency of exacerbations in patients with smoking related COPD. Thorax 56: 36–41, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goncalves RB, Coletta RD, Silverio KG, Benevides L, Casati MZ, da Silva JS, Nociti FH., Jr Impact of smoking on inflammation: overview of molecular mechanisms. Inflamm Res 60: 409–424, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Greene CM, McElvaney NG, O'Neill SJ, Taggart CC. Secretory leucoprotease inhibitor impairs Toll-like receptor 2- and 4-mediated responses in monocytic cells. Infect Immun 72: 3684–3687, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregersen LH, Jacobsen AB, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS One 5: e8836, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinzel-Wieland R, Steffens GJ, Flohe L. Inhibitory characteristics and oxidant resistance of site specific variants of recombinant human antileukoproteinase (ALP). Biomed Biochim Acta 50: 677–681, 1991 [PubMed] [Google Scholar]
- 19.Horvath KM, Brighton LE, Herbst M, Noah TL, Jaspers I. Live attenuated influenza virus (LAIV) induces different mucosal T cell function in nonsmokers and smokers. Clin Immunol 142: 232–236, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horvath KM, Herbst M, Zhou H, Zhang H, Noah TL, Jaspers I. Nasal lavage natural killer cell function is suppressed in smokers after live attenuated influenza virus. Respir Res 12: 102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang S, Bucana CD, Van Arsdall M, Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene 21: 2504–2512, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Jaspers I, Horvath KM, Zhang W, Brighton LE, Carson JL, Noah TL. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am J Respir Cell Mol Biol 43: 368–375, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kersul AL, Iglesias A, Rios A, Noguera A, Forteza A, Serra E, Agusti A, Cosio BG. Molecular mechanisms of inflammation during exacerbations of chronic obstructive pulmonary disease. Arch Bronconeumol 47: 176–183, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Kesic MJ, Meyer M, Bauer R, Jaspers I. Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza A infection. PLoS One 7: e35108, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kido H, Okumura Y, Yamada H, Mizuno D, Higashi Y, Yano M. Secretory leukoprotease inhibitor and pulmonary surfactant serve as principal defenses against influenza A virus infection in the airway and chemical agents up-regulating their levels may have therapeutic potential. Biol Chem 385: 1029–1034, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Kramps JA, Franken C, Dijkman JH. ELISA for quantitative measurement of low-molecular-weight bronchial protease inhibitor in human sputum. Am Rev Respir Dis 129: 959–963, 1984 [DOI] [PubMed] [Google Scholar]
- 27.Kunigal S, Ponnusamy MP, Momi N, Batra SK, Chellappan SP. Nicotine, IFN-gamma and retinoic acid mediated induction of MUC4 in pancreatic cancer requires E2F1 and STAT-1 transcription factors and utilize different signaling cascades. Mol Cancer 11: 24, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Louhelainen N, Rytila P, Haahtela T, Kinnula VL, Djukanovic R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm Med 9: 25, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallia P, Footitt J, Sotero R, Jepson A, Contoli M, Trujillo-Torralbo MB, Kebadze T, Aniscenko J, Oleszkiewicz G, Gray K, Message SD, Ito K, Barnes PJ, Adcock IM, Papi A, Stanciu LA, Elkin SL, Kon OM, Johnson M, Johnston SL. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in COPD. Am J Respir Crit Care Med 186: 1117–1124, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masuda K, Suga T, Takeuchi A, Kanesaki M, Imaizumi A, Suzuki Y. Specific cleavage of secretory leukoprotease inhibitor by neutrophil elastase and saliva. Biochem Pharmacol 48: 651–657, 1994 [DOI] [PubMed] [Google Scholar]
- 31.McNeely TB, Shugars DC, Rosendahl M, Tucker C, Eisenberg SP, Wahl SM. Inhibition of human immunodeficiency virus type 1 infectivity by secretory leukocyte protease inhibitor occurs prior to viral reverse transcription. Blood 90: 1141–1149, 1997 [PubMed] [Google Scholar]
- 32.Mehta H, Nazzal K, Sadikot RT. Cigarette smoking and innate immunity. Inflamm Res 57: 497–503, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Meyer M, Kesic MJ, Clarke J, Ho E, Simmen RC, Diaz-Sanchez D, Noah TL, Jaspers I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respir Med 107: 472–475, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulligan MS, Lentsch AB, Huber-Lang M, Guo RF, Sarma V, Wright CD, Ulich TR, Ward PA. Anti-inflammatory effects of mutant forms of secretory leukocyte protease inhibitor. Am J Pathol 156: 1033–1039, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen H, Teskey L, Lin R, Hiscott J. Identification of the secretory leukocyte protease inhibitor (SLPI) as a target of IRF-1 regulation. Oncogene 18: 5455–5463, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Noah TL, Zhou H, Monaco J, Horvath K, Herbst M, Jaspers I. Tobacco smoke exposure and altered nasal responses to live attenuated influenza virus. Environ Health Perspect 119: 78–83, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pace E, Ferraro M, Di Vincenzo S, Bruno A, Giarratano A, Scafidi V, Lipari L, Di Benedetto DV, Sciarrino S, Gjomarkaj M. Cigarette smoke increases BLT2 receptor functions in bronchial epithelial cells: in vitro and ex vivo evidence. Immunology 139: 245–255, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parameswaran GI, Wrona CT, Murphy TF, Sethi S. Moraxella catarrhalis acquisition, airway inflammation and protease-antiprotease balance in chronic obstructive pulmonary disease. BMC Infect Dis 9: 178, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rager JE, Bauer R, Muller LL, Smeester L, Carson JL, Brighton LE, Fry RC, Jaspers I. DNA methylation in nasal epithelial cells from smokers: identification of ULBP3-related effects. Am J Physiol Lung Cell Mol Physiol 305: L432–L438, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramadas RA, Wu L, LeVine AM. Surfactant protein A enhances production of secretory leukoprotease inhibitor and protects it from cleavage by matrix metalloproteinases. J Immunol 182: 1560–1567, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Reed KL, Badinga L, Davis DL, Chung TE, Simmen RC. Porcine endometrial glandular epithelial cells in vitro: transcriptional activities of the pregnancy-associated genes encoding antileukoproteinase and uteroferrin. Biol Reprod 55: 469–477, 1996 [DOI] [PubMed] [Google Scholar]
- 42.Reviglio VE, Sambuelli RH, Olmedo A, Falco M, Echenique J, O'Brien TP, Kuo IC. Secretory leukocyte protease inhibitor is an inducible antimicrobial peptide expressed in Staphylococcus aureus endophthalmitis. Mediators Inflamm 2007: 93857, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med 160: S49–S52, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O'Neill SJ, McElvaney NG. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med 202: 1659–1668, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taggart CC, Greene CM, McElvaney NG, O'Neill S. Secretory leucoprotease inhibitor prevents lipopolysaccharide-induced IkappaBalpha degradation without affecting phosphorylation or ubiquitination. J Biol Chem 277: 33648–33653, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Taggart CC, Lowe GJ, Greene CM, Mulgrew AT, O'Neill SJ, Levine RL, McElvaney NG. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem 276: 33345–33352, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis 5: 255–273, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Tournier JM, Jacquot J, Sadoul P, Bieth JG. Noncompetitive enzyme immunoassay for the measurement of bronchial inhibitor in biological fluids. Anal Biochem 131: 345–350, 1983 [DOI] [PubMed] [Google Scholar]
- 49.Velarde MC, Parisek SI, Eason RR, Simmen FA, Simmen RC. The secretory leukocyte protease inhibitor gene is a target of epidermal growth factor receptor action in endometrial epithelial cells. J Endocrinol 184: 141–151, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Wahl SM, McNeely TB, Janoff EN, Shugars D, Worley P, Tucker C, Orenstein JM. Secretory leukocyte protease inhibitor (SLPI) in mucosal fluids inhibits HIV-I Oral diseases. Oral Dis 3, Suppl 1: S64–S69, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Walters DM, Antao-Menezes A, Ingram JL, Rice AB, Nyska A, Tani Y, Kleeberger SR, Bonner JC. Susceptibility of signal transducer and activator of transcription-1-deficient mice to pulmonary fibrogenesis. Am J Pathol 167: 1221–1229, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weldon S, McNally P, McElvaney NG, Elborn JS, McAuley DF, Wartelle J, Belaaouaj A, Levine RL, Taggart CC. Decreased levels of secretory leucoprotease inhibitor in the Pseudomonas-infected cystic fibrosis lung are due to neutrophil elastase degradation. J Immunol 183: 8148–8156, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wewers MD, Herzyk DJ, Gadek JE. Comparison of smoker and nonsmoker lavage fluid for the rate of association with neutrophil elastase. Am J Respir Cell Mol Biol 1: 423–429, 1989 [DOI] [PubMed] [Google Scholar]