

Abstract
The activity of glucose-6-phosphate dehydrogenase (G6PD) controls a vascular smooth muscle relaxing mechanism promoted by the oxidation of cytosolic NADPH, which has been associated with activation of the 1α form of protein kinase G (PKG-1α) by a thiol oxidation-elicited subunit dimerization. This PKG-1α-activation mechanism appears to contribute to responses of isolated endothelium-removed bovine pulmonary arteries (BPA) elicited by peroxide, cytosolic NADPH oxidation resulting from G6PD inhibition, and hypoxia. Dehydroepiandrosterone (DHEA) is a steroid hormone with pulmonary vasodilator activity, which has beneficial effects in treating pulmonary hypertension. Because multiple mechanisms have been suggested for the vascular effects of DHEA and one of the known actions of DHEA is inhibiting G6PD, we investigated whether it promoted relaxation associated with NADPH oxidation, PKG-1α dimerization, and PKG activation detected by increased vasodilator-stimulated phosphoprotein (VASP) phosphorylation. Relaxation of BPA to DHEA under aerobic or hypoxic conditions was associated with NADPH oxidation, PKG-1α dimerization, and increased VASP phosphorylation. The vasodilator activity of DHEA was markedly attenuated in pulmonary arteries and aorta from a PKG knockin mouse containing a serine in place of a cysteine involved in PKG dimerization. DHEA promoted increased PKG dimerization in lungs from wild-type mice, which was not detected in the PKG knockin mouse model. Thus PKG-1α dimerization is a major contributing factor to the vasodilator actions of DHEA and perhaps its beneficial effects in treating pulmonary hypertension.
Keywords: cGMP, glucose-6-phosphate dehydrogenase, pulmonary hypertension
early studies by Archer and Weir on the mechanism of hypoxic pulmonary vasoconstriction suggested that the oxidation of cytosolic NAD(P)H promoted vasorelaxation of pulmonary arterial smooth muscle by opening potassium channels (3, 27). Observations that inhibitors of glucose-6-phosphate dehydrogenase (G6PD) promote vascular relaxation through mechanisms including opening various potassium channels (10, 16) evolved into evidence for cytosolic NADPH oxidation being a coordinator of mechanisms mediating vascular relaxation (13, 17, 25). Recent studies from our laboratory in bovine pulmonary arteries (BPA) suggested that cytosolic NADPH oxidation resulting from inhibition of G6PD by 6-aminonicotinamide promotes relaxation through activation of protein kinase G 1α (PKG-1α) by a thiol oxidation-mediated subunit dimerization (25). Vascular relaxation through this cGMP-independent mechanism of PKG activation by dimerization was initially reported by the Eaton laboratory to function as a novel and major contributing mechanism to the relaxation of systemic arteries to peroxide (5). Earlier studies from our laboratory provided evidence that hydrogen peroxide (H2O2) stimulates soluble guanylate cyclase (sGC) under conditions where it relaxes isolated endothelium-removed BPA (7). Subsequently, our laboratory documented evidence that peroxide appears to relax BPA by both a sGC/cGMP-dependent and a dimerization-dependent activation of PKG and that decreases in both of these mechanisms appear to contribute to the contraction of BPA elicited by acute hypoxia (23, 24). It is now known that the oxidation of cytosolic NADPH appears to both activate and coordinate multiple relaxing mechanisms in pulmonary and systemic arteries across several animal species (13, 14, 16, 17, 25). Thus cGMP-independent activation of PKG dimerization may be a major factor in the coordination of multiple processes contributing to vascular relaxation resulting from cytosolic NADPH oxidation.
Dehydroepiandrosterone (DHEA) is a steroid hormone that protects animals from the development of pulmonary hypertension, and it is currently being investigated in clinical trials for the treatment of patients with pulmonary hypertension (2, 4, 9, 18, 20, 26). Although it is known that DHEA functions as an inhibitor of G6PD (16, 31), it has also been suggested to promote vasodilation through several different mechanisms including opening voltage-regulated potassium and/or calcium channels (10, 16), upregulating sGC (26), and by decreasing Rho kinase activity (20). In this study, we used a “redox-dead” PKG knockin (PKG-KI, 28) mouse model to provide novel evidence that could define the role of PKG dimerization in the vasodilator actions of DHEA. This mouse model was generated by the Eaton laboratory based on the properties of PKG dimerization (5, 28). In this mouse model, cysteine that is involved in PKG dimerization by peroxide is replaced by serine, preventing both dimerization and PKG activation. Thus, if DHEA promoted relaxation through PKG activation by dimerization, its vasodilator actions should be attenuated along with effects of peroxide in arteries from this mouse model. In addition, we also examined the effects of DHEA on PKG dimerization and activation in BPA under both aerobic and hypoxic conditions to determine whether PKG dimerization is a major factor in the actions of DHEA. Based on our previous studies with the G6PD inhibitor 6-aminonicotinamide, we hypothesized that DHEA may promote a PKG dimerization-mediated relaxation by oxidizing cytosolic NADPH as a result of inhibiting G6PD.
MATERIALS AND METHODS
Materials.
All salts used for making physiological solutions were analyzed reagent grade from Baker Chemical (Phillipsburg, NJ). All gasses were purchased from Tech Air (White Plains, NY). Acetylcholine, DHEA, H2O2, and maleimide were obtained from Sigma Aldrich Chemical (St. Louis, MO). Spermine-NONOate was purchased from Cayman Chemical (Ann Arbor, MI). cGMP-dependent PKG-1α antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-Actin antibody was purchased from Sigma Chemical. Vasodilator-stimulated phosphoprotein (VASP) and total-VASP antibodies were purchased from Cell Signaling (Beverly, MA). NADPH concentrations were determined by using NADP/NADPH quantification kit (BioVision, Mountain View, CA). Fresh slaughterhouse-derived bovine lungs were purchased from Max Cohen, as previously described (23). PKG-KI (redox-dead) mice originally generated by Dr. Philip Eaton's laboratory at Kings' College, London (28) were generously provided to us through David Kass at John Hopkins University, Baltimore, MD. All animal protocols were IACUC approved at New York Medical College.
Preparation of isolated BPA.
Bovine lungs were obtained from a slaughterhouse in ice-cold buffered physiological saline. The second and third branches of main lobar pulmonary artery were used for BPA studies (23). Pulmonary arteries were cleaned of their connective tissue and cut into rings of 2–3-mm diameter and width. Endothelium was removed by rubbing the lumen. Freshly isolated blood vessel rings were used in studies for vascular reactivity and Western blot protein analysis.
Measurement of vascular reactivity in BPA.
Endothelium-removed artery rings were mounted on Grass FT-03 or Coulborne Instruments force displacement transducers for recording isometric force development through a Powerlab data acquisition system obtained from ADInstruments (Colorado Springs, CO), as previously described (23). Arterial rings were incubated at 37°C in Krebs-bicarbonate buffer (pH 7.4) containing 118 mM NaCl, 4.7 mM KCl, 1.5 mM CaCl2, 25 mM NaHCO3, 1.1 mM MgSO4, 1.2 mM KH2PO4, and 5.6 mM glucose under an atmosphere of 21% O2-5% CO2-74% N2 (pH 7.4) for 1 h under resting tension of 5 g. In all studies, arterial rings were depolarized with 123 mM KCl containing Krebs bicarbonate buffer, and the rings were then reequilibrated with Krebs-bicarbonate buffer for 30 min and subsequently contracted with Krebs bicarbonate containing 30 mM KCl. Some arteries were pretreated with 50 μM of the inhibitor of Nox oxidase activation gp91dstat, as previously described (1). Freshly isolated BPA rings were treated with 1 μM, 10 μM, and 100 μM DHEA or H2O2 doses under aerobic (21% O2) and/or hypoxic (95% N2-5% CO2, Po2 ∼8–10 Torr) conditions, as described in results. Some of the arteries from the DHEA protocol were subsequently flash frozen with liquid nitrogen for Western blot analysis when changes in force reached a steady state under conditions described in results. For NADPH measurements, BPA rings were also flash frozen with liquid nitrogen in the absence or presence of 100 μM DHEA treatment under aerobic and hypoxic conditions.
Isolation of pulmonary arteries and aorta from mice.
PKG-KI mice and their genetic background matched mice (WT) were initially anesthetized with 50 mg/kg pentobarbital for removal of their hearts, lungs, and aorta. Pulmonary arteries and the aorta were isolated carefully from each mouse in ice-cold buffer and maintained in cold Krebs-bicarbonate buffer. After isolation of tissues, small rings were prepared from each aorta and pulmonary artery (180 μm internal diameter), and these vascular segments were used for vascular reactivity studies.
Measurement of vascular reactivity in mouse aorta and pulmonary arteries.
Freshly prepared rings were used for force measurement studies. Isometric force was studied under an atmosphere of 21% O2-5% CO2-74% N2 (pH 7.4) in a temperature-controlled Krebs solution. Aortic rings were mounted on WPI force displacement transducers, which were attached to a Powerlab data acquisition system from ADInstruments. Pulmonary arterial rings were mounted on DMT myographs, which were also attached to a Powerlab data acquisition system. Aortic rings were initially incubated at a passive tension of 2 g, and pulmonary rings were initially incubated at a passive tension of 0.5 g for 1 h in Krebs-bicarbonate buffer solution gassed with 21% O2-5% CO2-74% N2. The temperature was maintained at 37°C in the individually thermostated baths containing the rings from aorta, while DMT myographs were also maintained at 37°C. Following this 1 h of incubation, the rings were depolarized with 123 mM KCl containing Krebs-bicarbonate buffer, and the rings were again reequilibrated with normal Krebs-bicarbonate buffer for another 30 min.
Western blot analysis.
PKG dimer expression was detected in mouse lung tissue or BPA by running a Western blot under nonthiol-reducing conditions as published previously by our laboratory (23). The phosphorylation of VASP at a PKG-selective site traditionally used as an indicator of extent of PKG activation (19) was measured to also help document stimulation of this system, also using previously employed methods (23). Frozen lung tissues or BPAs were pulverized and then homogenized in lysis buffer containing protease and phosphatase inhibitors, as previously described (24). Maleimide (100 mM) was included in the lysis buffer to alkylate the thiols to avoid artifactual disulfide bond formation during homogenization, as published previously (5, 23). Bradford method was used for protein quantification assay, and samples were prepared for gel electrophoresis. Proteins were separated using a 10% SDS-polyacrylamide gel. Gels were transferred to PVDF membranes, and the membranes were blocked with Tris-buffered saline with Tween-20 + 5% milk for 1 h. After this the membranes were exposed to primary and secondary antibodies per the manufacturer's protocol. Protein bands were visualized with an enhanced chemiluminescence kit (Pierce, Rockford, IL) on X-OMAT autoradiography paper (Kodak, Rochester, NY) in a dark room. Relative changes in PKG-1α monomer and dimer forms are reported as the percentage of the total PKG-1α. Changes in PKG-1α monomer and dimer expression were quantified after normalization to β-actin, and phosphorylated VASP was normalized to total VASP in each individual artery studied. Protein levels were measured using densitometry analysis with the UN-SCAN-IT gel software by Silk Scientific (Orem, UT). Molecular weights of PKG monomer and dimer are 75 KDa and 150 KDa, respectively.
Statistical analysis.
Data values are means ± SE of the number of arterial segments (n) from different animals. Statistical analyses between two groups were performed with paired and unpaired Student's t-test, and a one-way ANOVA with Newman Keuls correction was used for comparison between multiple groups. A value of P < 0.05 was used to establish statistical significance.
RESULTS
Acetylcholine relaxation and NONOate relaxation are not impaired in PKG-KI mice.
Isolated pulmonary arterial rings from each group were contracted with 100 nM phenylephrine and then relaxed with increasing cumulative concentrations of acetylcholine (10−8 to 10−5 M) or NONOate (10−9 M to 10−5 M) (Fig. 1, A and B). We found that there is no difference in relaxation to acetylcholine or a nitric oxide (NO) donor, suggesting that the NO-associated endothelium-mediated relaxation was not affected in PKG-KI mouse pulmonary arteries. Similarly, isolated aorta rings from each group were contracted with 100 nM phenylephrine and then relaxed with various concentrations of acetylcholine (10−8 to 10−5 M) or NONOate (10−9 M to 10−5 M) (Fig. 1, C and D). Again, in aorta also, there is no difference in relaxation to acetylcholine or an NO donor, suggesting that the NO-associated endothelium-mediated relaxation was not affected in PKG-KI mice aorta.
Fig. 1.

Effects of various doses of acetylcholine (A) and NONOate (B) in “redox dead” protein kinase G knockin (PKG-KI, 28) and genetic background-matched wild-type (WT) mouse pulmonary arteries as well as effects of various doses of acetylcholine (C) and NONOate (D) in WT and PKG-KI mouse aorta. Pulmonary arterial and aortic rings from each group were precontracted with 100 nM phenylephrine (n = 4).
DHEA relaxation is impaired in PKG-KI mice.
Mouse pulmonary arteries and aorta from each group were contracted with 100 nM phenylephrine and then relaxed with increasing cumulative doses of DHEA (1 μM, 10 μM, and 100 μM). The 10 μM and 100 μM concentrations of DHEA caused significant relaxation in wild-type (WT) mouse pulmonary arteries and aorta, but this relaxation by DHEA was markedly attenuated in PKG-KI mouse pulmonary arteries (Fig. 2A) and aorta (Fig. 2B). This suggests that DHEA may function by dimerization of PKG-1α because it was inhibited in PKG-KI mouse pulmonary arteries and aorta.
Fig. 2.

Effects of various doses of dehydroepiandrosterone (DHEA) in WT and PKG-KI mouse pulmonary arteries (A) and aorta (B). Pulmonary arterial and aortic rings from each group were precontracted with 100 nM phenylephrine. *P < 0.05 vs. WT 10 μM; #P < 0.05 vs. WT 100 μM doses of DHEA (n = 4).
Peroxide relaxation is impaired in PKG-KI mice.
Mouse pulmonary arteries and aorta from each group were contracted with 100 nM phenylephrine and then relaxed with increasing cumulative doses of H2O2 (1 μM, 10 μM, and 100 μM). Relaxation to 100 μM concentration of H2O2 was significantly inhibited in PKG-KI mouse pulmonary arteries (Fig. 3A). Relaxation to 10 μM and 100 μM concentrations of H2O2 was significantly inhibited in PKG-KI mouse aorta (Fig. 3B). The contractile responses of pulmonary arteries (Fig. 3C) and aorta (Fig. 3D) to 100 nM phenylephrine also did not appear to differ in these vascular segments that were obtained from the PKG-KI mice.
Fig. 3.
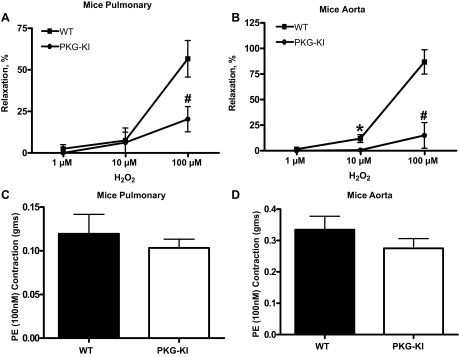
Effects of various doses of H2O2 in WT and PKG-KI mouse pulmonary arteries (A) and aorta (B). Pulmonary arterial and aortic rings from each group were precontracted with 100 nM phenylephrine. *P < 0.05 vs. WT 10 μM; #P < 0.05 vs. WT 100 μM doses of H2O2. Contraction of pulmonary arterial (C) and aortic rings (D) to 100 nM phenylephrine were not altered in vascular tissue from PKG-KI mice (n = 4). PE, phenylephrine.
PKG dimer expression was not detected in lung tissues of PKG-KI mice.
Lung tissues from both WT and PKG-KI mouse groups treated with 100 μM DHEA and 100 μM H2O2 were used for determining PKG dimer expression. Exposure to 100 μM H2O2 causes a significant increase in PKG dimerization in WT mice but not in PKG-KI mice, suggesting the role of peroxide in activation of PKG (Fig. 4, A and B). In addition to this, 100 μM DHEA also increased PKG dimerization in WT mice tissue but not in the PKG-KI mice lung tissue. Dimerization of PKG was essentially not detected in lung tissue from PKG-KI animals under any of the conditions examined.
Fig. 4.

Effects of 100 μM DHEA and 100 μM H2O2 on PKG (A and B) dimerization in WT and PKG-KI mouse lung tissue. Total PKG was normalized relative to actin, and then PKG-1α dimer was normalized to total PKG. Here WT control dimer is set to 100% to indicate percentage changes in remaining treatment in WT and PKG-KI mice tissue. *P < 0.05 vs. WT control dimer (n = 4).
PKG dimer expression and VASP phosphorylation were increased in BPA by DHEA under acute hypoxia.
BPAs were treated with 100 μM DHEA under aerobic and hypoxic conditions for determining whether it altered PKG dimer expression (Fig. 5A) and VASP phosphorylation (Fig. 5B). Although the VASP phosphorylation studied is thought to be a reliable indicator of PKG activation, the actions of this phosphorylated form of VASP are currently not known to be directly associated with major mechanisms directly controlling acute changes in contractile force (19). DHEA under aerobic conditions showed increases in PKG dimer (Fig. 5A) expression and VASP phosphorylation (Fig. 5B). Acute hypoxia caused significant decreases in the detection of PKG dimer (Fig. 5A) and VASP phosphorylation either in the absence or presence of DHEA (Fig. 5B). DHEA under acute hypoxic conditions also caused a significant increases in the detection of PKG dimer (Fig. 5A) and VASP phosphorylation (Fig. 5B).
Fig. 5.

Effects of 100 μM DHEA on expression of PKG dimer (A) and vasodilator-stimulated phosphoprotein (VASP)-phosphorylation (B) in bovine pulmonary arteries (BPAs). *P < 0.05 vs. control; #P < 0.05 vs. hypoxia; @P < 0.05 vs. DHEA (n = 6).
Effect of various doses of DHEA under aerobic and hypoxic conditions in BPAs.
BPAs were studied under aerobic and hypoxic conditions. Precontracted BPAs were relaxed with increasing cumulative doses of DHEA (1 μM, 10 μM, and 100 μM). The 100 μM concentration of DHEA caused significant relaxation in BPAs under aerobic condition, but this relaxation by DHEA was inhibited in BPAs under hypoxic condition (Fig. 6A). As shown in Fig. 6B, the contractile force to 30 mM KCl is similar under the aerobic and hypoxic conditions that were used. Thus the impaired relaxation to DHEA under hypoxia is not due to differences in force between aerobic and hypoxic conditions, and it was hypothesized that this suppression of relaxation might originate from the elevation (12, 25) of NADPH under hypoxia.
Fig. 6.
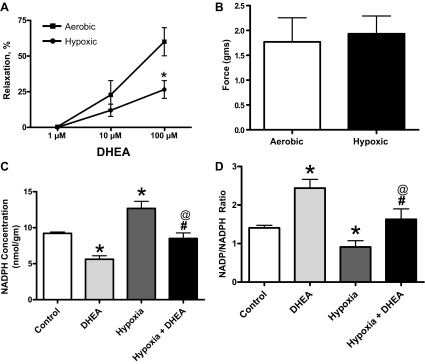
A: effect of various doses of DHEA in BPAs under aerobic and hypoxic conditions. *P < 0.05 vs. aerobic 100 μM; n = 6. B: force generation is similar in BPAs under aerobic and hypoxic conditions. Effects of hypoxia and 100 μM DHEA under hypoxia on NADPH levels (C), and NADP/NADPH ratios (D) in BPA. *P < 0.05 vs. control; #P < 0.05 vs. hypoxia; @P < 0.05 vs. DHEA (n = 6).
Effects of DHEA on NADPH levels and NADP/NADPH ratios in BPA under hypoxia.
NADPH levels and NADP/NADPH ratios were measured to document how they are altered by inhibition of G6PD with 100 μM DHEA under aerobic and hypoxic conditions. As shown in Fig. 6C, hypoxia increases NADPH and DHEA decreases NADPH under aerobic conditions compared with the aerobic control. DHEA also decreases NADPH under the hypoxic conditions compared with the hypoxic control. The data in Fig. 6D show that hypoxia caused a decrease in the NADP/NADPH ratio. Under these hypoxic conditions, DHEA caused an increased NADP/NADPH ratio or an oxidation of this pyridine nucleotide. The levels of NADPH in the presence of DHEA remained higher under hypoxia compared with aerobic conditions. In addition, the NADP/NADPH ratio in the presence of DHEA remained lower under hypoxia compared with aerobic conditions. Thus changes in NADPH related to its elevation under hypoxia were associated with impairment of its vasodilator actions.
Relaxation to DHEA is not altered by an inhibitor of Nox oxidase activation.
Because the control of NADPH redox by G6PD influences basal Nox oxidase activity in BPA (15), the effects of an inhibitor of Nox oxidase activation, gp91ds-tat, on relaxation to DHEA was examined under conditions previously demonstrated to lower superoxide in BPA (1). The data in Fig. 7A show that 50 μM gp91ds-tat did not alter relaxation of BPA to DHEA. Data in this figure also indicate that this inhibitor of Nox oxidase activation did not alter relaxation of BPA to H2O2 (Fig. 7B) or the force generated by 30 mM KCl (Fig. 7C) used to contract BPA.
Fig. 7.
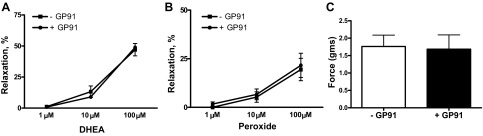
Absence of an effect of the inhibitor of Nox oxidase activation 50 μM gp91ds-tat (GP91) on relaxation of BPA to DHEA (A), H2O2 (B), or force generation (C) to the 30 mM dose of KCl used to contract the arteries under aerobic conditions (n = 6).
DISCUSSION
Data reported in the present study provide evidence that activation of PKG-1α by dimerization, potentially through processes shown in Fig. 8, has a major role in the vasodilator actions of DHEA based on a marked attenuation of relaxation responses in pulmonary arteries and aorta from PKG-KI mice (Fig. 2), which lack the ability to undergo dimerization (28). Studies in BPA (Fig. 5) support this mechanism by demonstrating relaxation to DHEA is associated with increased PKG dimerization and PKG-associated VASP phosphorylation. On the basis of 1) the detection DHEA increasing the NADP/NADPH ratio in BPA (Fig. 6), 2) evidence for inhibitors of G6PD promoting PKG dimerization by increasing the ratio of cytosolic NADP/NADPH in BPA (25), and 3) previous reports (16, 31) that DHEA is an inhibitor of G6PD, cytosolic NADPH oxidation resulting from an inhibition of G6PD by DHEA is likely to be a primary factor in promoting relaxation via increased dimerization activation of PKG.
Fig. 8.

Model showing how DHEA is hypothesized to play a role in thiol redox-mediated regulation of PKG dimerization in PKG-KI mice and in BPAs as a result of promoting cytosolic NADPH oxidation by inhibiting glucose-6-phosphate dehydrogenase (G6PD). The potential roles of NADPH redox regulation of PKG dimerization via thioredoxin reductase-1 (TrxR-1) and thioredoxin-1 (Trx-1) and sites where peroxide (H2O2) potentially regulates PKG dimerization are also shown (25).
In this study, we used a mouse model constitutively expressing the PKG-1α Cys42Ser mutation in place of PKG-1α to define the role of PKG dimerization in vasodilator responses because these redox-dead PKG-KI mice do not form the disulfide bond that normally activates PKG (28). Rings from pulmonary arteries and aorta from PKG-KI mice showed decreases in relaxation to peroxide compared with wild-type mice (Fig. 2). In contrast, relaxation to acetylcholine and a nitric oxide donor (Fig. 1) and contraction to phenylephrine (Fig. 3) were not detectibly altered in aorta and pulmonary arterial rings from PKG-KI mice under the conditions studied. Thus these studies in vascular tissue from PKG-KI mice confirm previously reported observations in aorta (28) and extend evidence for activation of PKG-1α by dimerization being a prominent mechanism in the relaxation of pulmonary arteries to peroxide. Although there is evidence for PKG dimerization being increased in BPAs under aerobic conditions (24) and for inhibitory effects of cGMP stimulation vs. dimerization stimulation of PKG with each other (6), the data in this study did not detect evidence for the functional effects of these interactions under the aerobic conditions used to study vascular tissue from PKG-KI mice. The absence of altered vasodilator responses to acetylcholine and a nitric oxide donor also supports previous evidence for nitric oxide/cGMP-associated vasodilator responses not being altered in systemic vascular tissue from PKG-KI mice (28). Relaxation to DHEA was observed to be markedly attenuated in both aortic and pulmonary arterial rings from PKG-KI mice, suggesting that PKG-1α dimerization was a prominent mechanism in these responses. As expected, we found that expression of PKG dimer was not detected in PKG-KI mouse lung tissue compared with wild-type. DHEA and peroxide both increased PKG dimerization in lung tissue from wild-type but not PKG-KI mice. These data suggest that PKG dimerization has a prominent role in the vasodilator response to DHEA in both mouse aorta and pulmonary arteries.
The actions of DHEA on isolated BPAs were studied further to define processes potentially contributing to its mechanism of vasodilation. DHEA was observed to decrease the detection of NADPH (Fig. 6C) associated with increased NADP/NADPH ratios (Fig. 6D), presumably as a result of its ability to inhibit G6PD. However, hypoxia increases NADPH levels, and this may have contributed to a suppression of relaxation to DHEA under hypoxic conditions because NADPH levels in the presence of hypoxia remained elevated compared with the levels observed with DHEA under aerobic conditions. Because the 30 mM KCl contractile conditions used for studying responses to DHEA showed only minimal levels of increased force under hypoxia, increased force under hypoxia does not explain the decreased relaxation that is observed. The levels of PKG dimerization and PKG activation demonstrated by VASP phosphorylation in BPAs were observed to be increased by DHEA under both aerobic and hypoxic conditions (Fig. 5), further supporting this mechanism of PKG activation in promoting vasodilation. Although DHEA increases PKG dimerization as well as PKG activity under hypoxia, the levels of dimerization and PKG activity do not appear to reach those seen under aerobic conditions. Thus there appears to be less inhibition of PKG dimerization by DHEA under hypoxia. These observations may help explain why there seems to be a decreased vasodilator response to DHEA under hypoxia compared with aerobic conditions (Fig. 6A).
It is likely that mechanisms in addition to PKG activation by dimerization contribute to the vasodilator actions of DHEA. Although the vasodilator actions of DHEA are markedly attenuated in pulmonary arteries and aorta from PKG-KI mice, a distinct vasodilator response to these agents remains detectible (Fig. 2), suggesting that additional processes potentially contribute to the actions of DHEA. Redox changes caused by NADPH oxidation can readily be linked to actions on other signaling systems because the availability of NADPH controls the generation of superoxide and additional ROS such as peroxide as a result of its use by Nox oxidases for the generation of these species (15, 29). In addition, the NADPH dependence of enzymes such as glutathione reductase and thioredoxin reductases are likely to have major effects on controlling the redox status of glutathione and thioredoxin, which are key regulators of the redox status of protein thiols involved in multiple cellular regulatory systems beyond those associated with PKG dimerization (11, 13). The data in Fig. 7 indicate that inhibition of Nox oxidase activation did not alter force generation or relaxation to DHEA and H2O2, suggesting that modulation of endogenous ROS generation or action by an oxidase such as Nox 2 (1) did not have detectible interactions with the vasoactive actions of these agents under the conditions examined. Mechanisms previously suggested to participate in the actions of DHEA such as the opening voltage-regulated potassium and/or calcium channels (10, 16), upregulating sGC (26), and decreasing Rho kinase activity (20) could be regulated by PKG and/or redox processes influenced by NADPH oxidation not involving PKG. In addition, DHEA could have actions independent of the consequences of its inhibitory effects on G6PD. Further studies are needed to define the importance of these alternative mechanisms.
Activation of PKG signaling by DHEA demonstrated in the present study has properties consistent with its vasodilator action and known beneficial effects in the treatment of pulmonary hypertension in animals and humans (2, 4, 9, 18, 20, 26). It is well established that PKG is a coordinator of multiple processes contributing to vasodilation (19, 22). Some of the systems regulated by PKG that contribute to its vasodilator action include multiple potassium channels, processes regulating intracellular levels, and actions of calcium, including Rho kinase (19, 22). The therapeutic effects of agents promoting PKG signaling, including inhibitors of the type 5 phosphodiesterase such as sildenafil (21) and direct activators of sGC (30), indicate that PKG signaling has beneficial effects in the treatment of pulmonary hypertension, which appear to be associated with PKG regulation of both vascular tone and remodeling. Although the effects of DHEA were observed to be decreased under hypoxic conditions, it is important to note that the data in Figs. 5 and 6 indicate that DHEA remains an effective vasodilator and activator of PKG by dimerization under hypoxic conditions. There is also preliminary evidence that pulmonary G6PD and NADPH levels appear to be elevated in an animal model of hypoxia-induced pulmonary hypertension (8). Although PKG-KI mice have been reported to show evidence of systemic hypertension (28), little appears to be known about the effects of a loss of PKG activation by dimerization on pulmonary circulatory function. Thus the G6PD inhibitory action of DHEA and its associated cGMP-independent activation of PKG by subunit dimerization are likely to be important factors in the vasodilator actions of DHEA and its beneficial effects in treating pulmonary hypertension.
GRANTS
This study was supported by NIH grants R01HL031069, P01HL043023, R01HL066331, and R01HL115124.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.P. and M.S.W. conception and design of research; D.P., S.K., M.K., and B.H.N. performed experiments; D.P., S.K., M.K., B.H.N., and M.S.W. analyzed data; D.P., S.K., B.H.N., and M.S.W. interpreted results of experiments; D.P. and S.K. prepared figures; D.P. and M.S.W. drafted manuscript; D.P., S.K., and M.S.W. edited and revised manuscript; D.P., S.K., M.K., B.H.N., and M.S.W. approved final version of manuscript.
REFERENCES
- 1.Ahmad M, Kelly MR, Zhao X, Kandhi S, Wolin MS. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol 298: H1879–H1888, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 304: H1708–H1713, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Archer SL, Will JA, Weir EK. Redox status in the control of pulmonary vascular tone. Herz 11: 127–141, 1986 [PubMed] [Google Scholar]
- 4.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, Santos PD, Savineau J-P, Baulieu E-E. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA 100: 9488–9493, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science 317: 1393–1397, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne JR, Prysyazhna O, Rudyk O, Eaton P. cGMP-dependent activation of protein kinase G precludes disulfide activation: Implications for blood pressure control. Hypertension 60: 1301–1308, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Burke TM, Wolin MS. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am J Physiol Heart Circ Physiol 252: H721–H732, 1987 [DOI] [PubMed] [Google Scholar]
- 8.Chettimada S, Oka M, McMurtry IF, Gupte SA. Role of glucose-6-phosphate dehydrogenase (G6PD) in chronic hypoxia-induced pulmonary hypertension (Abstract). FASEB J 24: 1023.–2., 2010. 19940258 [Google Scholar]
- 9.Dumas-de-La-Roque E, Savineau JP, Metivier AC, Billes MA, Kraemer JP, Doutreleau S, Jougon J, Marthan R, Moore N, Fayon M, Baulieu EE, Dromer C. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol (Paris) 73: 20–25, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Farrukh IS, Peng W, Orlinska U, Hoidal JR. Effect of dehydroepiandrosterone on hypoxic pulmonary vasoconstriction: a Ca2+-activated K+-channel opener. Am J Physiol Lung Cell Mol Physiol 274: L186–L195, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 287: C246–C256, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, Koller A, Wolin MS. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol 285: H2316–H2326, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 14: 543–558, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 301: 299–305, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Gupte SA, Wolin MS. Hypoxia promotes relaxation of bovine coronary arteries through lowering cytosolic NADPH. Am J Physiol Heart Circ Physiol 290: H2228–H2238, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Hampl V, Bíbová J, Povysilová V, Herget J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur Respir J 21: 862–865, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 86: 1–23, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Homma N, Nagaoka T, Karoor V, Imamura M, Taraseviciene-Stewart L, Walker LA, Fagan KA, McMurtry IF, Oka M. Involvement of RhoA/Rho kinase signaling in protection against monocrotaline induced pulmonary hypertension in pneumonectomized rats by dehydroepiandrosterone. Am J Physiol Lung Cell Mol Physiol 295: L71–L78, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5: expanding roles in cardiovascular regulation. Circ Res 101: 1084–1095, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91: 1421–1430, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Neo BH, Kandhi S, Wolin MS. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol 299: H1235–H1241, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neo BH, Kandhi S, Wolin MS. Roles for redox mechanisms controlling protein kinase G in pulmonary and coronary artery responses to hypoxia. Am J Physiol Heart Circ Physiol 301: H2295–H2304, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neo BH, Patel D, Kandhi S, Wolin MS. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of Protein Kinase G 1α. Am J Physiol Heart Circ Physiol 305: H330–H343, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, Limbird J, Imamura M, Gebb SA, Fagan KA, McMurtry IF. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res 74: 377–387, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olschewski A, Hong Z, Peterson DA, Nelson DP, Porter VA, Weir EK. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol 286: L15–L22, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18: 286–290, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorescu D, Somers MJ, Lassegue B, Grant S, Harrison DG, Griendling KK. Electron spin resonance characterization of the NAD(P)H oxidase in vascular smooth muscle cells. Free Radic Biol Med 30: 603–612, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123: 2263–2273, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsutsui EA, Marks PA, Reich P. Effect of dehydroepiandrosterone on glucose 6-phosphate dehydrogenase activity and reduced triphosphopyridine nucleotide formation in adrenal tissue. J Biol Chem 237: 3009–3013, 1962 [PubMed] [Google Scholar]