

Abstract
Inward remodeling is the most prevalent structural change found in the resistance arteries and arterioles of hypertensive individuals. Separate studies have shown that the inward remodeling process requires transglutaminase activation and the polymerization of actin. Therefore, we hypothesize that inward remodeling induced via endogenous transglutaminase activation requires and depends on actin cytoskeletal structures. To test this hypothesis, isolated and cannulated rat cremaster arterioles were exposed to dithiothreitol (DTT) to activate endogenous transglutaminases. DTT induced concentration-dependent vasoconstriction that was suppressed by coincubation with cystamine or cytochalasin-D to inhibit tranglutaminase activity or actin polymerization, respectively. Prolonged (4 h) exposure to DTT caused arteriolar inward remodeling that was also blocked by the presence of cystamine or cytochalasin-D. DTT inwardly remodeled arterioles had reduced passive diameters, augmented wall thickness-to-lumen ratios and altered elastic characteristics that were reverted upon disruption of the actin cytoskeleton with mycalolide-B. In freshly isolated arterioles, exposure to mycalolide-B caused no changes in their passive diameters or their elastic characteristics. These results suggest that, in arterioles, the early stages of the inward remodeling process induced by prolonged endogenous transglutaminase activation require actin dynamics and depend on changes in actin cytoskeletal structures.
Keywords: hypertension, vasoconstriction, stiffness, elasticity, passive diameter
inward remodeling of arterioles is the most prevalent structural change of the resistance vasculature observed in patients with hypertension and diabetes (14, 28). Its presence is associated with an increased risk for life-threatening cardiovascular events including stroke and myocardial infarction (22, 27). However, despite its clinical importance, the mechanism(s) responsible for its development have not been completely elucidated.
Cumulative evidence indicates that prolonged exposure to vasoconstrictor agonists causes inward remodeling of arterioles. Ex vivo, prolonged vasoconstriction induced by exposure of isolated arterioles to endothelin-1, angiotensin II, norepinephrine, or serum causes inward remodeling (4, 5, 19). In vivo, vasoconstriction also appears to be the primary stimulus causing inward remodeling in hypertension, as vasodilation and not a mere reduction in blood pressure is needed to prevent or revert inward remodeling in hypertensive individuals (8, 21). The mechanism(s) responsible for inducing remodeling during prolonged vasoconstriction, however, remain elusive.
A series of recent studies indicates that inward remodeling of arterioles requires transglutaminase activity. In 2005 Bakker et al. (1) showed that the inward remodeling induced by prolonged exposure of isolated arterioles to endothelin-1 was prevented by incubation with the inhibitors of transglutaminase, cystamine, or 5-(biotinamido) pentylamide. In vivo, it has also been shown that inhibition of transglutaminase activity with cystamine prevents the inward remodeling observed in mesenteric arterioles of rats subjected to prolonged infusion with phenylephrine or a local reduction in blood flow (1, 10). In conduit arteries, Santhanam et al. (31) showed that the reduced distensibility observed in carotid arteries of mice treated with the nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester was greater in wild-type animals than in those not expressing tissue-type transglutaminase. They also observed that the increased pulse wave velocity observed in old rats was diminished after treatment with cystamine, suggesting that inhibition of transglutaminase activity reduces aortic stiffness (31). As transglutaminase activity is known to participate in the cross-linking of extracellular matrix proteins, it has been proposed that inward remodeling and vascular stiffness occur as transglutaminase cross-links extracellular matrix proteins of vessels with reduced diameters and thus prevents subsequent vessel diameter expansion (1, 31).
Evidence is also accumulating that indicates the process of vasoconstriction involves the activation of small GTP binding proteins and the polymerization of actin within vascular smooth muscle cells (18). Recently, we demonstrated that inhibition of actin polymerization or the signaling pathways associated with the activity of the small GTP binding proteins Rho and Rac prevents prolonged vasoconstriction from inducing inward remodeling in isolated arterioles (34). Moreover, we showed that nearly 75% of the structural reduction in passive diameter observed in the early stages of vasoconstriction-induced inward remodeling is reversible upon the enzymatic depolymerization of F-actin (34). Other studies have shown that dithiothreitol (DTT) induces inward remodeling in isolated arterioles via the activation of endogenous transglutaminases (37) and that DTT induces vasoconstriction in isolated arteries (11, 12). Moreover, transglutaminase activity is known to activate RhoA (15). Therefore, we designed the present study to test the hypothesis that prolonged activation of endogenous transglutaminase with DTT reduces the passive diameter of arterioles through processes that depend on actin cytoskeletal structures and require actin polymerization and vasoconstriction.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (∼200 g) were used in all experiments. All animal protocols and procedures implemented during these studies were approved by the Animal Care Quality Assurance Office and the Animal Care and Use Committee at the University of Missouri-Columbia. Before experimentation rats were housed in pairs under a 12 h per day illumination regimen and provided with ad libitum access to standard rat chow and water.
Vessel Isolation
Rats were anesthetized by means of an intraperitoneal injection of pentobarbital sodium at a dose of 100 mg/kg. After confirmation that spinal reflexes were lost, both cremaster muscles were excised and placed in a cold (∼4°C) physiological saline solution (PSS) containing the following: 145.0 NaCl, 4.7 KCl, 2.0 CaCl2, 1.0 MgSO4, 1.2 NaH2PO4, 0.02 EDTA, 2.0 pyruvic acid, 5.0 glucose, and 3.0 MOPS (all concentrations are given in mM) with a final pH of 7.4. First order (1A) feed arterioles from each cremaster were isolated, cannulated, and pressurized for experimentation as previously described (19). Briefly, arteriolar segments of ∼1 mm in length were cannulated onto glass micropipettes within an observation chamber (Living Systems Instrumentation, Burlington, VT) filled with PSS. The arterioles were pressurized without flow to 60 mmHg using a Pressure Servo System (Living Systems Instrumentation) and PSS containing 0.15 mM BSA. The observation chamber with the cannulated vessel was transferred to an inverted microscope equipped with a video display and video caliper system (Living Systems Instrumentation) to record measurements of wall thickness and luminal diameter. All experiments were performed at 34.5°C.
Experimental Protocols
Assessment of endogenous transglutaminase activation by DTT in isolated arterioles.
To determine relative transglutaminase activity, isolated arteriolar segments were incubated with the transglutaminase substrate Alexa Fluor 488 cadaverine. Rat cremaster 1A arterioles were isolated and then transferred to 1.5-ml tubes containing 10 μM cadaverine in PSS and incubated over night at 4°C. The next day, vessels were warmed to 37°C and incubated in one of the following treatments: PSS + 10 μM cadaverine (control), PSS + 200 μM DTT + 10 μM cadaverine (DTT), and PSS + 200 μM DTT + 1.0 mM cystamine + 10 μM cadaverine (DTT + cystamine) for 4 h at 37°C. All subsequent steps were performed at 4°C. Vessels were washed twice in PBS and then fixed for 1 h in 4% paraformaldehyde. Vessels were washed twice in PBS and then incubated for 1 h in 2.0 μM 4′,6-diamidino-2-phenylindole (DAPI) in PBS + 10% BSA to visualize nuclei. Vessels were washed three times in wash buffer followed by two-time wash in PBS. Vessels were then imaged using a Leica SP5 confocal microscope with a ×63/1.2 numerical aperture water objective. Cadaverine was excited with an Argon laser at 488 nm. DAPI was excited with a multiphoton laser at 720 nm. Images were processed and quantified using Imaris software. For quantification, a region of interest (ROI) was selected containing predominantly vascular smooth muscle cells: ROI ≅ 40 × 40 × 15 μm. The mean fluorescent intensity per micrometers cubed was determined for the ROI; n ≥ 5 for all treatments.
Effects of acute activation of endogenous transglutaminase on arteriolar tone.
To determine the acute effect of endogenous transglutaminase activation on arteriolar function, isolated arterioles with spontaneous myogenic tone were exposed abluminally to increasing concentrations of DTT (10−6.5 to 10−3 M) in the absence or presence of the transglutaminase inhibitor cystamine (1 mM). Cystamine has been previously used at concentrations of 0.1 and 1 mM to inhibit the activity of transglutaminase in vascular tissues (1, 16). In our experiments designed to inhibit transglutaminase activity, cystamine was added to the superfusate 20 min before and at all times during exposure to DTT. In an additional series of experiments, arterioles were incubated with 500 nM cytochalasin-D 20 min before and at all times during exposure to increasing concentrations of DTT to block actin polymerization (34). To construct vessel diameter response curves, each concentration of DTT was maintained for 5 min in the absence or presence of cystamine or cytochalasin-D.
Effects of prolonged activation of endogenous transglutaminase on arteriolar remodeling.
To confirm previous observations that prolonged activation of endogenous transglutaminase induces arteriolar inward remodeling (1, 37), isolated arterioles pressurized to 60 mmHg were exposed to 200 μM DTT for 4 h in the absence or presence of 500 nM cytochalasin-D. We chose a concentration of 200 μM DTT because it induced a submaximal constriction of arterioles that is completely blocked by 1 mM cystamine (Fig. 1A). Before and after the prolonged exposure to DTT, arterioles were exposed to adenosine (10−4 M) and then to Ca2+-free PSS containing adenosine (10−4 M) and EGTA (2 mM) to obtain maximum passive diameter. A reduction in the maximal luminal diameter obtained after the prolonged exposure to DTT was considered evidence that inward remodeling had occurred. The presence of cytochalasin-D caused the prolonged vasoconstriction induced by DTT to wane over time. Therefore, as both cytochalasin-D and cystamine prevented DTT from maintaining vasoconstriction, additional experiments were conducted in vessels pressurized to 5 mmHg to maintain a reduced diameter during the prolonged exposure to DTT in combination with cytochalasin-D or cystamine. In all experiments where cytochalasin-D or cystamine was used, arterioles were preincubated for 20 min with the inhibitor and at all times during exposure to DTT.
Fig. 1.
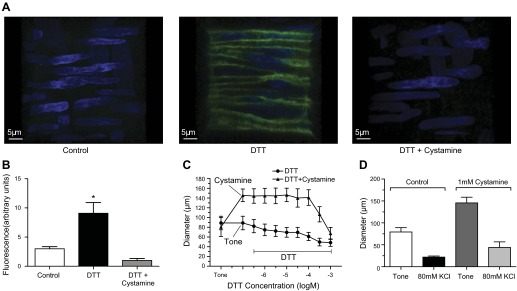
Cystamine blocks dithiothreitol (DTT)-dependent activation of endogenous transglutaminases and DTT-induced vasoconstriction but not depolarization-induced vasoconstriction. A: confocal images of rat cremaster arteriolar walls incubated with Alexa Fluor 488-cadaverine (green) and exposed for 4 h to vehicle control (left), 200 μM DTT (middle), or DTT in the presence of 1mM cystamine (right). Blue shows smooth muscle nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI). B: mean fluorescence intensity of Alexa Fluor 488 cadaverine in control (n = 5), DTT (n = 7)-, and DTT + cystamine (n = 5)-treated arterioles. *P ≤ 0.05 vs. control or DTT + cystamine. C: percent control diameter of isolated arterioles exposed to incremental concentrations of DTT in the absence (n = 5) or presence (n = 5) of 1 mM cystamine. Diameters in the presence of cystamine at DTT 10−6.5 to 10−3.5 M were significantly greater than those with DTT alone (*P ≤ 0.05). D: internal diameter (μm) of isolated and pressurized (60 mmHg) arterioles before (tone) and after exposure to 80 mM KCl in the absence (n = 5) or presence (n = 5) of 1 mM cystamine. Data are means ± SE.
Effect of prolonged activation of endogenous transglutaminase on actin polymerization.
To determine the effect of prolonged activation of endogenous transglutaminase on actin polymerization, we measured the amount of F- and G-actin present in isolated arterioles exposed for 4 h to DTT or vehicle control as previously described (34). Freshly isolated rat cremasteric 1A arterioles were incubated in 1.5-ml vials at 34°C for 4 h in either PSS (control) or PSS + 200 μM DTT. Immediately after treatment, arterioles were homogenized in an F-actin stabilization buffer (no. LAS01; Cytoskeleton) with ATP, halt protease and phosphatase inhibitor cocktail, and EDTA (no. 78440; Thermo Scientific). Subsequently, the homogenized samples were centrifuged for 1 h at 100,000 g. After centrifugation, the supernatants were transferred into new labeled 1.5-ml vials, while the pellets were incubated and resuspended for 1 h in an F-actin depolymerization buffer (no. FAD02; Cytoskeleton) at 4°C. The supernatant and pellet portions of the samples were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membranes (no. 162–0147; Bio-Rad). The blots were probed with rabbit polyclonal anti-actin antibody (no. AAN01 at a 1:1,000 dilution; Cytoskeleton) and protein bands visualized with a ChemiDOC XRS+ (Bio-Rad). Images were analyzed, and the signals for supernatants (G-actin) and pellets (F-actin) for each treatment were quantified, and F-actin/total-actin ratios were computed and compared (34).
Effect of F-actin cytoskeleton disruption on control and remodeled arterioles.
To test the effect of F-actin cytoskeleton disruption on the vasculature, and to determine the role of F-actin cytoskeletal structures in vascular remodeling, freshly isolated arterioles and DTT-remodeled arterioles were subjected to a 1-h incubation with the actin-depolymerizing agent mycalolide-B (2 μM) in Ca2+-free PSS (29, 34). Subsequently, passive pressure-diameter relationships were determined for each arteriole.
Determination of arteriolar elastic characteristics.
To study the elastic characteristics of the arteriolar wall, and to determine the effect of actin cytoskeleton disruption on remodeled arterioles, pressure-diameter curves were obtained under passive conditions (Ca2+-free PSS) before and after every main treatment in the experimental protocols. Pressurizations were performed in steps covering a range between 5 and 120 mmHg. Maximum internal diameter and wall (left and right) thicknesses were recorded at each pressure. This information was later used to determine the circumferential stress, strain, and moduli of elasticity curves for each group of vessels.
Chemicals.
All chemicals and drugs used in this study were purchased from Sigma (St. Louis, MO), except for mycalolide-B, which was acquired from Wako (Wako Chemicals, Richmond, VA); Alexa Fluor 488 cadaverine was from Invitrogen; and the chemicals and anti-bodies used for F- and G-actin separation were purchased from Bio-Rad, KPL, Cytoskeleton, Chemicon, and Thermo Scientific. In preparation for the experiments, stock solutions of adenosine, cystamine, and DTT were prepared in PSS (without glucose and pyruvate) at a concentration of 10−2 M, 10−1 M and 10−1 M, respectively. Stock solutions of cytochalasin-D and mycalolide-B were made in DMSO at a concentration of 1 and 2 mM, respectively. Stock solutions were diluted in the buffer solution used as superfusate (i.e., PSS or Ca2+-free PSS). The final concentrations reported refer to concentrations in the superfusate.
Data Analyses
Data are presented as mean values of multiple experiments (the number of experiments is reported for each experimental series in results and figures) ± SE. Diameter (μm), wall thickness (μm), pressure (mmHg), circumferential stress (dyne/cm2), strain, and modulus of elasticity (dyne/cm2) were calculated as previously described (25, 33), and are expressed in absolute values. The circumferential stress was calculated at every pressure as σi = PiDi/(τL,i + τR,i), where Pi is the intraluminal pressure, Di is the internal diameter, and, τL,i and τR,i are the left and right wall thicknesses obtained from the two-dimensional projection of the video caliper. The strain was calculated as εi =(Di − D0)/D0, where D0 represents the internal diameter measured at the lowest intraluminal pressure (5 mmHg) and Di represents the internal diameter measured at greater intraluminal pressures. We calculated the modulus of elasticity as the ratio between stress and strain (Ei = σi/εi) to obtain a measure of the wall stiffness. Repeated-measures ANOVA and paired or unpaired t-tests were used to make statistical comparisons between means. Differences were considered significant at values of P ≤ 0.05.
RESULTS
DTT-Induced Activation of Transglutaminases Is Blocked by Cystamine
To assess the activation of endogenous transglutaminases by DTT and to determine the effectiveness of cystamine to block this activation, we utilized a previously described (37) fluorescent assay that quantifies transglutaminase activity by the incorporation of transglutaminase substrate Alexa Fluor 488 cadaverine in isolated arterioles. The vessels not exposed to DTT had a modest amount of fluorescence indicative of minimal incorporation of cadaverine via transglutaminase cross-linking (Fig. 1, A and B). In contrast, DTT-treated vessels displayed a roughly threefold increase in fluorescent intensity indicative of cadaverine incorporation into arteriolar structures. The addition of the transglutaminase inhibitor cystamine (1 mM) completely abolished the marked increase in cadaverine incorporation induced by DTT.
Transglutaminase Inhibition with Cystamine Blocks DTT-Induced Arteriolar Constriction
Exposure of isolated and pressurized (60 mmHg) arterioles to DTT induced concentration-dependent vasoconstriction with a maximal constriction of 43.8 ± 5.8% from original spontaneous tone at 10−3 M DTT (n = 5). Inhibition of transglutaminase activity with 1 mM cystamine caused vessels to lose 85.7 ± 11.5% of spontaneous myogenic tone and prevented DTT at all concentrations up to 200 μM from inducing vasoconstriction (n = 5; Fig. 1C). Exposure to cystamine did not prevent a solution containing 80 mM KCl from inducing depolarization-dependent vasoconstriction (Fig. 1D).
Prolonged Exposure to DTT Causes Inward Eutrophic Remodeling in Isolated Arterioles
Exposure of isolated and pressurized (60 mmHg) arterioles to DTT (200 μM) for 4 h caused a strong (39.5 ± 5.1% from spontaneous myogenic tone) and continuous (entire exposure time) vasoconstriction (Fig. 2A). Arterioles remained constricted by 31.8 ± 6.3% of spontaneous myogenic tone after DTT was removed from the perfusate for 10 min and by 19.7 ± 8.9% after exposure to the vasodilator adenosine (10−4 M). Once arterioles were exposed to Ca2+-free solution, their maximal passive diameter attained was significantly smaller and their media/lumen ratio was significantly larger than those observed before exposure to DTT (Fig. 2, A and B). Passive pressure-diameter profiles obtained before and after exposure to DTT showed that the reduction in inner passive diameter was evident at all pressures (Fig. 2C). Strain-stress profiles showed that the remodeled arterioles had decreased circumferential stress but no major changes in distensibility (Fig. 2D). Remodeled arterioles had a tendency towards a reduced modulus of elasticity at high pressures that was not significantly different (P > 0.05) from controls (Fig. 2E).
Fig. 2.
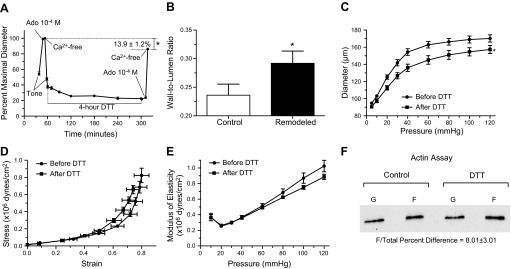
Prolonged exposure to DTT induces inward remodeling in isolated arterioles. A: arterioles were exposed to 200 μM DTT for 4 h. Before and after the 4-h exposure to DTT, arterioles were allowed to develop spontaneous tone and subsequently exposed to 10−4 M adenosine (Ado) and then to calcium-free solution. Data are means ± SE of the maximal passive diameter obtained during the first exposure to calcium-free conditions. After the second exposure to calcium-free conditions, maximal passive diameter was significantly reduced (*P ≤ 0.05; n = 5). B: wall/lumen ratios of arterioles in calcium-free conditions before (control) and after a prolonged (4-h) exposure to DTT (n = 5). *P ≤ 0.05 vs. control. C: passive pressure-diameter curves of arterioles obtained before (before DTT, n = 5) and after (after DTT, n = 5) exposure (4 h) to 200 μM DTT. *P ≤ 0.05 vs. before DTT. D: passive strain-stress relationships of isolated arterioles before (before DTT, n = 5) and after (after DTT, n = 5) exposure (4 h) to 200 μM DTT. E: incremental modulus of elasticity vs. pressure in isolated arterioles under passive conditions before (before DTT, n = 5) and after (after DTT, n = 5) exposure (4 h) to 200 μM DTT. F: representative immunoblot of the F- and G-actin portions of the cytoskeleton from arterioles treated with vehicle control or DTT (200 μM) for 4 h. Exposure to DTT increased (P ≤ 0.05) the F-actin/total actin by 8.01 ± 3.01% vs. controls (n = 6 for each treatment).
Prolonged Exposure to DTT Increases the F-Actin/Total Actin Ratio in Isolated Arterioles
Use of a differential centrifugation assay to quantify F- and G-actin proportions revealed that a 4-h incubation in DTT (200 μM) increased the F-actin/total actin ratio by 8.01 ± 3.01% (n = 6) compared with control arterioles kept in vehicle control for 4 h (Fig. 2F).
F-Actin Disruption Does Not Affect the Passive Diameter or Elastic Properties of Freshly Isolated Arterioles
To determine the role that disruption of the F-actin cytoskeleton has on the elastic characteristics of freshly isolated arterioles, we first confirmed that a 1-h incubation in mycalolide-B completely destroyed F-actin fibers in isolated arterioles using confocal microscopy. Three-dimensional reconstruction images of arterioles exposed for 1 h to vehicle control or mycalolide-B stained to visualize F-actin fibers with Alexa Phalloidin 546 are shown in Fig. 3A. Notice that incubation with mycalolide-B caused a complete obliteration of F-actin fibers contained within cells of the arteriolar wall. In comparison, intact F-actin cytoskeletal structures are evident in freshly isolated arterioles exposed to vehicle (Fig. 3A).
Fig. 3.
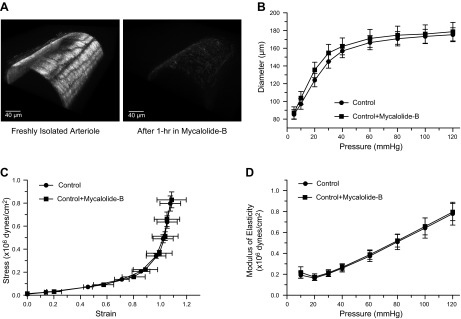
Actin cytoskeletal disruption does not affect the passive diameter or elastic characteristics of freshly isolated arterioles. A: 3-dimensional confocal images of isolated arterioles exposed to vehicle control (left) or 2 μM mycalolide-B (right) and subsequently stained with phalloidin-Alexa 546 to visualize the actin cytoskeleton. B: pressure-diameter curves of freshly isolated arterioles before (control, n = 7) and after (control + mycalolide-B, n = 7) exposure (1 h) to 2 μM mycalolide-B. C: strain-stress relationships of freshly isolated arterioles before (control, n = 7) and after (control + mycalolide-B, n = 7) exposure (1 h) to 2 μM mycalolide-B. D: incremental modulus of elasticity vs. pressure in freshly isolated arterioles before (control, n = 7) and after (control + mycalolide-B, n = 7) exposure (1 h) to 2 μM mycalolide-B.
Although a 1-h incubation in mycalolide-B (2 μM) caused complete disruption of F-actin fibers in freshly isolated (nonremodeled) arterioles, no significant changes in the internal passive diameter or the elastic properties of the vessel wall were evident (Fig. 3, B–D).
Disruption of the F-Actin Cytoskeleton Reverts the Inward Remodeling Induced by DTT
The reduction in passive internal diameter induced by prolonged (4-h) exposure to DTT (200 μM) was completely reverted (97.3 ± 7.2%) after mycalolide-B-induced disruption of the F-actin cytoskeleton. In comparison, exposure to vehicle control only partially reversed the DTT-induced inward remodeling by 57.1 ± 8.0%. Consequently remodeled arterioles exposed to mycalolide-B had significantly greater (P < 0.05) passive diameters than those exposed to vehicle control (Fig. 4, A and B). Pressure diameter curves showed that inwardly remodeled arterioles had greater passive diameters after vs. before exposure to mycalolide-B at all pressures in the curve (5–120 mmHg). In comparison inwardly remodeled arterioles treated with vehicle control had only greater passive diameters at pressures >20 mmHg (Fig. 4, C and D). With regard to the elastic properties of the arteriolar wall, exposure of remodeled arterioles to mycalolide-B caused reduced distensibility and greater circumferential stress, as well as a 36.7 ± 12.65% increase in stiffness (Fig. 4, E and G). In comparison, the stress, strain and stiffness of remodeled arterioles treated with vehicle control were not significantly affected (Fig. 4, F and H).
Fig. 4.

Disruption of the actin cytoskeleton reverts the inward remodeling caused by prolonged exposure of isolated arterioles to DTT. A: arterioles were exposed to 200 μM DTT for 4 h. Before and after the 4-h incubation with DTT, arterioles were allowed to develop spontaneous tone and subsequently exposed to 10−4 M adenosine and then to calcium-free solution. After 5 min in the second exposure to calcium-free solution, arterioles were exposed for 1 h to vehicle control (n = 6) or 2 μM mycalolide-B (n = 6). Data are means ± SE of the maximal passive diameter obtained during the first exposure to calcium-free conditions. *P ≤ 0.05 vs. vehicle control. B: change in diameter caused by 1-h exposure to mycalolide-B (2 μM) or its vehicle control in arterioles whose passive diameter had been reduced by a 4-h exposure to 200 μM DTT. The percent reversal change was significantly greater in vessels exposed to mycalolide-B vs. vehicle controls (*P ≤ 0.05). C and D: pressure-diameter curves of DTT-inwardly remodeled arterioles before (remodeled, n = 7 in C and n = 5 in D) and after exposure (1 h) to 2 μM mycalolide-B (remodeled + mycalolide-B, n = 7) or its vehicle control (remodeled + vehicle, n = 5). *P ≤ 0.05 vs. remodeled + mycalolide-B or remodeled + vehicle. E and F: strain-stress relationships of DTT-inwardly remodeled arterioles before (remodeled, n = 7 in C and n = 5 in D) and after exposure (1 h) to 2 μM mycalolide-B (remodeled + mycalolide-B, n = 7) or its vehicle control (remodeled + vehicle, n = 5). G and H: incremental modulus of elasticity vs. pressure in DTT-inwardly remodeled arterioles before (remodeled, n = 7 in G and n = 5 in H) and after exposure (1 h) to 2 μM mycalolide-B (remodeled + mycalolide-B, n = 7) or its vehicle control (remodeled + vehicle, n = 5). *P ≤ 0.05 vs. remodeled + mycalolide-B.
Inhibition of Actin Polymerization Blocks DTT-Induced Arteriolar Constriction
Exposure of isolated and pressurized (60 mmHg) arterioles to cytochalasin-D (500 nM) did not significantly affect vascular tone during the preincubation period but over time caused vessels to lose myogenic tone and dilate. The presence of cytochalasin-D also prevented DTT at all concentrations used from inducing vasoconstriction (Fig. 5A).
Fig. 5.
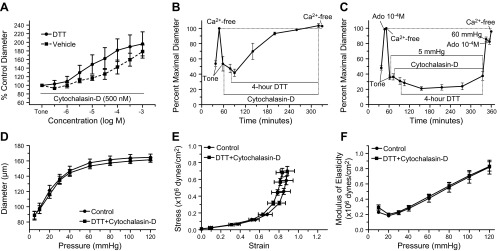
Cytochalasin-D inhibits concentration-dependent DTT constriction responses and prevents DTT-induced inward remodeling in isolated arterioles. A: percent control diameter of isolated arterioles exposed to incremental concentrations of DTT (n = 7) or vehicle control (n = 5) in the presence of cytochalasin-D (500 nM). B: arterioles were incubated for 20 min with 500 nM cytochalasin-D and then exposed to 200 μM DTT for 4 h in the presence of cytochasin-D. Before and after the incubation with cytochalasin-D and DTT, arterioles were allowed to develop spontaneous tone and subsequently exposed to 10−4 M adenosine and then to calcium-free solution (n = 4). Data are means ± SE of the maximal passive diameter obtained during the first exposure to calcium-free conditions. C: arterioles with intraluminal pressure set at 5 mmHg were incubated for 20 min with 500 nM cytochalasin-D and then exposed to 200 μM DTT for 4 h in the presence of cytochasin-D. Before and after the incubation with cytochalasin-D and DTT, intraluminal pressure was increased to 60 mmHg and arterioles were allowed to develop spontaneous tone followed by exposure to 10−4 M adenosine and then to calcium-free solution (n = 6). Data are means ± SE of the maximal passive diameter obtained during the first exposure to calcium-free conditions. D: passive pressure-diameter curves of arterioles obtained before (control, n = 6) and after (DTT + cytochalasin-D, n = 6) exposure (4 h) to cytochalasin-D (500 nM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg. E: passive strain-stress relationships of isolated arterioles obtained before (control, n = 6) and after (DTT + cytochalasin-D, n = 6) exposure (4 h) to cytochalasin-D (500 nM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg. F: incremental modulus of elasticity vs. pressure in isolated arterioles under passive conditions obtained before (control, n = 6) and after (DTT + cytochalasin-D, n = 6) exposure (4 h) to cytochalasin-D (500 nM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg.
Inhibition of Actin Polymerization Prevents DTT-Induced Inward Remodeling
In isolated arterioles pressurized to 60 mmHg, the presence of cytochalasin-D (500 nM) inhibited the constriction induced by 200 μM DTT and caused arterioles to dilate over the 4-h exposure to the agents (Fig. 5B). At the end of the prolonged exposure to cytochalasin-D and DTT, the passive diameter of arterioles did not differ from that obtained before exposure to the agents (Fig. 5B). To ensure that vasodilation or the lack of constriction (reduction in arteriolar diameter) was not an influencing factor in preventing remodeling, an additional series of experiments were performed in arterioles maintained at 5 mmHg of intraluminal pressure. In these vessels, exposure to DTT with cytochalasin-D for 4 h caused a small but significant constriction. Nonetheless, DTT in the presence of cytochalasin-D failed again to cause a significant reduction in maximal passive diameter measured at 60 mmHg (Fig. 5C). Consequently, no significant changes were observed after vs. before the prolonged exposure to DTT and cytochalasin-D in pressure-diameter profiles, strain-stress curves, and elastic moduli of arterioles (Fig. 5, D–F).
Transglutaminase Inhibition with Cystamine Blocks DTT-Induced Inward Remodeling
Since cystamine also blocked the vasoconstriction induced by DTT, a series of experiments were performed in arterioles maintained at 5 mmHg of intraluminal pressure to maintain vascular diameter reduced in the presence of cystamine and DTT. In these vessels, exposure to DTT with cystamine induced no significant changes in vascular diameter (Fig. 6A). In addition, prolonged exposure to these agents caused no significant reduction in the maximal passive diameter measured at 60 mmHg (Fig. 6, A and B). No significant changes were observed either in the pressure-diameter profiles, strain-stress curves and elastic moduli obtained after the prolonged exposure to DTT and cystamine when compared with those obtained before exposure to the agents (Fig. 6, B–D).
Fig. 6.
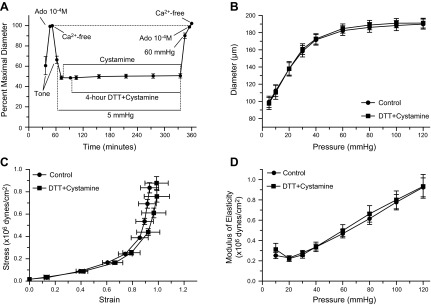
Cystamine prevents DTT from causing inward remodeling in isolated arterioles. A: arterioles with intraluminal pressure set at 5 mmHg were incubated for 20 min with 1 mM cystamine and then exposed to 200 μM DTT for 4 h in the presence of cystamine. Before and after the incubation with cystamine and DTT, intraluminal pressure was increased to 60 mmHg and arterioles were allowed to develop spontaneous tone followed by exposure to 10−4 M adenosine and then to calcium-free solution (n = 6). Data are means ± SE of the maximal passive diameter obtained during the first exposure to calcium-free conditions. B: passive pressure-diameter curves of arterioles obtained before (control, n = 6) and after (DTT + cystamine, n = 6) exposure (4 h) to cystamine (1 mM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg. C: passive strain-stress relationships of isolated arterioles obtained before (control, n = 6) and after (DTT + cystamine, n = 6) exposure (4 h) to cystamine (1 mM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg. D: incremental modulus of elasticity vs. pressure in isolated arterioles under passive conditions obtained before (control, n = 6) and after (DTT + cystamine, n = 6) exposure (4 h) to cystamine (1 mM) and DTT (200 μM), while at an intraluminal pressure of 5 mmHg.
DISCUSSION
The primary finding of the present study is that endogenous transglutaminase activation with DTT in isolated arterioles induces vasoconstriction and inward remodeling that are dependent on actin cytoskeletal dynamics. Based on previous publications that indicated DTT induces vasoconstriction in dog coronary arteries (11, 12) and activation of transglutaminase in mesenteric resistance arteries (37), we first confirmed that DTT induced endogenous transglutaminase activation as determined by the incorporation of Alexa Fluor cadaverine into smooth muscle arteriolar structures. This cadaverine incorporation induced by DTT was inhibited by the presence of the tranglutaminase inhibitor cystamine (Fig. 1, A and B). Similar results using a different more specific transglutaminase inhibitor (i.e., L682777) have been previously reported (37). Our current results, therefore, indicate that in isolated rat-cremaster arterioles DTT induces endogenous smooth muscle transglutaminase activation.
Next, we performed a series of experiments to determine the effect of acute DTT exposure on vascular function in isolated cremaster arterioles. DTT caused a concentration-dependent vasoconstriction that was completely blocked or diminished by the transglutaminase inhibitor cystamine (Fig. 1C). In comparison, cystamine did not block the vasoconstriction induced by membrane depolarization with KCl (Fig. 1D). This suggests that DTT induces arteriolar vasoconstriction for the most part through the activation of endogenous transglutaminases. The transglutaminases are a group of enzymes whose primary function is to deamidate, transamidate, and cross-link free amine groups with a protein glutamine (13, 24). Of the eight transglutaminases known to exist in humans, transglutaminase 1, 2, and 4 have been found in blood vessels, where their primary location is within the cytosol of smooth muscle and endothelial cells (3, 16). It has been previously shown that the vasoconstriction induced by a number of agonists depends on transglutaminase activity (9, 17, 38). Putatively, transglutaminases, in particular transglutaminase 2, could exert vasoconstriction via their activity as G proteins, as activators of Rho, or via their transamidation process of a number of cytoskeletal molecules associated with vascular smooth muscle contraction, including α actin, filamin A, and myosin (24, 38). Our results showing that inhibition of actin polymerization with cytochalasin-D blocked the constriction induced by increasing concentrations of DTT (Fig. 5A) suggest that indeed endogenous transglutaminase activation causes vasoconstriction through a process that requires actin dynamics.
Next, we determined that a prolonged (4-h) exposure of isolated and pressurized arterioles to DTT induced a strong and sustained vasoconstriction that resulted in the vessels becoming inwardly remodeled, that is, their maximal passive diameter became smaller (Fig. 2). We also determined that prolonged exposure to DTT induced actin polymerization, as determined by the increased F-actin/total-actin ratio observed in DTT-exposed arterioles compared with controls. Previously we showed that a similar 4-h exposure to the vasoconstrictor agonists norepinephrine and angiotensin-II induces inward remodeling in isolated arterioles (19, 34) and that the remodeling caused by those vasoconstrictor agonists is reversible upon disruption of the actin cytoskeleton (34). Prolonged exposure to vasoconstrictor agonists has also been previously reported to diminish the response of arterioles to adenosine or sodium nitroprusside (19). We have interpreted these reduced responses to endothelium-independent vasodilators as indication that inward remodeling processes are taking place (20). Similarly, in the present study, prolonged exposure to DTT diminished the vasodilatory response of arterioles to adenosine, but the mechanisms responsible for this effect of prolonged vasoconstriction remain to be determined.
To determine whether the inward remodeling induced by the 4-h exposure to DTT was also dependent on actin cytoskeletal structures, we exposed DTT-remodeled arterioles to the actin-depolymerizing agent, mycalolide-B. Actin cytoskeletal disruption completely reversed the remodeling induced by DTT (Fig. 4, A and B), suggesting that changes in actin cytoskeletal structures are responsible for reducing the passive diameter of arterioles during the early stages of the inward remodeling process induced by transglutaminase activation. The observation that incubation with cytochalasin-D also prevented the passive diameter of arterioles exposed for 4 h to DTT from becoming reduced (Fig. 5, B and C) further suggests that actin polymerization is needed during the inward remodeling process induced by transglutaminase activation. Incubation of remodeled arterioles with the vehicle control for mycalolide-B also reversed the remodeling induced by DTT but to a lesser extent than the complete reversal achieved by the actin-depolymerizing agent (Fig. 4, A and B). We previously reported that the vehicle control for mycalolide-B, which contains no calcium and the vasodilator adenosine, does not reverse the inward remodeling induced by a 4-h exposure of isolated arterioles to norepinephrine and angiotensin-II (34), suggesting that the presence of these vasoconstrictor agonists causes a more permanent modification of cytoskeletal structures than that achieved by the mere DTT-dependent activation of endogenous transglutaminases. The observation that the reversal of inward remodeling induced by mycalolide-B or its vehicle control differed in their effects on the elastic characteristics of the vascular wall further suggests that vasodilation alone reverts only a portion of the cytoskeletal changes induced by transglutaminase activation. This is evident by the capacity of mycalolide-B to increase the passive diameter of remodeled arterioles even at intraluminal pressures of 5 and 10 mmHg (Fig. 4C). Disruption of the actin cytoskeleton also reduced the distensibility of remodeled arterioles and made them stiffer (Fig. 4, E and G), while vessels exposed to vehicle control had no changes in these parameters (Fig. 4, F and H). These results support our hypothesis that in the early stages of the inward remodeling process induced by prolonged exposure to DTT, changes in the actin cytoskeleton are in part responsible for modifying the elastic properties of the vascular wall. They are also consistent with data indicating that actin fibers are more elastic (pliable) than extracellular matrix structures (35, 36), which more likely determine the elastic characteristics of the arterioles after disruption of the actin cytoskeleton.
To determine whether the involvement of actin cytoskeletal structures in the elastic characteristics of arterioles is only a feature present in remodeled arterioles, we compared pressure-diameter curves, strain-stress relationships, and elastic moduli of freshly isolated arterioles before and after exposure to mycalolide-B (Fig. 3). Our results indicate that actin cytoskeletal structures have no significant role on the passive elastic characteristics of nonremodeled arterioles and that involvement of the actin cytoskeleton on reducing the passive diameter of arterioles is a common feature in the early stages of the inward remodeling induced by direct activation of endogenous transglutaminase or exposure to prolonged vasoconstrictor agonists.
Previously, van den Akker et al. (37) reported that exposure of nonpressurized resistance arteries to DTT for 24 h induces inward remodeling through processes dependent on the activation of transglutaminase 2. Their data suggest that the reduction in passive diameter occurs as extracellular matrix components of the vascular wall are cross-linked by transglutaminase activity around a reduced vascular diameter. Because our experiments in which DTT was incubated with cytochalasin-D caused vessels to dilate to near maximal passive diameter during the 4-h exposure to the agents, we performed a series of experiments in which vessel diameter was maintained reduced at 5 mmHg of intravascular pressure during the exposure to DTT with cytochalasin-D (Fig. 5, B and C). Results from those experiments indicate that inhibition of actin dynamics and not vasodilation or the lack of constriction was the reason why vessels did not remodeled inwardly. We also performed experiments in which vessels were incubated for 4 h in DTT with the transglutaminase inhibitor cystamine while maintained at 5 mmHg of intravascular pressure to keep a reduced vascular diameter (Fig. 6). Those vessels also failed to remodel inwardly, indicating that indeed endogenous transglutaminase activation was responsible for changing actin cytoskeletal structures and reducing the passive diameter of arterioles exposed for 4 h to DTT. Overall, these results suggest that in the early stages of the inward remodeling process induced by endogenous transglutaminase activation, reduction of the passive diameter requires changes in actin cytoskeletal structures. Our results do not rule out that later on in the remodeling process, extracellular transglutaminase activity may solidify the reduced structural diameter of vessels by cross-linking extracellular components of the vascular wall. An additional potential explanation of our results could be that disruption of actin cytoskeletal structures prevents transglutaminase translocation to the cell membrane and extracellular environment. However, this is highly unlikely based on our observation that once inward remodeling induced by DTT had occurred, it was completely reversed upon actin disruption with mycalolide-B.
The physiological relevance of our findings rely on previous reports indicating that inhibition of transglutaminase 2 activity blocks or retards the development of inward remodeling in resistance arteries exposed to low blood flow (1, 2) and reduces the stiffness of conduit arteries that occurs in advanced age (31). The novelty of our findings is that changes in vascular structure associated with endogenous transglutaminase activation start at the level of the cytoskeleton and not with extracellular matrix components. This is particularly important in light of recent reports that indicate vascular smooth muscle cellular stiffness plays a more important role in hypertension than previously thought (26, 32, 40). Although the methods of activation and inhibition of transglutaminase used in our experiments have been used extensively in the past (1, 10, 17, 31, 37–39), a limitation of our study rests on the potential nonspecific effects of DTT and cystamine. Nonetheless, our results are consistent with an increasing body of evidence that indicates intracellular transglutaminase activation participates in vascular pathology (6, 7, 23, 30). A better understanding of the pathways associated with transglutaminase-dependent cytoskeletal changes should provide new therapeutic avenues for controlling vascular remodeling and the adverse cardiovascular events associated with it.
GRANTS
This study was supported by the National Heart, Lung, and Blood Institute Grant R01-HL-088105 (to L. A. Martinez-Lemus).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.A.C.-G., M.C.S., and C.A.F. performed experiments; J.A.C.-G., M.C.S., and C.A.F. analyzed data; J.A.C.-G., C.A.F., L.P.P., and L.A.M.-L. interpreted results of experiments; J.A.C.-G., M.C.S., and C.A.F. prepared figures; J.A.C.-G. drafted manuscript; J.A.C.-G., M.C.S., C.A.F., L.P.P., and L.A.M.-L. approved final version of manuscript; C.A.F., L.P.P., and L.A.M.-L. edited and revised manuscript; L.A.M.-L. conception and design of research.
ACKNOWLEDGMENTS
We thank Guiling Zhao for excellent technical assistance.
REFERENCES
- 1.Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 96: 119–126, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase: possible compensation by macrophage-derived factor XIII. Circ Res 99: 86–92, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Bakker EN, Pistea A, VanBavel E. Transglutaminases in vascular biology: relevance for vascular remodeling and atherosclerosis. J Vasc Res 45: 271–278, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Bakker EN, van Der Meulen ET, Spaan JA, VanBavel E. Organoid culture of cannulated rat resistance arteries: effect of serum factors on vasoactivity and remodeling. Am J Physiol Heart Circ Physiol 278: H1233–H1240, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodeling follows chronic vasoconstriction in isolated resistance arteries. J Vasc Res 39: 12–20, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Beazley KE, Banyard D, Lima F, Deasey SC, Nurminsky DI, Konoplyannikov M, Nurminskaya MV. Transglutaminase inhibitors attenuate vascular calcification in a preclinical model. Arterioscler Thromb Vasc Biol 33: 43–51, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen NX, O′Neill K, Chen X, Kiattisunthorn K, Gattone VH, Moe SM. Transglutaminase 2 accelerates vascular calcification in chronic kidney disease. Am J Nephrol 37: 191–198, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Christensen KL, Mulvany MJ. Vasodilatation, not hypotension, improves resistance vessel design during treatment of essential hypertension: a literature survey. J Hypertens 19: 1001–1006, 2001 [DOI] [PubMed] [Google Scholar]
- 9.del Campo L, Guvenc Tuna B, Ferrer M, van Bavel E, Bakker EN. Testosterone and beta-oestradiol prevent inward remodelling of rat small mesenteric arteries: role of NO and transglutaminase. Clin Sci (Lond) 124: 719–728, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Eftekhari A, Rahman A, Schaebel LH, Chen H, Rasmussen CV, Aalkjaer C, Buus CL, Mulvany MJ. Chronic cystamine treatment inhibits small artery remodelling in rats. J Vasc Res 44: 471–482, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Fujioka H, Horiike K, Takahashi M, Ishida T, Kinoshita M, Nozaki M. Dithiothreitol-induced triphasic response of dog coronary arteries. Eur J Pharmacol 166: 13–22, 1989 [DOI] [PubMed] [Google Scholar]
- 12.Fujioka H, Horiike K, Takahashi M, Ishida T, Kinoshita M, Nozaki M. Triphasic vascular effects of thiol compounds and their oxidized forms on dog coronary arteries. Experientia 49: 47–50, 1993 [DOI] [PubMed] [Google Scholar]
- 13.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. Biochem J 368: 377–396, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension 21: 391–397, 1993 [DOI] [PubMed] [Google Scholar]
- 15.Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell 17: 1606–1619, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson KB, Petersen-Jones H, Thompson JM, Hitomi K, Itoh M, Bakker EN, Johnson GV, Colak G, Watts SW. Vena cava and aortic smooth muscle cells express transglutaminases 1 and 4 in addition to transglutaminase 2. Am J Physiol Heart Circ Physiol 302: H1355–H1366, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson KB, Thompson JM, Watts SW. Modification of proteins by norepinephrine is important for vascular contraction. Front Physiol 1: 131, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim H, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med 12: 2165–2180, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Lemus LA. Persistent agonist-induced vasoconstriction is not required for angiotensin ii to mediate inward remodeling of isolated arterioles with myogenic tone. J Vasc Res 45: 211–221, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda) 24: 45–57, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Mathiassen ON, Buus NH, Larsen ML, Mulvany MJ, Christensen KL. Small artery structure adapts to vasodilatation rather than to blood pressure during antihypertensive treatment. J Hypertens 25: 1027–1034, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Mathiassen ON, Buus NH, Sihm I, Thybo NK, Morn B, Schroeder AP, Thygesen K, Aalkjaer C, Lederballe O, Mulvany MJ, Christensen KL. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens 25: 1021–1026, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Matlung HL, Neele AE, Groen HC, van Gaalen K, Tuna BG, van Weert A, de Vos J, Wentzel JJ, Hoogenboezem M, van Buul JD, VanBavel E, Bakker EN. Transglutaminase activity regulates atherosclerotic plaque composition at locations exposed to oscillatory shear stress. Atherosclerosis 224: 355–362, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 294: 1–97, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R522–R530, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107: 615–619, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation 108: 2230–2235, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Rizzoni D, Porteri E, Guelfi D, Muiesan ML, Valentini U, Cimino A, Girelli A, Rodella L, Bianchi R, Sleiman I, Rosei EA. Structural alterations in subcutaneous small arteries of normotensive and hypertensive patients with non-insulin-dependent diabetes mellitus. Circulation 103: 1238–1244, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Saito S, Watabe S, Ozaki H, Fusetani N, Karaki H. Mycalolide B, a novel actin depolymerizing agent. J Biol Chem 269: 29710–29714, 1994 [PubMed] [Google Scholar]
- 30.Sane DC, Kontos JL, Greenberg CS. Roles of transglutaminases in cardiac and vascular diseases. Front Biosci 12: 2530–2545, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res 107: 117–125, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, Vatner DE, Meininger GA, Vatner SF. Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol 305: H1281–H1287, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Souza-Smith FM, Katz PS, Trask AJ, Stewart JA, Jr, Lord KC, Varner KJ, Vassallo DV, Lucchesi PA. Mesenteric resistance arteries in type 2 diabetic db/db mice undergo outward remodeling. PLoS One 6: e23337, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staiculescu MC, Galinanes EL, Zhao G, Ulloa U, Jin M, Beig MI, Meininger GA, Martinez-Lemus LA. Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc Res 98: 428–436, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unterberger MJ, Schmoller KM, Bausch AR, Holzapfel GA. A new approach to model cross-linked actin networks: multi-scale continuum formulation and computational analysis. J Mech Behav Biomed Mater 22: 95–114, 2013 [DOI] [PubMed] [Google Scholar]
- 36.van den Akker J, Schoorl MJ, Bakker EN, Vanbavel E. Small artery remodeling: current concepts and questions. J Vasc Res 47: 183–202, 2009 [DOI] [PubMed] [Google Scholar]
- 37.van den Akker J, VanBavel E, van Geel R, Matlung HL, Guvenc Tuna B, Janssen GM, van Veelen PA, Boelens WC, De Mey JG, Bakker EN. The redox state of transglutaminase 2 controls arterial remodeling. PLoS One 6: e23067, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watts SW, Priestley JR, Thompson JM. Serotonylation of vascular proteins important to contraction. PLoS One 4: e5682, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang G, Matsumura Y, Matsumoto S, Hayashi Y, Mori T. Effects of Ca2+ and sulfhydryl reductant on the polymerization of soybean glycinin catalyzed by mammalian and microbial transglutaminases. J Agric Food Chem 51: 236–243, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Zhu Y, Qiu H, Trzeciakowski JP, Sun Z, Li Z, Hong Z, Hill MA, Hunter WC, Vatner DE, Vatner SF, Meininger GA. Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell 11: 741–750, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]