

Abstract
Protein S-palmitoylation describes the thioester linkage of long-chain fatty acids to cysteine residues in proteins. Traditional methods for analyzing S-palmitoylation require metabolic labeling with [3H]-palmitate, immunoprecipitation, and SDS-PAGE separation followed by long film exposures for days or weeks. New methods have been developed based on metabolic incorporation of alkynyl fatty acid analogues. The commercially available analog 17-octadecynoic acid (17-ODYA) is readily accepted by the endogenous cellular palmitoylation machinery and transferred to endogenous sites of modification. After metabolic labeling, the extent of protein palmitoylation can be monitored by simple copper-catalyzed click chemistry to azide-linked reporter tags for in-gel fluorescence analysis or affinity enrichment. This approach is especially useful for monitoring the dynamics of palmitoylation using classic pulse-chase approaches. This protocol describes the metabolic labeling and in-gel fluorescence analysis of dynamic palmitoylation in mammalian cells.
Basic Protocol
Introduction
Protein S-palmitoylation describes the thioester linkage of long-chain fatty acids to cysteine residues in proteins. Traditional methods for analyzing S-palmitoylation require metabolic labeling with [3H]-palmitate, immunoprecipitation, and SDS-PAGE separation followed by long film exposures for days or weeks. New methods have been developed based on metabolic incorporation of alkynyl fatty acid analogues. The commercially available analog 17-octadecynoic acid (17-ODYA) is readily accepted by the endogenous cellular palmitoylation machinery and transferred to endogenous sites of modification. After metabolic labeling, the extent of protein palmitoylation can be monitored by simple copper-catalyzed click chemistry to azide-linked reporter tags for in-gel fluorescence analysis or affinity enrichment. This approach is especially useful for monitoring the dynamics of palmitoylation using classic pulse-chase approaches. This protocol describes the metabolic labeling and in-gel fluorescence analysis of dynamic palmitoylation in mammalian cells (Figure 1).
Figure 1.

Schematic of 17-ODYA metabolic labeling and detection of palmitoylated proteins. The commercially available alkynyl fatty acid 17-ODYA is added to cells in culture. Over the course of several hours, the probe is conjugated to CoA, and can serve as a substrate for protein palmitoyl transferases. The cells are then lysed and mixed with click chemistry reagents. The TCEP reduces the Cu(II) to Cu(I), which is bound to the TBTA ligand. Addition of azide-linked reporters, such as rhodamine-azide or biotin-azide, facilitate click chemistry conjugation and triazole formation, covalently linking the reporter to 17-ODYA thioesters on proteins. The labeled palmitoylated proteins are then separated by SDS-PAGE separation or affinity purified for mass spectrometry-based proteomics.
Materials
Cultured mammalian cells. The number of cells varies depending on the application.
For gel-based analysis, a 6 cm culture dish of confluent 293T is more than sufficient.
The membrane protein yield will vary between cell lines.
25 mM 17-octadecynoic acid (Cayman Chemical cat. no. 90270) in DMSO
Dialyzed FBS (Gemini Bio-Products, cat. no. 100-108)
DMSO
Culture media (DMEM, Invitrogen, cat. no. 10564-011) (Other media are also OK).
Penicillin-Streptomycin-Glutamine (PSQ) (Invitrogen, cat. no. 10378-016)
D-PBS, Calcium and Magnesium free (Invitrogen, cat. no. 14190-136)
PMSF (Sigma, cat. no. P7626)
HDSF (Calbiochem, cat. no. 373250) or hexadecylfluorophosphonate (HDFP) (optional).
500 mM Palmitic acid in DMSO
1 mM Rhodamine-azide = tetramethylrhodamine (TAMRA) azide (tetramethylrhodamine 5-carboxamido-(6-azidohexanyl)) *5-isomer* (Invitrogen, cat. no. T10182) in DMSO
Tris 2-carboxyethyl phosphine (Sigma-Aldrich, cat. no. 93284)
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (Sigma-Aldrich, cat. no. 678937)t-Butanol (Sigma-Aldrich, cat. no. 360538)
50 mM Copper (II) sulfate (Sigma-Aldrich, cat. no. 451657) in water
Hydroxylamine (Alfa Aesar, cat no. B22202 or L16990)
Beckman Optima Max Ultracentrifuge with TLA-100.3 rotor, or equivalent
Ultracentrifuge tubes (cat. no. 357448)
Branson Sonifier cell disruptor with 1/8” tapered microtip
Tris-Glycine SDS-PAGE gel
4X SDS PAGE loading buffer
Flatbed Fluorescence gel scanner, Hitachi FMBIO II or equivalent
Protein Quantification assay
17-ODYA Metabolic Labeling
Grow mammalian cells to desired density in standard culture media. For gel-based analysis, plan to harvest enough cells for approximately 1 mg of proteome. For Jurkat cells, this is approximately 2×107 cells in 20 mL media, or approximately 4×106 293T cells on a 6 cm culture dish in 4 mL media.
Wash cells with warm D-PBS (37°C) to remove residual growth media. For suspension cells, collect cells and centrifuge at 500 g for 5 minutes at room temperature. Gently resuspend cell pellet in 10 mL D-PBS, centrifuge again, and repeat one or two times. For adherent cells, gently aspirate media and add a sufficient volume of D-PBS to cover cells for 1 minute. Aspirate D-PBS and repeat 2 times. If adherent cells detach, collect cells and centrifuge as described above, then add back to dish.
Add warm 17-ODYA (37°C) labeling media (see recipe) to the cells. For suspension cells, add enough media to support log phase growth. For Jurkat cells, dilute cells to 1×106 cells / mL. For adherent cells, add labeling media equal to the original volume of media. Return cells to the CO2 incubator. You may need to do a time course to determine the optimal labeling time, but 2 hours labeling is typically sufficient for pulse-chase methods.
Optional: Add warm chase media (see recipe) for predetermined lengths of time, such as 2 hours, 4 hours, and 6 hours.
After incubation, wash cells 3 times with cold D-PBS (4°C). Harvest adherent cells by scraping. Do not use trypsin. Pellet cells at 500 g for 2 minutes (4°C). Freeze cell pellet at −80°C or continue. The frozen cell pellet is stable for several months.
Preparation of membrane lysates
-
6
On ice, add D-PBS (without EDTA) to cell pellets supplemented with 1 mM PMSF, HDSF, or hexadecylfluorophosphonate (HDFP) and lyse cells by sonication. Do not add EDTA. These inhibitors inactivate potential protein thioesterase activities, although the lysosomal thioesterase PPT1 is resistant to PMSF. Avoid the use of Tris buffers or additives with free amines, which can interfere with click chemistry. Sonicate insoluble pellet for 10 seconds on ice.
-
7
In thick walled ultracentrifuge tubes, separate soluble and insoluble fractions by ultracentrifugation at 100,000 g for 45 minutes at 4°C.
-
8
Collect supernatant and set aside in a new 1.5 mL tube. Add fresh cold D-PBS (without EDTA) to the pellet at an appropriate volume to yield approximately 1–2 mg / mL proteome. This is approximately 0.5 mL for a standard 10 cm plate of cells. Sonicate for 10 seconds on ice for 5 seconds, or until the pellet is dispersed.
-
9
Measure protein concentration by Bradford or equivalent assay. Normalize concentrations to 1.1 mg / mL with D-PBS (without EDTA).
Optional: Immunoprecipate a specific target. Use standard immunoprecipitation protocol in phosphate buffer without EDTA.
Click Chemistry
-
10
Dissolve TCEP in D-PBS at a concentration of 14.35 mg/mL to make a 50 mM stock. Do not reuse.
-
11
Add 50 μg of the 1.1 mg / mL membrane lysate in 44 μL of total volume with PBS. Add 1 μL of 50 mM CuSO4, 1 μL 50 mM TCEP, 3 μL of 1× ligand, and 1 μL of Rhodamine-azide. Vortex and let sit at room temperature for 30 minutes, then vortex and let sit another 30 minutes. The copper precipitates proteins during reaction in the absence of detergents. This reaction also works well with immunoprecipitated proteins on beads with continuous mixing.
-
12
Add 1X SDS gel loading buffer. Adding β-mercaptoethanol (150 mM) to the loading buffer gives better resolution, but do not use DTT and do not boil. Remove half of the sample and place in a separate tube. Add 1.25 μL of 50% stock of hydroxylamine (pH 7) to the 25 μL reaction to check for thioester-dependent labeling and incubate at room temperature for 1 hour, or boil 5 minutes.
-
13
Load samples for standard SDS-PAGE gel electrophoresis. Scan the gel fluorescence. Alternatively, if the lysate was reacted with biotin-azide, the gel should be transferred to nitrocellulose and blocked with BSA (Nonfat dry milk contains endogenous biotin). Probe blot with streptavidin-HRP for chemiluminescence detection. [*Gwen to add appropriate CPPS cross-refs]
REAGENTS AND SOLUTIONS
17-ODYA Labeling media
Cell culture media (DMEM, RPMI)
1% PSQ
10% dialyzed FCS
25 μM 17-ODYA. Prepare a 25 mM stock of 17-ODYA in DMSO. Add 1:1000 to labeling media.
Prepare fresh just before use. Warm labeling media to 37°C before adding to cells.
Sonicate or vortex to mix the probe in the media. Discard after use.
Palmitic acid Chase Media
Cell culture media (DMEM, RPMI)
1% PSQ
10% dialyzed FCS
250 μM Palmitic acid
Prepare fresh just before use. Sonicate thoroughly dissolve palmitate acid to fine particulates in media. Discard after use.
TBTA ligand solution
50× TBTA
8.85 mg of TBTA in 200 μL DMSO
Store at room temperature for up to 6 months.
1× TBTA
20 μL 50× ligand
180 μl DMSO
800 μl t-butanol
Store at room temperature for up to 6 months.
COMMENTARY
Background Information
The analysis of protein S-palmitoylation has traditionally utilized radioisotope labeling with [3H]-palmitate (Schlesinger et al., 1980; Schmidt et al., 1979). This procedure requires additional safety precautions for handling radioactivity and demands film exposures of days to weeks. Acyl biotin exchange (ABE) is a popular method to enrich and analyze palmitoylated proteins (Drisdel and Green, 2004; Kang et al., 2008; Roth et al., 2006). This approach involves thioester hydrolysis with hydroxylamine, followed by free thiol capture with disulfide-biotin derivatives or thiol resin (Forrester et al., 2011). This method is technically challenging, and requires complete reduction and alkylation of all thiols before hydroxylamine addition. This is accomplished by multiple washes and precipitation steps, which can take several hours alone. Furthermore, this approach has no specificity for palmitoyl-thioesters, and leads to hydrolysis of all thioesters in a proteome sample. This leads to the non-specific enrichment of other protein thioesters, including enzymes with lipoic acid cofactors and enzymes with stable acyl-intermediates, such as ubiquitin ligases (Kang et al., 2008; Wan et al., 2007). Furthermore, ABE is not compatible with pulse-chase approaches to analyze palmitoylation stability and dynamics.
To circumvent these issues, bioorthogonal, metabolically incorporated fatty acids have been introduced for the rapid analysis of protein palmitoylation (Hang et al., 2007; Kostiuk et al., 2008; Martin and Cravatt, 2009). Since palmitoylation a labile post-translational modification, there is a continual cycle of enzymatic and non-enzymatic hydrolysis, followed by re-acylation. This provides a unique opportunity to metabolically incorporate modified acyl chains into existing proteins. The commercially available alkynyl fatty acid analogue 17-ODYA is incorporated by the endogenous cellular machinery to native sites of palmitoylation. This non-radioactive analogues enables pulse-chase labeling, which has led to the identification of dynamic sites of palmitoylation with rapid turnover kinetics (Martin et al., 2012; Zhang et al., 2010). Using these methods, it was observed that the majority of palmitoylation sites are relatively stable, yet some palmitoylated proteins are enzymatically depalmitoylated and highly dynamic (Martin et al., 2012).
This unit provides a straightforward protocol for metabolic labeling and analysis of protein palmitoylation using click chemistry (Fig. 1). The choice of the azide-modified reporter depends on the downstream application (Charron et al., 2009; Martin and Cravatt, 2009). In-gel fluorescence is routinely used for simple analysis of global palmitoylation profiles. The choice of fluorophore depends on the availability of excitation wavelengths for the flatbed fluorescence gel scanner. Biotin-azide is also useful for streptavidin enrichment or blots. In comparison with in-gel fluorescence approaches, streptavidin blots require additional steps for transfer and detection, and also highlight endogenous biotinylated proteins, such as carboxylase enzymes. These background biotinylated proteins can saturate the signal and prevent highly sensitive detection. This can be resolved by pre-clearing lysates with avidin beads, but this adds additional purification steps. Biotin-azide is especially useful for enrichment of palmitoylated proteins for downstream proteomics applications for analysis of tryptic peptides (Martin and Cravatt, 2009; Yount et al., 2010).
Critical Parameters and Troubleshooting
Several factors will determine the quality of 17-ODYA labeling and detection. Before performing more challenging pulse-chase experiments, first optimize the concentration and time of 17-ODYA required to achieve maximal metabolic incorporation. Different cell lines have different metabolic rates, which can directly affect the efficiency of incorporation. After careful optimization of the labeling conditions, then chase conditions should also be carefully tested. The time and concentration of the pulse labeling is essential for achieving any detectible palmitoylation turnover. If the labeling period is too long, 17-ODYA is metabolized into cellular lipid pools, and is especially difficult to washout. There is a careful balance between longer labeling times required for robust detection and sufficient chase times to detect dynamic sites of palmitoylation. Current reported methods generally use 1 or 2 hours of pulse labeling (Martin et al., 2012; Zhang et al., 2010). When using [3H]-palmitate, much shorter pulse labeling protocols are especially useful for detecting rapid turnover (Ahearn et al., 2011). This suggests that similar rapid methods could be coupled with immunoaffinity enrichment of select targets for non-radioactive detection using 17-ODYA.
During lysis, it is useful to include non-selective serine hydrolase inhibitors to block any potential thioesterase activities. Reports have demonstrated that the lysosomal thioesterase PPT1 is resistant to PMSF inhibition, yet sensitive to hexadecylsulfonyl fluoride (HDSF) inhibition (Das et al., 2000). This inhibitor is commercially available and potentially useful in samples with high PPT1 activities. Palmostatin B has also been shown to inhibit APT1, APT2, and PPT1, which covers all three known candidate protein palmitoyl thioesterases (Dekker et al., 2010; Rusch et al., 2011). The non-selective lipase inhibitor hexadecylfluorophosphonate (HDFP) has been demonstrated to sufficiently inhibit protein palmitoyl thioesterase activity in lysates by blocking more than 20 serine hydrolases, including PPT1 (Martin et al., 2012). It is important to test whether any of these inhibitors have any role in stabilizing palmitoyl thioesters in different biological samples.
Early reported methods for bioorthogonal detection used azido fatty acids. Significant background labeling occurs in the presence of excess alkyne-reporters, which is thought to be thiol dependent. It is therefore highly recommended to avoid azido-fatty acids, since alkyne and strained-alkyne reporters demonstrate significantly higher non-specific background labeling. Copper-catalyzed click chemistry is highly sensitive to amines, which are present in buffers such as Tris, and should be avoided. It is also essential to avoid using buffers with EDTA, which can affect the copper reactivity. When adding the click chemistry reagents, it is recommended to add them each separately to each tube, rather than making a master mix.
For more sensitive labeling, the concentration of rhodamine-azide can be increased at least 10-fold. For gel-based experiments, the typical final concentration of rhodamine-azide is 20 μM, yet for proteomics experiments, the concentration of biotin-PEG3-azide required for saturated labeling was experimentally determined to be 500 μM (Martin et al., 2012). During the click chemistry reaction, in the absence of any detergent, the proteome is precipitated by the excess copper. Excess unreacted fluorophore can easily be removed by centrifugation and cold methanol washes. If the reaction is carried out in the presence of detergents, it can still be cleaned up by performing chloroform/methanol extractions (Wan et al., 2007).
Finally, when preparing samples for SDS-PAGE, it is important to think about possible steps that could lead to thioester hydrolysis. Beta-mercaptoethanol is known to exchange with thioesters, yet does not significantly affect 17-ODYA stability. On the other hand, dithiolthreitol (DTT) rapidly hydrolyzes palmitoyl thioesters. It is important to test gel loading buffers with various reducing agents to ensure that palmitoyl thioesters are not sacrificed. It is also important to take precautions if boiling samples, since thioesters are easily susceptible to hydrolysis. Traditional Tris-Glycine gels separate protein samples at pH 8.8 or higher, which are not favorable conditions for thioester stability. Bis-Tris gels have several advantages over traditional Tris-Glycine gels, since they run samples at a neutral pH. While these issues may be important under certain circumstances, but practically the final separation and data quality is very similar for each type of gel chemistry when performing 17-ODYA in-gel fluorescence analysis.
Anticipated Results
Using the protocols described, protein palmitoylation can easily be analyzed by gel-based fluorescence. If there is no fluorescence flatbed gel scanner, it is possible to transfer gels to membranes for streptavidin blots. Without pre-clearing, there will be a significant number of endogenous biotinylated proteins detected in the absence of 17-ODYA. This is not the case with rhodamine-azide labeling. As discussed above, because fluorescent labeling is more sensitive, lower concentrations of fluorescent azide reporters are needed for detection (Figure 2). We suspect this 200-fold concentration difference accounts for the different proteins detected by biotin-azide and rhodamine-azide. Overall, it is recommended that these concentrations be optimized individually for detection of specific palmitoylated proteins.
Figure 2.
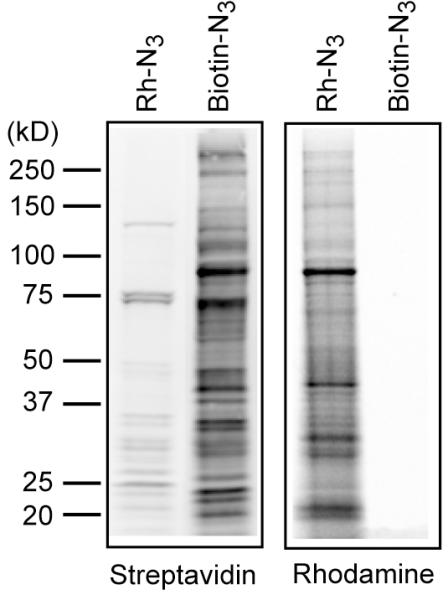
Comparison of biotin-azide detection and rhodamine-azide detection of 17-ODYA labeling. BW5147 cells were metabolically labeled with 17-ODYA for 4 hours, then lysed and the insoluble fraction was reacted with either 20 μM rhodamine-azide or 400 μM biotin-azide. The resulting samples were separated using a 4–20% Tris-Glycine SDS precast mini-gel. The gel was first scanned using a Hitachi FMBioII flatbed fluorescence scanner to detect rhodamine fluorescence. The gel was then transferred to nitrocellulose, blocked with 5% BSA, and probed with Streptavidin 800 (LiCOR), and scanned using a LiCOR fluorescence scanner. The gel demonstrates the relative sensitivity of both approaches for detection.
The turnover of protein palmitoylation can be detected using 17-ODYA pulse-chase labeling strategies (Figure 3). After 2 hours of 17-ODYA labeling, incorporation has reached equilibrium. Replacing the 17-ODYA labeling media with excess palmitic acid for several hours reveals a subset of proteins with rapid palmitoylation turnover. Shorter labeling periods show more significant turnover, but weaker signal. The total protein abundance is not changed during the timecourse of the experiment, as shown by coomassie staining.
Figure 3.
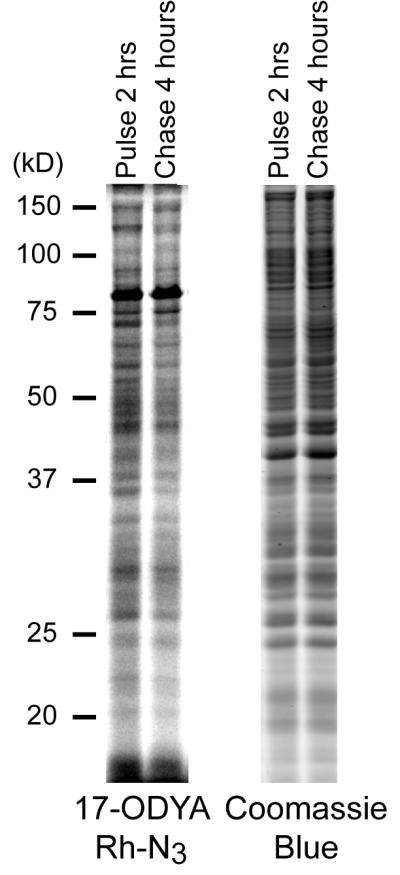
Pulse-chase analysis of dynamic protein palmitoylation. BW5147-derived T-cell hybridoma cells were labeled with 20 μM 17-ODYA for 2 hours, and an aliquot was washed and frozen. The remaining cells were washed and placed in media containing 250 μM palmitic acid for 4 hours, then washed and frozen. Cells pellets were lysed by sonication in the presence of 20 μM HDFP, then separated into soluble and insoluble fractions by ultracentrifugation at 100,000 g for 45 minutes. The insoluble fraction was diluted to 1 mg/ml, and click chemistry reagents (TCEP, TBTA, and CuSO4) with rhodamine-azide were added for 1 hour at room temperature. The samples were separated by SDS-PAGE using a Tris-Glycine system with an 11 cm separating gel. The gel was analyzed using a Hitachi FMBIOII fluorescence gel scanner, then transferred to coomassie blue for staining total protein. The coomassie stained gel was scanned at 700 nm using a LiCOR fluorescence gel scanner, and demonstrates equivalent loading.
Time Considerations
17-ODYA metabolically labeling takes several hours. This is lengthened if performing a pulse-chase experiment, but can still be easily accomplished in an afternoon. After harvesting the labeled cells, the cell pellets can be frozen at −80°C for experiments on another day. After thawing cells and sonication, it is preferred to enrich the membrane fraction by ultracentrifugation. This procedure takes less than one hour, but enhances the sensitivity by adding an additional enrichment and eliminating soluble proteins. Further density fractionization is also compatible, so long as it is carried out in phosphate buffers and with amine-free detergents. After protein quantification, the click chemistry reaction takes a few minutes to assemble and is left at room temperature for 1 hour. Following click chemistry, the sample is separated by SDS-PAGE and analyzed using a fluorescence gel scanner. It is preferred to complete the entire analysis after the initial thaw of the cell pellet to avoid freeze-thaw cycles.
References
- Ahearn IM, Tsai FD, Court H, Zhou M, Jennings BC, Ahmed M, Fehrenbacher N, Linder ME, Philips MR. FKBP12 Binds to Acylated H-Ras and Promotes Depalmitoylation. Molecular Cell. 2011;41:173–185. doi: 10.1016/j.molcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. Journal of the American Chemical Society. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- Das AK, Bellizzi JJ, 3rd, Tandel S, Biehl E, Clardy J, Hofmann SL. Structural basis for the insensitivity of a serine enzyme (palmitoyl-protein thioesterase) to phenylmethylsulfonyl fluoride. J Biol Chem. 2000;275:23847–23851. doi: 10.1074/jbc.M002758200. [DOI] [PubMed] [Google Scholar]
- Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Schölermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PIH, Waldmann H. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–456. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Green WN. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res. 2011;52:393–398. doi: 10.1194/jlr.D011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. Chemical probes for the rapid detection of Fatty-acylated proteins in Mammalian cells. J Am Chem Soc. 2007;129:2744–2745. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, Iii, Davis NG, El-Husseini A. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, Bertozzi CR, Rajaiah G, Falck JR, Berthiaume LG. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008;22:721–732. doi: 10.1096/fj.07-9199com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods. 2012;9:84–89. doi: 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch M, Zimmermann TJ, Burger M, Dekker FJ, Gormer K, Triola G, Brockmeyer A, Janning P, Bottcher T, Sieber SA, Vetter IR, Hedberg C, Waldmann H. Identification of Acyl Protein Thioesterases 1 and 2 as the Cellular Targets of the Ras-Signaling Modulators Palmostatin B and M. Angew Chem Int Ed Engl. 2011 doi: 10.1002/anie.201102967. [DOI] [PubMed] [Google Scholar]
- Schlesinger M, Magee A, Schmidt M. Fatty acid acylation of proteins in cultured cells. J. Biol. Chem. 1980;255:10021–10024. [PubMed] [Google Scholar]
- Schmidt MF, Bracha M, Schlesinger MJ. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc Natl Acad Sci U S A. 1979;76:1687–1691. doi: 10.1073/pnas.76.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- Yount JS, Moltedo B, Yang Y-Y, Charron G, Moran TM, López CB, Hang HC. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, Tsou LK, Charron G, Raghavan AS, Hang HC. Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proceedings of the National Academy of Sciences. 2010;107:8627–8632. doi: 10.1073/pnas.0912306107. [DOI] [PMC free article] [PubMed] [Google Scholar]