

Abstract
Study Objectives:
During normal sleep several neuroplasticity changes occur, some of which are considered to be fundamental to strengthen memories. Given the evidence linking sleep to neuroplasticity, it is conceivable that individuals with chronic sleep disruption, such as patients with chronic insomnia (CI), would experience abnormalities in neuroplastic processes during daytime. Protocols testing use-dependent plasticity (UDP), one of the mechanisms underlying formation of motor memories traces, provide a sensitive measure to assess neuroplasticity in the context of motor training.
Design and Participants:
A well-established transcranial magnetic stimulation (TMS) paradigm was used to evaluate the ability of patients with CI and age-matched good sleeper controls to undergo UDP. We also investigated the effect of insomnia on intracortical motor excitability measures reflecting GABAergic and glutamatergic mechanisms.
Setting:
Human Brain Physiology Laboratory, Johns Hopkins Medical Institutions.
Measurements and Results:
We found that patients with CI experienced increased UDP changes relative to controls. This effect was not due to differences in motor training. In addition, patients with CI showed enhanced intracortical facilitation relative to controls, in the absence of changes in intracortical inhibitory measures.
Conclusion:
This study provides the first evidence that patients with chronic insomnia have an increased plasticity response to physical exercise, possibly due to larger activation of glutamatergic mechanisms. This suggests a heightened state of neuroplasticity, which may reflect a form of maladaptive plasticity, similar to what has been described in dystonia patients and chronic phantom pain after amputation. These results could lead to development of novel treatments for chronic insomnia.
Citation:
Salas RE; Galea JM; Gamaldo AA; Gamaldo CE; Allen RP; Smith MT; Cantarero G; Lam BD; Celnik PA. Increased use-dependent plasticity in chronic insomnia. SLEEP 2014;37(3):535-544.
Keywords: Insomnia, plasticity, memory, transcranial magnetic stimulation, motor training
INTRODUCTION
Sleep has been shown to have a role in normal neurophysiological processes such as memory and neuroplasticity.1–3 For instance, animal studies support the role of sleep in homeostatic neuroplasticity, a dynamic process of maintaining relative synaptic strength and neuronal excitability.4,5 Given the emerging evidence linking sleep to neuroplasticity and sleep to memory processes, it seems conceivable that deficits in neuroplasticity during wake time would occur in individuals who experience chronic sleep disruption as a result of conditions such as insomnia.
Insomnia is clinically defined as difficulty with sleep initiation, sleep maintenance, or overall diminished sleep quality in association with reports of poor daytime functioning (i.e., fatigue, decreased memory and concentration, daytime distress).6 Recently, the new DSM criteria recognize insomnia as a clinically relevant disorder (i.e., insomnia disorder) warranting independent clinical attention.7 Chronic insomnia (CI) has a prevalence of up to 15% in the general population and is a major public health problem that negatively affects both morbidity and mortality.8 While the exact mechanisms underlying CI remain unclear, it has been postulated that CI is a manifestation of a dysregulation of autonomic and central nervous system arousal networks producing hyperarousal.9–11 Indeed, some studies suggest that this hyperarousal, rather than the chronic loss of sleep, serves as the neurobiological mechanism behind the functional daytime complaints commonly encountered in patients with CI.9,11–13 In this line it has been postulated that hyperactivated brain networks in patients with CI may disproportionately utilize CNS metabolic resources at the expense of other networks critical for memory,8 and potentially other forms of neuroplasticity.
Use-dependent plasticity (UDP) is a form of neuroplasticity that has been extensively studied in healthy young and older individuals14–18 as well as in stroke patients.19 Here, using transcranial magnetic stimulation (TMS), it is possible to observe how execution of voluntary repetitive thumb movements modify the strength of the agonist and antagonist corticomotor representations involved in the training. Thus, this form of neuroplasticity, following motor training, is thought to represent the initial steps of encoding simple motor memories.14,15,20
While some investigations in patients with insomnia have shown impaired sleep-related motor memory (i.e., percentage of improvement from the learning to the retrieval session) using a mirror-tracing task,3 others have failed to find motor learning deficits in these patients.21 Relatively few studies have demonstrated cognitive impairments using standardized neurobehavioral tests despite reported complaints from individuals with insomnia.22 However, methodological issues, heterogeneity of participants, and use of insensitive measures may have accounted for results not revealing major effects.23 In a recent meta-analysis, insomnia was found to be in fact associated with mild to moderate impairments on specific cognitive functions (tasks assessing working memory, episodic memory and problem solving, and some attentional processes), but not on other aspects of attention, perceptual and psychomotor processes, verbal function, procedural memory and certain domains of executive function.23
From a societal impact, individuals with insomnia have been found to have increased work-related errors,24 motor vehicle accidents,25 and decreased work productivity,24,25 which some have suggested may be mediated by cognitive impairment. While the underlying mechanisms for these daytime deficits remain unclear, it is likely that factors such as sleep loss, fatigue, and mood disturbances contribute in some manner. However, since neuroplasticity plays a functional role in cognitive processing, one is left to wonder if abnormal neuroplasti-city may also play a role in the daytime deficits experienced by patients with insomnia. Here, we assessed UDP, a form of plasticity that might involve mechanisms dependent on sleep.26 We evaluated whether patients with CI experience abnormalities of UDP when performing a repetitive thumb movement task. In other words, we did not focus on how sleep shapes neuroplasticity, but rather whether patients with insomnia demonstrate deficits in neuroplasticity during wakefulness.
Since the pathophysiology of CI seems to involve a dysregulation of arousal mechanisms, it is possible that patients have an imbalance of the neurotransmitters responsible for sleep and wakefulness. One particular neurotransmitter is γ-aminobutyric acid (GABA), long speculated and recently implicated to be involved in CI.27–30 Another neurotransmitter that may be involved in CI is glutamate, well known to contribute to the regulation of sleep and wakefulness through arousal.31,32 Given that GABA and glutamate have also been shown to affect UDP,15,33 we assessed neurophysiological measures linked to these neurotransmitters in the same people undergoing the UDP paradigm. Specifically, we tested short- and long-interval intra-cortical inhibition (SICI and LICI), known to reflect GABAA and GABAB mechanisms, respectively.34–36 Additionally, we quantified intracortical facilitation (ICF), thought to reflect excitability-mediated predominately by glutamatergic, but also GABAergic processes.37 We predicted that patients with CI would show decreased UDP relative to healthy matched control individuals, an effect that would be associated to abnormalities in GABAergic and glutamatergic measures.
METHODS
Participants
We recruited a total of 28 participants over 50 years of age who met criteria for CI (n = 18, 6 males, 58 ± 2 years, range 50-71years) and good sleeper controls (GS, n = 10, 4 males; 60 ± 3 years, range 50-74 years). The participants were recruited from a study addressing sleep in osteoarthritis (R01AR054871MTS). Participants with and without knee osteoarthritis completed diagnostic interviews and questionnaires to establish CI and GS status38 according to methods previously described in detail.39 All patients with insomnia had insomnia for longer than one year (Mean = 9.35, SD = 9.75, range 1 to 25 years). Briefly, we excluded patients with unstable major medical and psychiatric disorders that would likely impact sleep, including congestive heart failure, history of cerebral vascular accident, insulin dependent diabetes, cancer, seizure disorder, history of psychotic disorders, dementia, and substance abuse. To rule out psychopathology and substance abuse, participants completed a computerized Structured Clinical Interview screening for DSM-IV Psychiatric Disorders,40 the Center for Epidemiologic Studies Depression Scale (CES-D), and the Mini Mental Status Exam. Participants also completed urine toxicology screening. Following review of the questionnaires and the SCID report, a psychologist conducted a follow-up clinical diagnostic interview selecting relevant full SCID-IV Axis I40 modules. All participants in the related project completed a portable polysomnography (PSG) study to rule out sleep disorders other than insomnia. This data was used for a secondary exploratory analysis. We excluded patients with apnea/hypopnea and/or periodic limb movement (with arousals) indices > 15, derived according to American Academy of Sleep Medicine Criteria.38
All participants signed an informed consent approved by Johns Hopkins Medical Institutional Review Board and in accordance with the declaration of Helsinki. Participants also completed the Pittsburgh Sleep Quality Index (PSQI),41–43 Epworth Sleepiness Scale (ESS),44 and Insomnia Severity Index (ISI)45 before each TMS session and underwent a brief clinical evaluation to confirm they still met the criteria for insomnia or good sleeper. Lastly, a medical history and neurological examination was performed on each participant by a board-certified neurologist.
All participants were right- or left- (not ambidextrous) handed as determined by the Oldfield handedness inventory.46 Menopausal status, use of hormone therapy, and menstrual cycle phase were balanced between groups for women since estradiol and the follicular phase of the menstrual cycle may affect glutamatergic47,48 and GABAergic activity,49,50 as well as change cortical excitability during the menstrual cycle.51 Participants were balanced for osteoarthritis in both groups. Participants were not permitted to use alcohol, caffeine, or tobacco within 24 hours of the TMS study. Additionally, none of the participants were taking opioids, sedative hypnotics, antipsychotics, antidepressants, β-blockers, stimulants, or mood stabilizers.
Study Design
The study included 2 sessions. In the first session we assessed the UDP paradigm in all participants. In the second session, completed within a year from session 1, we tested the neurophysiological markers linked to GABA A, GABA B and glutamatergic activity in 21 participants from the original group (CI: n = 12; 4 males, 56 ± 2 years, range 50-64 years; GS: n = 9, 4 males; 60 ± 3 years, range 50-74 years). Seven participants from the first session (5 with CI) could not be contacted or were not able to participate in the second session. We performed all TMS experiments in the morning (between 9 and 11AM) to control for time of day effects.
At the beginning of first and second TMS sessions, the psychomotor vigilance test (PVT), a validated 10-min task sensitive to sleepiness and alertness, was administered to each participant.52 Participants also recorded the degree of sleepiness on a sleepiness scale pre and post PVT.52
Recording Procedures
Using disposable surface electrodes we recorded EMG activity from the dominant flexor pollicis brevis and extensor pollicis brevis muscles. We set the sampling rate at 2 kHz and monitored the signals online. Using MATLAB (Mathworks Inc. Natick, MA) we analyzed EMG activity offline. To record kinematic measures and calculate movement direction we used a 3-D accelerometer (Kistler Instruments, Amherst, NY) placed over the dorsal aspect of the proximal thumb phalanx.14,16,53
Session 1: Use-Dependent Plasticity (UDP)
Participants sat on a comfortable chair with their dominant arm supported in a neutral position by a splint. A 70-mm figure-of-eight coil (Magstim BiStim2, Whitland, UK) was held over the dominant primary motor cortex (M1) to deliver TMS pulses generating consistent involuntary thumb movements in a consistent direction. This site was recorded as “hot spot” using a frameless neuro-navigation system (BrainSight, Rogue Research, Montreal, Quebec, Canada), which ensured accurate positioning of the TMS coil during the entire session. In this location, we determined the resting motor threshold (rMT) for the muscle agonist during the training (see below), which was defined as the minimum TMS intensity output that evoked motor-evoked potentials (MEPs) of 50 μV in ≥ 5 of 10 trials.54
UDP was assessed using a previously described protocol.14–16,53,55,56 Briefly, each participant received 65 TMS stimuli delivered at 0.1 Hz over the “hot spot” to determine the baseline (Pre) TMS-evoked thumb movement direction (Figure 1). The intensity of the stimulation was set above rMT and titrated to evoked thumb movements in a consistent direction. After this, participants were instructed to perform brisk, voluntary thumb movements for 30 min (3 blocks of 10 min separated by a 2-min rest period) in the direction opposite to the baseline TMS-evoked motions. During training, participants counted the number of movements they performed to ensure that all participants maintained a consistent level of attention.57 After the 30-min training period, another 65 pulses of TMS were delivered over the hotspot as done at baseline to elicit focal, involuntary thumb movements (Post 1, P1). To assess longevity of the effects this assessment was repeated 15 min after training (Post 2, P2). During the Pre, Post 1, Post 2, and in between thumb voluntary movements, we monitored muscle relaxation by EMG signals.
Figure 1.
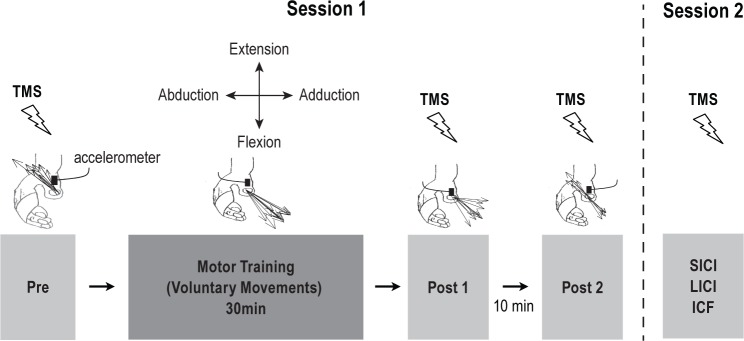
Schematic representation of the experiment setup. Session 1 (i.e., UDP Measures): at baseline (Pre) TMS-evoked movement directions were derived from the first-peak acceleration in the 2 major axes of the movement (extension/flexion and abduction/adduction) recorded by an accelerometer mounted on the proximal phalanx of the thumb. Black arrows indicate the direction of individual TMS-evoked thumb movements (in this case extension and abduction). Motor training was then completed for 30 min, where voluntary thumb movements were performed in a direction opposite to the baseline TMS-evoked movement direction (in this case: flexion and adduction). The direction of TMS-evoked thumb movements was determined again at Post 1 as done during Pre. After a 10-min rest period, a second assessment of TMS-evoked thumb movements was repeated (Post 2). Session 2 (i.e., Paired-Pulse Measures): a subgroup of participants returned on a separate day to undergo paired-pulse TMS measures (i.e., SICI, LICI, and ICF).
To determine changes in TMS-evoked movement direction after the training, we calculated a training target zone (TTZ), defined as a ± 20° window of the mean direction of the training movements.15,55,56 Using this, we calculated our primary outcome measure as the percentage of TMS evoked thumb movements falling within the TTZ. Additionally, we calculated the deltas of percent TMS-evoked movements falling within the TTZ before relative to after the training (Pre to P1 and Pre to P2). To test cortical excitability changes related to the training we assessed the MEP amplitudes in the agonist and antagonist muscles to the trained movement direction. Lastly, we calculated the delta rMT before, relative to after the training (Pre to P1) for the training (agonist) muscle.
Session 2: TMS Measures of Intracortical Inhibition and Excitation
To elucidate potential substrates underlying differences in UDP, we performed a second session in a subset of participants (n = 21). Participants returned to the lab within a year of the first session to be tested for measures of short-interval intracortical inhibition (SICI), long-interval intracortical inhibition (LICI), and intracortical facilitation (ICF)37 of the flexor pollicis brevis muscle. After determining the rMT we established the stimulator intensity required to produce 1 mV MEP responses. SICI and ICF were assessed using paired-pulse TMS with a subthreshold conditioning stimulus (CS) set at 80% of rMT, preceding a supra-threshold test stimulus to elicit ∼1 mV MEPs. LICI was assessed using paired-pulse TMS with a suprathreshold CS followed by a suprathreshold TS. SICI was tested with a 2-msec inter-stimulus interval, LICI with 100 msec, and ICF with 10 msec.58 For each measurement we recorded and then averaged 10 MEPs. SICI, LICI, and ICF were expressed as a ratio of the conditioned MEPs over the mean of the MEP of the test stimulus.
Statistical Analysis
We used independent t-tests and χ2 tests to analyze potential group (CI vs. GS) differences in demographics, sleep surveys (PSQI, ISI, and ESS), PVT performance, motor training counts, delta rMT (pre and post training), and TMS intensity output. A multivariate analysis of variance (MANOVA) was conducted to explore for potential group differences in 4 measures of training kinematics (compound acceleration, radial distance, angular variance, and distance from baseline).
Independent sample t-tests were conducted on Pre measures of TTZ and MEP agonist and antagonist amplitudes to ensure that the groups (CI vs. GS) were not significantly different at baseline. To account for within subject baseline levels, we calculated delta values for our primary outcome measure (the percent of TMS-evoked movements falling in TTZ) and MEP amplitudes (agonist and antagonist) by subtracting baseline values (Pre) from values taken at time point 1 (Post 1). This new variable was referred to as Delta 1 (Post 1 − Pre). To assess our primary outcome measure, we conducted an independent t-test to evaluate group differences in TTZ Delta 1. To examine the longevity of the UDP effect between groups, we examined Delta 2 (Post 2 − Pre). Lastly, we used independent t-tests to test group differences in delta values of MEP amplitudes.
For the second session data, we assessed group differences for ICF, SICI, and LICI using separate independent t-tests for each measurement. All analyses were conducted using the SPSS version 17.0 statistical software package. Data are expressed as means ± SEM and the statistical significance defined as P < 0.05.
RESULTS
Demographic and Training Group Differences
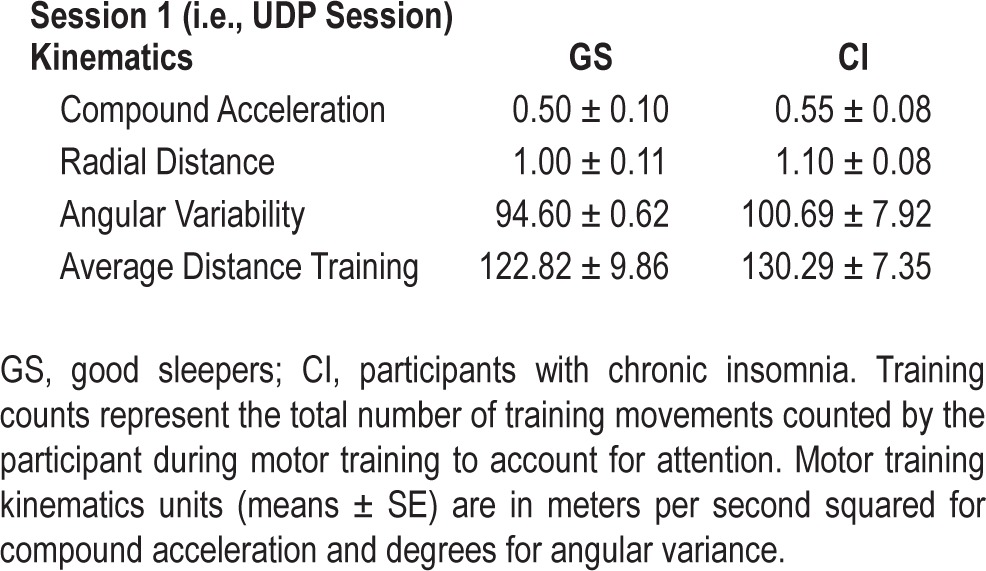
There were no significant differences between groups in age, race, PSG standard indices (i.e., sleep efficiency, total sleep time, sleep latency, and wake after sleep onset), or education level for the UDP experiment (session 1) or for those participating on the second session (Table 1). Participants' psycho-motor vigilance measures were comparable across both days of testing for number of lapses and fastest 10% of responses on the PVT (Table 1). There were no significant differences in pre and post reports of subjective sleepiness or performance on the PVT. Groups performed similarly on motor training counts (the number of training movements each subject counted; Table 1) and kinematic parameters (compound acceleration, radical distance, angular variability, and average distance training; F4,23 = 0.23, P = 0.90; Table 2).
Table 1.
Group demographics and clinical characteristics
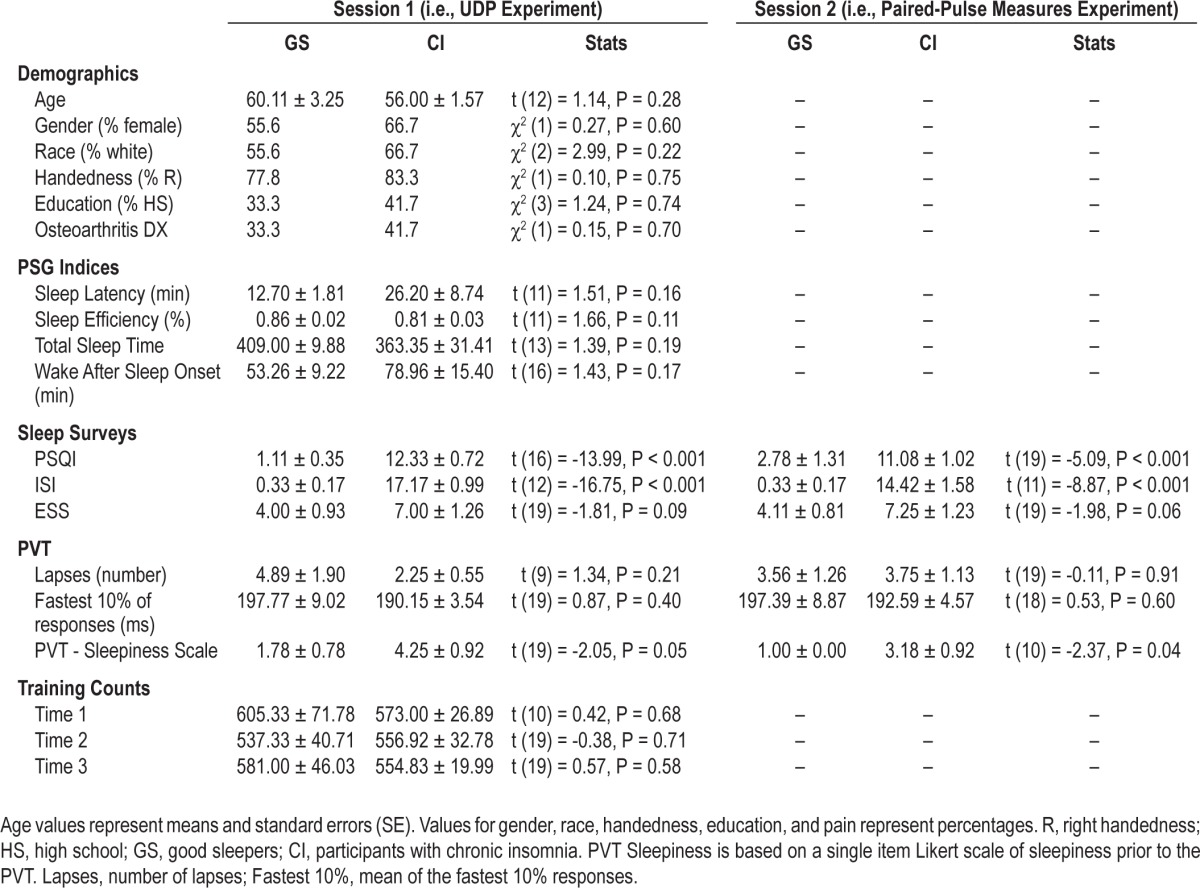
Table 2.
Group differences in motor training counts and kinematics
Session 1: Use Dependent Plasticity Is Increased in Patients with Chronic Insomnia
At baseline, the percentage of TMS-evoked movements falling within the TTZ was similar across groups (Pre) (CI: 0.48% ± 0.23% vs. GS: 0.92% ± 0.52%; t26 = 0.89, P = 0.380). After training, the CI group had significantly greater TMS-evoked movements falling within the TTZ (18% ± 5.81%) than the GS group (3% ± 0.74%; TTZ Delta 1: t20 = -2.51, P = 0.021). However, there were no significant group differences in the TTZ Delta 2 (CI: 7% ± 2.76% vs. GS: 7% ± 5.29%; t26 = -0.15, P = 0.88; see Figure 2), suggesting that the group differences observed after training were not long lasting.
Figure 2.
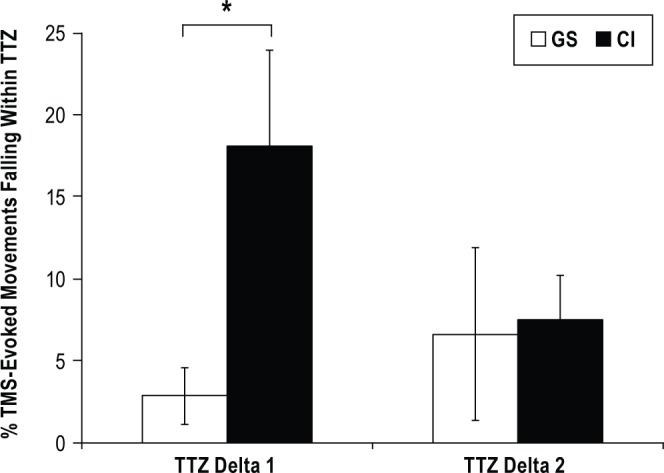
Percentage of movements falling in the training target zone (TTZ). There was a significant increase in movements falling within the TTZ, reflecting increased UDP, from Pre to Post 1 (TTZ Delta1) which was short lived (i.e., TTZ Delta 2, Pre-Post 2) for the chronic insomnia group (CI, black bars) but not for the good sleeper group (GS, white bars). Asterisk denotes P ≤ 0.03. Data are means ± SE.
TMS Measures of Excitability during the UDP Task: Motor Threshold and MEP Amplitudes
Patients with CI (45.83 ± 2.07) had a strong trend towards a higher Pre rMT than GS (39.90 ± 1.21; t26 = -2.02, P = 0.05). There was no significant difference between groups for delta rMT (CI : -0.56 ± 0.28; GS: -0.40 ± 0.64; t26 = 0.26, P = 0.799). There were no significant group differences for the MEP agonist or antagonist amplitudes at Pre (CI: 0.73 ± 0.09 vs. GS: 0.94 ± 0.12; t26 = 1.44, P = 0.163, and CI: 0.85 ± 0.13 vs. GS: 1.20 ± 0.20; t26 = 1.59, P = 0.123, respectively; Figure 3A). When assessing MEP Agonist Delta 1, the CI group had significantly greater amplitudes (0.55 ± 0.17) than the GS group (-0.08 ± 0.14; t25 = -2.39, P = 0.025; Figure 3B). However, these differences were not maintained over time, as determined by the MEP agonist Delta 2 amplitudes (CI: 0.30 ± 0.16 vs. GS: 0.05 ± 0.14; t26 = -1.04, P = 0.308; see Figure 3B). To the contrary, we found no significant group differences in the MEP Antagonist Delta 1 (CI: 0.36 ± 0.20 vs. GS: -0.20 ± 0.17; t25 = -1.84, P = 0.077; Figure 3C) or the MEP Antagonist Delta 2 (CI: 0.02 ± 0.18 vs. GS: 0.08 ± 0.14; t26 = 0.20, P = 0.845; Figure 3C). Lastly, the TMS intensity to evoke involuntary thumb movements was similar for groups (CI: 52.56 ± 2.62 vs. GS: 48.90 ± 1.74; t26 = -0.97, P = 0.342).
Figure 3.
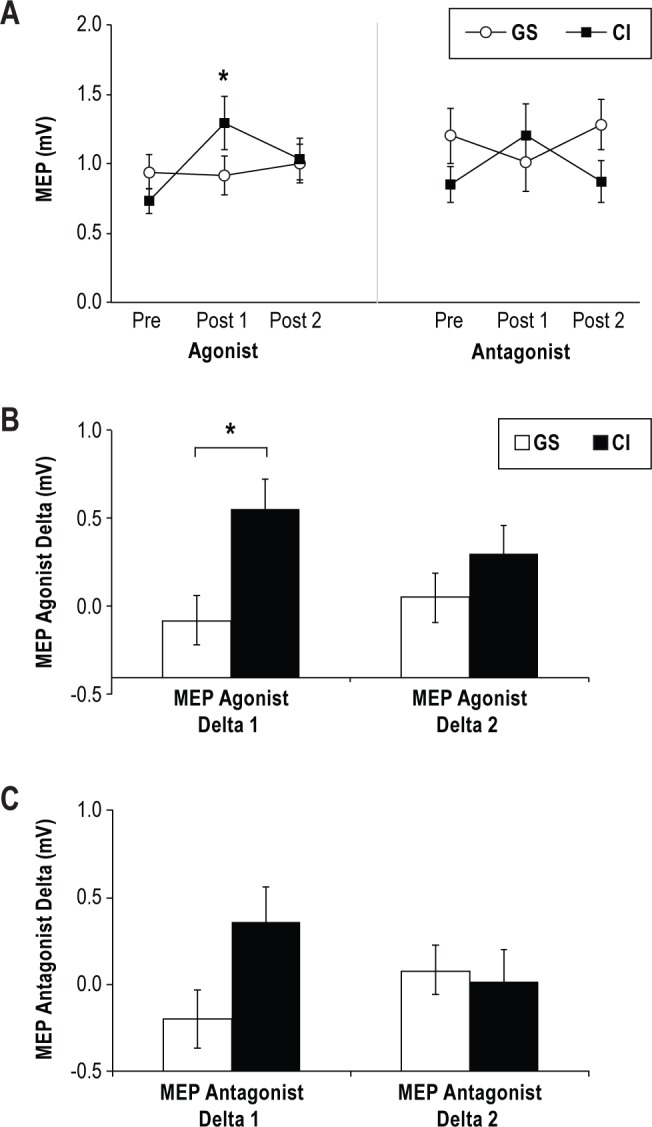
Corticomotor excitability, measured by motor-evoked potential (MEP) amplitude for the agonist and antagonist muscles involved in motor training. (A) Changes in the MEP amplitude recorded from the training for chronic insomnia group (CI, closed black squares) and good sleeper group (GS, open circles) for agonist and antagonist muscles. MEP amplitudes significantly increased in the CI group only. (B) The MEP Agonist Delta 1 (Pre-Post 1) was significantly greater for the CI than GS but there was no significant difference at MEP Agonist Delta 2 (Post 2-Pre). (C) The MEP Antagonist Delta 1 and Delta 2 were not significantly different between groups. Asterisks denote P ≤ 0.03. Data are means ± SE.
Session 2: Measures of ICF, SICI, and LICI
There were no significant group differences in baseline rMT (CI: 46.50 ± 2.98 vs. GS: 41.00 ± 2.30, t17 = -1.28, P = 0.218). We found that ICF was significantly increased in patients with CI relative to GS (CI: 180.77 ± 24.04 vs. GS: 74.42 ± 12.10; t16 = -3.95, P = 0.001; see Figure 4). However, we did not observe differences in SICI or LICI between patients with CI and GS (CI: 50.16 ± 9.70 vs. GS: 51.22 ± 10.33; t19 = 0.07, P = 0.942; and CI: 28.12 ± 9.34 vs. GS: 29.32 ± 11.95; t19 = 0.08, P = 0.937; Figure 4).
Figure 4.
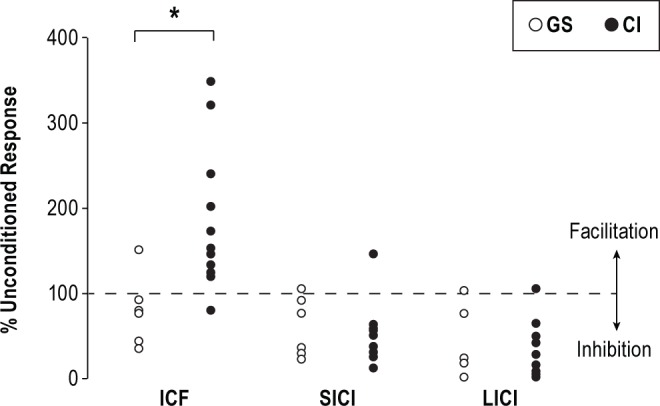
Group Differences in ICF, SICI, and LICI. The chronic insomnia group (CI, black circles) demonstrated a significant increase in intracortical facilitation (ICF), but no difference in short and long interval intracortical inhibition (SICI and LICI, respectively) relative to good sleepers (GS, white circles), supporting increased glutamatergic mechanisms. Asterisk denotes P ≤ 0.01. Data are means ± SE.
Correlation Analysis between Sleep and TMS Measures
We performed exploratory correlations analyses between sleep and neuroplastic measures. We found that greater UDP, particularly Delta 1 TTZ, was significantly associated with greater PSQI for both sessions (UDP Session: r = 0.46, P < 0.05; Paired-Pulse Session: r = 0.50, P < 0.05), ISI (UDP Session: r = 0.46, P < 0.05), and ESS (UDP Session: r = 0.57, P < 0.01) scores, suggesting that worse subjective sleep quality, increased insomnia severity, and increased sleepiness is associated with greater UDP. Greater UDP (Delta 1 TTZ) was also significantly associated with greater PSG sleep latency (r = 0.59, P < 0.05). Greater ICF was significantly associated with greater PSQI (both session 1 [r = 0.60, P < 0.01] and session 2 [r = 0.71, P < 0.01]) and ISI (both session 1 [r = 0.60, P < 0.01] and session 2 [r = 0.74, P < 0.01]). None of the other excitatory measures (SICI and LICI) were associated with the sleep indices.
DISCUSSION
The main result of this study is that patients with CI expressed increased use-dependent plasticity changes relative to GS controls. In addition, we found that patients with CI exhibited increased ICF with no abnormalities in SICI and LICI (i.e., GABAergic activity). Interestingly, a first exploratory analysis suggested that sleep abnormalities were proportional to these neuroplastic measures, where the more severe sleep disorder individuals had larger plastic changes. Altogether, the results point to an association between sleep abnormalities, glutamatergic mechanisms (i.e., ICF), and an increased plasticity response to training.
The general finding that CI is associated with increased UDP seems, at first glance, counterintuitive with the general concept that sleep supports memory and neuroplasticity as well as the clinical reports of memory and cognitive impairment in CI. Since patients with CI report daytime difficulty with memory and there is substantial evidence documenting that sleep is important for memory and plasticity, we originally predicted that patients with CI would experience low UDP changes. Our data however indicate that the relationship between chronic disrupted sleep experienced by those with insomnia and neuroplasticity is not as simple as we initially hypothesized. We attributed this discrepancy to be consistent with hyperarousal theories of insomnia positing that it is a disorder of heightened information processing that interferes with sleep. In other words, we think that the heightened neuroplasticity may be more directly linked with hyperarousal rather than the disrupted sleep itself. This issue will need to be explored in more detail in future studies.
Understanding the biological implications here requires first careful consideration of the classification of CI. Although the CI diagnosis requires a subjective functional impairment expected to affect learning and memory, such impairments have not been consistently documented on neuropsychological and behavioral measures.3,22,59–61 Patients with CI appear to exhibit increased deficits in tasks involving complex processes in attention and in tasks involving high working memory demand, but are able to do simpler tasks without much difficulty.8,24 This may indicate compensatory neuronal changes might occur (increased plasticity) that allow successful performance of simpler tasks (our UDP paradigm). However, these changes are not enough when increased cognitive demand is required to successfully complete a more complex task.8,24
While changes in UDP have been described in healthy individuals,14,16,56,62–65 it was unknown whether patients with chronic disrupted sleep have the capacity to undergo similar changes in response to motor training. Here, we found that patients with CI experience larger UDP changes relative to good sleepers despite performing similar motor training. Of note, this effect was observed only immediately after the training.
Previous work has supported that sleep facilitates memory formation through synaptic downscaling, resulting in a non-Hebbian reset of synaptic weights. Conversely, other work supports that sleep is important for Hebbian synaptic upscaling in order to consolidate new memories. In a recent review article, one new theory proposes that sleep involves neither synaptic downscaling nor upscaling exclusively, but instead a combination of both.66 It is reasonable to suggest that individuals with chronic disrupted sleep, such as those with insomnia, would demonstrate deficits in memory formation, especially since many report difficulty with memory and concentration during the day. Despite this, our results indicate that patients with insomnia show increased UDP. This suggests a defective down-scaling ability in patients with CI, consistent with the synaptic homeostatic hypothesis, resulting in increased excitability and UDP. Alternatively, it is possible that both processes are affected in CI leading to abnormally increased UDP, as observed in this study, while affecting other forms of memory and cognitive performance as described by other investigations.22,23 Interestingly, our findings in patients with CI indicating abnormal plasticity in the possible setting of a hyperaroual state may be relevant in the development of other mood disorders such as depression prevalent in this population.67
Although not significant, our results in the control group are consistent with previous work in older populations55,68,69 showing that healthy older adults have reduced UDP. It is important to stress that our healthy participants were all older than 50 years and underwent an extensive evaluation for sleep and mood disorders besides neurological evaluations. Thus, our data further support that advancing age, rather than other factors such as chronic disrupted sleep may account for the reduced UDP in the healthy individuals. Altogether, the results of the present study support the idea that patients with CI do not “lose” UDP with age. In fact, patients with CI experience similar UDP changes relative to younger healthy individuals.16
It remains unknown whether the increased UDP observed following training in patients with CI is behaviorally beneficial or not. Other pathological conditions have shown that more plasticity is not entirely beneficial.70 For instance, maladaptive plasticity has been observed in patients with phantom limb pain and dystonia.71–73 Therefore, it is possible that the hyperarousal state observed in CI might be linked to heightened conditions of cortical plasticity. Alternatively, it is possible that patients with CI demonstrate an enhanced capability for neuroplasticity as a compensatory mechanism to deal with chronic conditions of abnormal sleep. This may be useful to counteract the consequence of sleep deprivation such as decreased executive functions,23 poor motor performance,3 and daytime sleepiness.74–76 Regardless, it is unclear how heightened UDP may or may not impact other behaviors and learning.
Similar to what has been observed in young healthy individuals,14,16,48,62–65 patients with CI experienced a clear change in the excitability of the cortical representation of the muscles involved in the motor training. In particular, the agonist muscle excitability increased after the training without changes in the antagonist muscle representation. Not surprisingly, these findings were not present in the healthy control group, consistent with the lack of clear UDP changes. The specific excitability change in the representations involved in the task has been considered the physiological marker of this form of UDP, where the relative weight of the different representations change in association to the motor training.14,16,20,55,63
To further explore the potential substrates of the UDP findings from session 1, we invited the same group of participants to return for a follow-up experiment to assess levels of intra-cortical excitability. Since decreased GABA has been reported in CI27 and GABAergic and glutamatergic mechanisms have been shown to impact UDP,15 we investigated GABAergic (i.e., GABAA [SICI], GABAB [LICI]) and glutamatergic (i.e., ICF) mechanisms using paired-pulse TMS measures. Here, we found similar result levels of SICI and LICI in the CI and control groups suggesting that GABAergic neurotransmission cannot explain difference in UDP. Of note, two recent reports using magnetic resonance spectroscopy (MRS) described contradictory findings, where one study showed decreased cortical GABA levels in patients with insomnia,29 and the other found increased brain GABA levels.30 Therefore, further work is required at this time to elucidate the role of GABA neurotrans-mission in patients with CI.
We found that patients with CI have an increased ICF level relative to controls. While ICF reflects glutamatergic and GABAergic neurotransmission,37 our results point to glutamatergic mechanisms (and not GABAergic) as the distinct element in patients with CI, since SICI and LICI were similar across study groups. Glutamate, responsible for synaptic transmission, is important for neural development, neuroplasticity, learning, and memory.77–86 Excessive activation of glutamate receptors has been shown to result in excitotoxicity with irreversible disruption of ion homeostasis and cell death.87 This is in line with many studies reporting that excitotoxic mechanisms contribute to alterations in neuroplasticity88 and memory.89 Thus, our results may suggest that homeostatic mechanisms that keep cortical excitability within a normal physiological range are impaired in CI. Interestingly, increased glutamate and glutamine were found to be increased in the cerebral cortex of sleep deprived individuals,90 supporting the link between glutamatergic abnormalities and sleep loss. Of note, we found in the GS group that SICI and LICI had similar levels as young healthy adults, but exhibited decreased ICF. The effects of healthy aging on cortical excitability have been examined yielding conflicting results. While, one study reported decreased ICF in healthy older adults91 relative to younger individuals, two other studies found no difference.92,93 Differences in age, TMS paradigm, time of day, demographics (i.e., menstrual cycle), and sample sizes might be responsible for the discrepant results.91 Interestingly, human studies using MRS and rodent investigations have also found decreased glutamate with normal aging in the motor cortex,94–96 consistent with our results in healthy controls.
Patients with CI showed higher resting motor thresholds than the control group, yet they exhibited increased ICF. While motor thresholds are thought to reflect membrane excitability,34,97 paired-pulse measures (SICI, ICF, LICI) assess intracortical synaptic excitability.97 Therefore, our findings may suggest that physiological cortical excitability changes occur at different levels in patients with CI. For instance, it is possible that an increase in glutamatergic synaptic activity, as tested by ICF, may be compensated by altered rMT levels. In other words, some of the physiological abnormalities may be the primary process while others may be compensatory in nature. Of note, our findings contradict a report by van der Werf et al., where decreased ICF was found in individuals with insomnia.98 Differences may be attributable to methods or time of day effects, since we studied patients in the morning (08:00-12:00) and the aforementioned study performed their session in the early evening (16:00-18:00). Future work using TMS paradigms to explore diurnal variation in cortical excitability in CI and in normal aging is therefore needed.
This study has some limitations that need mentioning. First, although participants completed sleep surveys the day of the TMS experiments (ISI, PSQI, and EES), PSG data was captured at different times for participants and was not performed in immediate relation to the TMS sessions. As a result, we consider the analysis of PSG data in relation to the TMS measures to be only exploratory. Indeed, the correlations were not controlled for multiple comparisons because such an approach increases type II error or decreases the likelihood for observing a significant relationship.99 Thus, although these results are supportive of the association between sleep disorders and abnormally enhanced plasticity, they need to be taken with caution and should be corroborated in future studies.
Another limitation is that albeit we collected vigilance (i.e., PVT), attention (i.e., counts of TMS pulses), and degree of sleepiness (Likert scale and EES), we did not assess levels of fatigue and or mood changes at the time of the TMS experiments. Lastly, while we excluded participants with unstable major medical and psychiatric disorders that would affect sleep, we included participants who had osteoarthritis and stable medical conditions such as hypertension. As a result, generalizability issues may be potentially associated with this approach.
In summary, we found patients with CI experienced increased capacity to undergo neuroplasticity, at least in relation to use-dependent changes observed after performing a simple motor training. This phenomenon may be explained by the finding that these individuals have increased glutamatergic intracortical activity. Although the exact mechanisms behind the current enhancement of UDP in CI requires further investigations, the results point to potential heightened plasticity which may be linked to a hyperarousal state described in patients with CI. While our results point to differences in UDP in patients with insomnia, it is not possible to discern whether these findings are a consequence of chronic sleep disturbance present in different patients with disrupted sleep (i.e., restless legs syndrome, obstructive sleep apnea, sleep deprivation) or an abnormality that is unique to hyperarousal conditions. These results could lead to development of treatments targeting direct changes in excitability for CI. Future studies should address this intriguing possibility.
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by NICHD, NIH (R01 HD053793 Supplement). R01AR054871 (Sleep Disturbance, Central Pain Modulation, and Clinical Pain in Osteoarthritis); R01AR059410 (Effects of Insomnia on Pain and Function in Osteoarthritis: Role of Inflammation). The authors have indicated no financial conflicts of interest.
REFERENCES
- 1.Stickgold R, Walker MP. Sleep and memory: the ongoing debate. Sleep. 2005;28:1225–7. doi: 10.1093/sleep/28.10.1225. [DOI] [PubMed] [Google Scholar]
- 2.Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 3.Nissen C, Kloepfer C, Feige B, et al. Sleep-related memory consolidation in primary insomnia. J Sleep Res. 2011;20:129–36. doi: 10.1111/j.1365-2869.2010.00872.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci. 2011;34:452–63. doi: 10.1016/j.tins.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- 6.Iber C, Ancoli-Israel S, Cheeson A, Quan SF. 1st ed. Westchester, IL: American Academy of Sleep Medicine; 2007. The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. [Google Scholar]
- 7.American Psychiatric Association. Arlington, VA: American Psychiatric Publishing; 2013. Diagnostic and statistical manual of mental disorders, 5th ed. (DSM-5) [Google Scholar]
- 8.Bastien CH. Insomnia: Neurophysiological and neuropsychological approaches. Neuropsychol Rev. 2011;21:22–40. doi: 10.1007/s11065-011-9160-3. [DOI] [PubMed] [Google Scholar]
- 9.Perlis ML, Giles DE, Mendelson WB, Bootzin RR, Wyatt JK. Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res. 1997;6:179–88. doi: 10.1046/j.1365-2869.1997.00045.x. [DOI] [PubMed] [Google Scholar]
- 10.Monroe LJ. Psychological and physiological differences between good and poor sleepers. J Abnorm Psychol. 1967;72:255–64. doi: 10.1037/h0024563. [DOI] [PubMed] [Google Scholar]
- 11.Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161:2126–8. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- 12.Bonnet MH, Arand DL. Hyperarousal and insomnia. Sleep Med Rev. 1997;1:97–108. doi: 10.1016/s1087-0792(97)90012-5. [DOI] [PubMed] [Google Scholar]
- 13.Riemann D, Spiegelhalder K, Feige B, et al. The hyperarousal model of insomnia: a review of the concept and its evidence. Sleep Med Rev. 2010;14:19–31. doi: 10.1016/j.smrv.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Classen J, Liepert J, Wise SP, Hallett M, Cohen LG. Rapid plasticity of human cortical movement representation induced by practice. J Neurophysiol. 1998;79:1117–23. doi: 10.1152/jn.1998.79.2.1117. [DOI] [PubMed] [Google Scholar]
- 15.Butefisch CM, Davis BC, Wise SP, et al. Mechanisms of use-dependent plasticity in the human motor cortex. Proc Natl Acad Sci U S A. 2000;97:3661–5. doi: 10.1073/pnas.050350297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galea JM, Celnik P. Brain polarization enhances the formation and retention of motor memories. J Neurophysiol. 2009;102:294–301. doi: 10.1152/jn.00184.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawaki L, Yaseen Z, Kopylev L, Cohen LG. Age-dependent changes in the ability to encode a novel elementary motor memory. Ann Neurol. 2003;53:521–4. doi: 10.1002/ana.10529. [DOI] [PubMed] [Google Scholar]
- 18.Floel A, Rosser N, Michka O, Knecht S, Breitenstein C. Noninvasive brain stimulation improves language learning. J Cogn Neurosci. 2008;20:1415–22. doi: 10.1162/jocn.2008.20098. [DOI] [PubMed] [Google Scholar]
- 19.Celnik P, Paik NJ, Vandermeeren Y, Dimyan M, Cohen LG. Effects of combined peripheral nerve stimulation and brain polarization on performance of a motor sequence task after chronic stroke. Stroke. 2009;40:1764–71. doi: 10.1161/STROKEAHA.108.540500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celnik P, Stefan K, Hummel F, Duque J, Classen J, Cohen LG. Encoding a motor memory in the older adult by action observation. Neuroimage. 2006;29:677–84. doi: 10.1016/j.neuroimage.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 21.Backhaus J, Junghanns K, Born J, Hohaus K, Faasch F, Hohagen F. Impaired declarative memory consolidation during sleep in patients with primary insomnia: Influence of sleep architecture and nocturnal cortisol release. Biol Psychiatry. 2006;60:1324–30. doi: 10.1016/j.biopsych.2006.03.051. [DOI] [PubMed] [Google Scholar]
- 22.Shekleton JA, Rogers NL, Rajaratnam SM. Searching for the daytime impairments of primary insomnia. Sleep Med Rev. 2010;14:47–60. doi: 10.1016/j.smrv.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Fortier-Brochu E, Beaulieu-Bonneau S, Ivers H, Morin CM. Insomnia and daytime cognitive performance: a meta-analysis. Sleep Med Rev. 2012;16:83–94. doi: 10.1016/j.smrv.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Kessler RC, Berglund PA, Coulouvrat C, et al. Insomnia and the performance of US workers: Results from the america insomnia survey. Sleep. 2011;34:1161–71. doi: 10.5665/SLEEP.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daley M, Morin CM, LeBlanc M, Gregoire JP, Savard J. The economic burden of insomnia: Direct and indirect costs for individuals with insomnia syndrome, insomnia symptoms, and good sleepers. Sleep. 2009;32:55–64. [PMC free article] [PubMed] [Google Scholar]
- 26.Donlea JM, Ramanan N, Shaw PJ. Use-dependent plasticity in clock neurons regulates sleep need in drosophila. Science. 2009;324:105–8. doi: 10.1126/science.1166657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkelman JW, Buxton OM, Jensen JE, et al. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-MRS) Sleep. 2008;31:1499–506. doi: 10.1093/sleep/31.11.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winkelman J, Buxton O, Jensen J, Benson K, Wang W, Renshaw P. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-Mrs) Sleep. 2009;32:A251. doi: 10.1093/sleep/31.11.1499. (Abstract Supplement) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plante DT, Jensen JE, Winkelman JW. The role of GABA in primary insomnia. Sleep. 2012;35:741–2. doi: 10.5665/sleep.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan PT, Pace-Schott EF, Mason GF, et al. Cortical GABA levels in primary insomnia. Sleep. 2012;35:807–14. doi: 10.5665/sleep.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kodama T, Honda Y. Acetylcholine and glutamate release during sleep-wakefulness in the pedunculopontine tegmental nucleus and norepinephrine changes regulated by nitric oxide. Psychiatry Clin Neurosci. 1999;53:109–11. doi: 10.1046/j.1440-1819.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 32.Disbrow JK, Ruth JA. Differential glutamate release in brain regions of long sleep and short sleep mice. Alcohol. 1984;1:201–3. doi: 10.1016/0741-8329(84)90099-5. [DOI] [PubMed] [Google Scholar]
- 33.Ziemann U, Muellbacher W, Hallett M, Cohen LG. Modulation of practice-dependent plasticity in human motor cortex. Brain. 2001;124:1171–81. doi: 10.1093/brain/124.6.1171. [DOI] [PubMed] [Google Scholar]
- 34.Ziemann U, Lonnecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res. 1996;109:127–35. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]
- 35.Ilic TV, Meintzschel F, Cleff U, Ruge D, Kessler KR, Ziemann U. Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J Physiol. 2002;545:153–67. doi: 10.1113/jphysiol.2002.030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonnell MN, Orekhov Y, Ziemann U. The role of GABA(B) receptors in intracortical inhibition in the human motor cortex. Exp Brain Res. 2006;173:86–93. doi: 10.1007/s00221-006-0365-2. [DOI] [PubMed] [Google Scholar]
- 37.Ziemann U. Pharmacology of TMS. Suppl Clin Neurophysiol. 2003;56:226–31. [PubMed] [Google Scholar]
- 38.Edinger JD, Bonnet MH, Bootzin RR, et al. Derivation of research diagnostic criteria for insomnia: Report of an American Academy of Sleep Medicine work group. Sleep. 2004;27:1567–96. doi: 10.1093/sleep/27.8.1567. [DOI] [PubMed] [Google Scholar]
- 39.Smith MT, Wickwire EM, Grace EG, et al. Sleep disorders and their association with laboratory pain sensitivity in temporomandibular joint disorder. Sleep. 2009;32:779–90. doi: 10.1093/sleep/32.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spitzer R, Gibbon R, Williams J. Structured Clinical Interview for DSMIV Axis 1 Disorders. 1997 Version 2. [Google Scholar]
- 41.Buysse DJ, Reynolds CF, III, Monk TH, Hoch CC, Yeager AL, Kupfer DJ. Quantification of subjective sleep quality in healthy elderly men and women using the Pittsburgh Sleep Quality Index (PSQI) Sleep. 1991;14:331–8. [PubMed] [Google Scholar]
- 42.Cole JC, Motivala SJ, Buysse DJ, Oxman MN, Levin MJ, Irwin MR. Validation of a 3-factor scoring model for the Pittsburgh sleep quality index in older adults. Sleep. 2006;29:112–6. doi: 10.1093/sleep/29.1.112. [DOI] [PubMed] [Google Scholar]
- 43.Carpenter JS, Andrykowski MA. Psychometric evaluation of the Pittsburgh Sleep Quality Index. J Psychosom Res. 1998;45:5–13. doi: 10.1016/s0022-3999(97)00298-5. [DOI] [PubMed] [Google Scholar]
- 44.Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14:540–5. doi: 10.1093/sleep/14.6.540. [DOI] [PubMed] [Google Scholar]
- 45.Bastien CH, Vallieres A, Morin CM. Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med. 2001;2:297–307. doi: 10.1016/s1389-9457(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 46.Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- 47.Woolley CS. Electrophysiological and cellular effects of estrogen on neuronal function. Crit Rev Neurobiol. 1999;13:1–20. doi: 10.1615/critrevneurobiol.v13.i1.10. [DOI] [PubMed] [Google Scholar]
- 48.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–59. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christian CA, Moenter SM. Estradiol induces diurnal shifts in GABA transmission to gonadotropin-releasing hormone neurons to provide a neural signal for ovulation. J Neurosci. 2007;27:1913–21. doi: 10.1523/JNEUROSCI.4738-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59:851–8. doi: 10.1001/archpsyc.59.9.851. [DOI] [PubMed] [Google Scholar]
- 51.Smith MJ, Keel JC, Greenberg BD, et al. Menstrual cycle effects on cortical excitability. Neurology. 1999;53:2069–72. doi: 10.1212/wnl.53.9.2069. [DOI] [PubMed] [Google Scholar]
- 52.Drummond SP, Bischoff-Grethe A, Dinges DF, Ayalon L, Mednick SC, Meloy MJ. The neural basis of the psychomotor vigilance task. Sleep. 2005;28:1059–68. [PubMed] [Google Scholar]
- 53.Cantarero G, Galea JM, Ajagbe L, Salas R, Willis J, Celnik P. Disrupting the ventral premotor cortex interferes with the contribution of action observation to use-dependent plasticity. J Cogn Neurosci. 2011;23:3757–66. doi: 10.1162/jocn_a_00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossini PM, Barker AT, Berardelli A, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord and roots: basic principles and procedures for routine clinical application. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol. 1994;91:79–92. doi: 10.1016/0013-4694(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 55.Celnik P, Stefan K, Hummel F, Duque J, Classen J, Cohen LG. Encoding a motor memory in the older adult by action observation. Neuroimage. 2006;29:677–84. doi: 10.1016/j.neuroimage.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 56.Stefan K, Classen J, Celnik P, Cohen LG. Concurrent action observation modulates practice-induced motor memory formation. Eur J Neurosci. 2008;27:730–8. doi: 10.1111/j.1460-9568.2008.06035.x. [DOI] [PubMed] [Google Scholar]
- 57.Stefan K, Wycislo M, Classen J. Modulation of associative human motor cortical plasticity by attention. J Neurophysiol. 2004;92:66–72. doi: 10.1152/jn.00383.2003. [DOI] [PubMed] [Google Scholar]
- 58.Kujirai T, Sato M, Rothwell JC, Cohen LG. The effect of transcranial magnetic stimulation on median nerve somatosensory evoked potentials. Electroencephalogr Clin Neurophysiol. 1993;89:227–34. doi: 10.1016/0168-5597(93)90100-4. [DOI] [PubMed] [Google Scholar]
- 59.Mendelson WB, Garnett D, Linnoila M. Do insomniacs have impaired daytime functioning? Biol Psychiatry. 1984;19:1261–4. [PubMed] [Google Scholar]
- 60.Fortier-Brochu E, Beaulieu-Bonneau S, Ivers H, Morin CM. Insomnia and daytime cognitive performance: A meta-analysis. Sleep Med Rev. 2012;16:83–94. doi: 10.1016/j.smrv.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Hauri PJ. Cognitive deficits in insomnia patients. Acta Neurol Belg. 1997;97:113–7. [PubMed] [Google Scholar]
- 62.Butefisch CM, Davis BC, Sawaki L, et al. Modulation of use-dependent plasticity by d-amphetamine. Ann Neurol. 2002;51:59–68. doi: 10.1002/ana.10056. [DOI] [PubMed] [Google Scholar]
- 63.Butefisch CM, Khurana V, Kopylev L, Cohen LG. Enhancing encoding of a motor memory in the primary motor cortex by cortical stimulation. J Neurophysiol. 2004;91:2110–6. doi: 10.1152/jn.01038.2003. [DOI] [PubMed] [Google Scholar]
- 64.Floel A, Breitenstein C, Hummel F, et al. Dopaminergic influences on formation of a motor memory. Ann Neurol. 2005;58:121–30. doi: 10.1002/ana.20536. [DOI] [PubMed] [Google Scholar]
- 65.Floel A, Hummel F, Breitenstein C, Knecht S, Cohen LG. Dopaminergic effects on encoding of a motor memory in chronic stroke. Neurology. 2005;65:472–4. doi: 10.1212/01.wnl.0000172340.56307.5e. [DOI] [PubMed] [Google Scholar]
- 66.Ribeiro S. Sleep and plasticity. Pflugers Arch. 2012;463:111–20. doi: 10.1007/s00424-011-1031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baglioni C, Spiegelhalder K, Nissen C, Riemann D. Clinical implications of the causal relationship between insomnia and depression: How individually tailored treatment of sleeping difficulties could prevent the onset of depression. EPMA J. 2011;2:287–93. doi: 10.1007/s13167-011-0079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sawaki L, Cohen LG, Classen J, Davis BC, Butefisch CM. Enhancement of use-dependent plasticity by D-amphetamine. Neurology. 2002;59:1262–4. doi: 10.1212/wnl.59.8.1262. [DOI] [PubMed] [Google Scholar]
- 69.Rogasch NC, Dartnall TJ, Cirillo J, Nordstrom MA, Semmler JG. Corticomotor plasticity and learning of a ballistic thumb training task are diminished in older adults. J Appl Physiol. 2009;107:1874–83. doi: 10.1152/japplphysiol.00443.2009. [DOI] [PubMed] [Google Scholar]
- 70.Celnik PA, Cohen LG. Modulation of motor function and cortical plasticity in health and disease. Restor Neurol Neurosci. 2004;22:261–8. [PubMed] [Google Scholar]
- 71.Flor H, Nikolajsen L, Staehelin Jensen T. Phantom limb pain: a case of maladaptive CNS plasticity? Nat Rev Neurosci. 2006;7:873–81. doi: 10.1038/nrn1991. [DOI] [PubMed] [Google Scholar]
- 72.Quartarone A, Rizzo V, Bagnato S, et al. Rapid-rate paired associative stimulation of the median nerve and motor cortex can produce long-lasting changes in motor cortical excitability in humans. J Physiol. 2006;575:657–70. doi: 10.1113/jphysiol.2006.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flor H. Maladaptive plasticity, memory for pain and phantom limb pain: review and suggestions for new therapies. Expert Rev Neurother. 2008;8:809–18. doi: 10.1586/14737175.8.5.809. [DOI] [PubMed] [Google Scholar]
- 74.Stepanski E, Lamphere J, Badia P, Zorick F, Roth T. Sleep fragmentation and daytime sleepiness. Sleep. 1984;7:18–26. doi: 10.1093/sleep/7.1.18. [DOI] [PubMed] [Google Scholar]
- 75.Seidel WF, Ball S, Cohen S, Patterson N, Yost D, Dement WC. Daytime alertness in relation to mood, performance, and nocturnal sleep in chronic insomniacs and noncomplaining sleepers. Sleep. 1984;7:230–8. doi: 10.1093/sleep/7.3.230. [DOI] [PubMed] [Google Scholar]
- 76.Roehrs T, Diederichs C, Gillis M, et al. Nocturnal sleep, daytime sleepiness and fatigue in fibromyalgia patients compared to rheumatoid arthritis patients and healthy controls: a preliminary study. Sleep Med. 2013;14:109–15. doi: 10.1016/j.sleep.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 77.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 78.Takao M, Morigiwa K, Sasaki H, et al. Impaired behavioral suppression by light in metabotropic glutamate receptor subtype 6-deficient mice. Neuroscience. 2000;97:779–87. doi: 10.1016/s0306-4522(00)00053-1. [DOI] [PubMed] [Google Scholar]
- 79.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–7. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 80.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–9. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 81.Izquierdo I, Bianchin M, Silva MB, et al. CNQX infused into rat hippocampus or amygdala disrupts the expression of memory of two different tasks. Behav Neural Biol. 1993;59:1–4. doi: 10.1016/0163-1047(93)91061-q. [DOI] [PubMed] [Google Scholar]
- 82.Meldrum A, Page KJ, Everitt BJ, Dunnett SB. Age-dependence of malonate-induced striatal toxicity. Exp Brain Res. 2000;134:335–43. doi: 10.1007/s002210000465. [DOI] [PubMed] [Google Scholar]
- 83.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 84.Stephens ML, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Age-related changes in glutamate release in the CA3 and dentate gyrus of the rat hippocampus. Neurobiol Aging. 2011;32:811–20. doi: 10.1016/j.neurobiolaging.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanacora G, Gueorguieva R, Epperson CN, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–13. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 86.Bhagwagar Z, Wylezinska M, Jezzard P, et al. Reduction in occipital cortex gamma-aminobutyric acid concentrations in medication-free recovered unipolar depressed and bipolar subjects. Biol Psychiatry. 2007;61:806–12. doi: 10.1016/j.biopsych.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 87.Gleichmann M, Collis LP, Smith PJ, Mattson MP. Simultaneous single neuron recording of O2 consumption, [Ca2+]i and mitochondrial membrane potential in glutamate toxicity. J Neurochem. 2009;109:644–55. doi: 10.1111/j.1471-4159.2009.05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sapoznik S, Ivenshitz M, Segal M. Age-dependent glutamate induction of synaptic plasticity in cultured hippocampal neurons. Learn Mem. 2006;13:719–27. doi: 10.1101/lm.351706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bettendorff L, Sallanon-Moulin M, Touret M, Wins P, Margineanu I, Schoffeniels E. Paradoxical sleep deprivation increases the content of glutamate and glutamine in rat cerebral cortex. Sleep. 1996;19:65–71. doi: 10.1093/sleep/19.1.65. [DOI] [PubMed] [Google Scholar]
- 91.McGinley M, Hoffman RL, Russ DW, Thomas JS, Clark BC. Older adults exhibit more intracortical inhibition and less intracortical facilitation than young adults. Exp Gerontol. 2010;45:671–8. doi: 10.1016/j.exger.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peinemann A, Lehner C, Conrad B, Siebner HR. Age-related decrease in paired-pulse intracortical inhibition in the human primary motor cortex. Neurosci Lett. 2001;313:33–6. doi: 10.1016/s0304-3940(01)02239-x. [DOI] [PubMed] [Google Scholar]
- 93.Wassermann EM. Variation in the response to transcranial magnetic brain stimulation in the general population. Clin Neurophysiol. 2002;113:1165–71. doi: 10.1016/s1388-2457(02)00144-x. [DOI] [PubMed] [Google Scholar]
- 94.Kaiser LG, Schuff N, Cashdollar N, Weiner MW. Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4 T. Neurobiol Aging. 2005;26:665–72. doi: 10.1016/j.neurobiolaging.2004.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davis JM, Himwich WA. Neurochemistry of the developing and aging mammalian brain. In: Ordy JM, Brizzee KR, editors. Neurobiology of aging. New York: Plenum Press; 1975. pp. 329–57. [Google Scholar]
- 96.Strolin Benedetti M, Cini M, Fusi R, Marrari P, Dostert P. The effects of aging on MAO activity and amino acid levels in rat brain. J Neural Transm Suppl. 1990;29:259–68. doi: 10.1007/978-3-7091-9050-0_25. [DOI] [PubMed] [Google Scholar]
- 97.Conforto AB, Santos RL, Farias SN, Marie SK, Mangini N, Cohen LG. Effects of somatosensory stimulation on the excitability of the unaffected hemisphere in chronic stroke patients. Clinics (Sao Paulo) 2008;63:735–40. doi: 10.1590/S1807-59322008000600005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van der Werf YD, Altena E, van Dijk KD, et al. Is disturbed intracortical excitability a stable trait of chronic insomnia? A study using transcranial magnetic stimulation before and after multimodal sleep therapy. Biol Psychiatry. 2010;68:950–5. doi: 10.1016/j.biopsych.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 99.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1:43–6. [PubMed] [Google Scholar]