

Abstract
Polymeric microparticles have been used widely for sustained drug delivery. Current methods of microparticle production can be improved by making homogeneous particles in size and shape, increasing the drug loading, and controlling the initial burst release. In the current study, the hydrogel template method was used to produce homogeneous poly(lactide-co-glycolide) (PLGA) microparticles and to examine formulation and process-related parameters. Poly(vinyl alcohol) (PVA) was used to make hydrogel templates. The parameters examined include PVA molecular weight, type of PLGA (as characterized by lactide content, inherent viscosity), polymer concentration, drug concentration and composition of solvent system. Three model compounds studied were risperidone, methylprednisolone acetate and paclitaxel. The ability of the hydrogel template method to produce microparticles with good conformity to template was dependent on molecular weight of PVA and viscosity of the PLGA solution. Drug loading and encapsulation efficiency were found to be influenced by PLGA lactide content, polymer concentration and composition of the solvent system. The drug loading and encapsulation efficiency were 28.7% and 82% for risperidone, 31.5% and 90% for methylprednisolone acetate, and 32.2 % and 92 % for paclitaxel, respectively. For all three drugs, release was sustained for weeks, and the in vitro release profile of risperidone was comparable to that of microparticles prepared using the conventional emulsion method. The hydrogel template method provides a new approach of manipulating microparticles.
Keywords: PLGA Microparticle, PVA template method, controlled release, risperidone, methylprednisolone acetate, paclitaxel
1. Introduction
Polymeric microparticle drug delivery systems have been widely explored for controlled delivery of active pharmaceutical ingredients. Microparticles provide several advantages as drug delivery vehicles, such as protection of encapsulated drug from unfavorable environmental conditions and ability to control drug release profile for a specified period of time (1). In particular, the potential to control drug release profile over an extended period of time is one of the most desirable attributes (2). Suitable drug candidates that may benefit greatly from such controlled drug delivery systems based on polymeric microparticles include those that have a broad therapeutic window, require a low daily dose and are used for the long-term treatment of disease.
Poly(lactide-co-glycolide) (PLGA) is probably the most extensively used polymer in microparticle drug delivery systems (1). This copolymer of lactide and glycolide degrade by simple hydrolysis when exposed to an aqueous environment such as inside the human body. PLGA has been used in a host of drug products approved by Food and Drug Administration (FDA), such as Zoladex Depot® (goserelin), Lurpon Depot® (leuprolide), Sandostatin LAR® Depot (octreotide acetate), Nutropin Depot® (somatotropin), Trelstar® (triptorelin), Somatulin® Depot (lanreotide), Risperidal® Consta® (risperidone), Vivitrol® (naltrxone) and Bydureon® (exenatide). PLGAs are available at various molecular weights (or intrinsic viscosities) and lactide/glycolide ratios with either ester end-caps or free carboxylic acid end-caps. The properties of PLGA have been shown to influence important microparticle characteristics, such as the amount of drug loading, loading efficiency and drug release both in vitro and in vivo (3–5). Previous studies have demonstrated that the rate of hydrolysis, and therefore, drug release is heavily dependent on the PLGA molecular weight and monomer composition. Consequently, it is possible to design PLGA-based microparticle drug delivery systems with tailored polymer degradation characteristics and release patterns by varying the PLGA composition.
In addition to polymer composition and properties, there are other formulation- and process-related parameters that may affect microparticle performance. Formulation-related factors include type of organic solvent used, concentration of polymer used, and drug-polymer interactions (3, 6, 7). Various studies have shown that these formulation-related factors affect drug encapsulation efficiency and drug distribution within polymeric matrix, which in turn influences the initial burst release. The initial burst release is one of the major challenges in developing drug-encapsulated microparticle systems. The release of a large bolus of drugs before microparticles reach a steady state release is both therapeutically undesirable and economically ineffective. Therefore, the ability to control and limit the initial burst release is highly sought-after and extensively studied. In addition, there are process-related parameters that can affect the performance of microparticles produced using these methods. Currently, spray drying and emulsion-based methods are well-established and most commonly used to prepare drug-loaded PLGA microparticles. Process-related parameters in these methods that influence drug-loaded microparticle characteristics include the ratio of dispersed phase to continuous phase and the rate of solvent removal/extraction. The factors outlined above and their effects on microparticle performance, however, have been mostly studied in the emulsion-based methods only.
Although emulsion-based and spray-drying methods are widely used, their applicability is restricted by a number of limitations. Techniques such as spray drying may be unsuitable for substances sensitive to heating and mechanical shear of atomization, which narrows the field of applicability for this technique (8). Low product yield due to deposition of materials on the interior surface of drying chamber is yet another common concern for spray drying. For both spray drying and emulsion-based methods particle formation is random and results in microparticles with broad size distribution (9). Microparticle size is an important factor that affects the choice of administration route (10–12), drug encapsulation within the microparticle and therefore drug release profile from the delivery vehicle (13–15). Another common problem with spray drying and emulsion-based methods is low drug loading, often with an average of less than 10% (16–18). Certainly there is room for improvement in microencapsulation techniques.
To address limitations associated with conventional methods of microparticle preparation, we have developed a microfabrication technique for preparation of microparticles. The approach utilizes the unique properties of physical gels that can undergo sol-gel phase transitions or water-soluble polymers that do not dissolve in organic solvents. The approach is collectively called the hydrogel template method (19). The hydrogel template approach allows a more precise control of microparticle size and shape, which translates into narrow size distribution and increased microparticle homogeneity. In addition, the method provides flexibility in producing microparticles of various desirable size ranges. Another improvement over existing methods is the possibility of incorporating a higher amount of drug into the polymeric matrix, since the particle formation process is no longer random, thereby allowing more control over drug encapsulation. The hydrogel template approach does not require the application of excessive heat, mechanical force or any harsh treatment conditions. It is a simple and fast process.
Early method development of the hydrogel template technology and initial study on the effect of the particle size on drug release were discussed in previous publications (19, 20). The main objective of the present study is to evaluate the hydrogel template method for producing drug-loaded polymeric microparticles, with the goal of gaining a better understanding of this method that will ultimately aid in method optimization. Three drugs with different physicochemical properties were used as model compounds in this study. The data obtained were compared and contrasted to microparticles prepared using the conventional emulsion-based technique.
2. Materials and methods
2.1 Materials
Risperidone (RIS) and methylprednisolone acetate (MPA) were purchase from Sigma-Aldrich (St. Louis, MO), paclitaxel (PTX) was supplied by Samyang Genex Corporation (Republic of Korea). Poly(D,L-lactide-co-glycolide) (PLGA) 5050, 6535, 7525 and 8515 (corresponding to lactide:glycolide ratio of 50:50, 65:35, 75:25 and 85:15, respectively) were purchased from Lactel (Pelham, AL). Poly(vinyl alcohol) (PVA, 87~89%, 96%, 98~99% and >99% hydrolyzed) of various typical molecular weight was purchased from Sigma-Aldrich (St. Louis, MO). Benzyl alcohol (BA, analytical reagent grade), ethyl acetate (EA, analytical reagent grade) and methylene chloride (DCM, analytical grade) were purchased from VWR (Batavia, IL). All other chemicals or solvents were of reagent or analytical grade and used as received without further purification.
2.2 Preparation of hydrogel templates
The basic approach to producing gelatin-based templates containing an array of cylindrical posts with pre-determined diameters and heights was described before (19, 20). A similar method was adopted to produce templates in this particular study with some modifications. A silicon wafer master template was constructed by spin-coating with SU8 2010 photoresist (Microchem, Cambridge, MA) at 3500 rpm for 30 s and baking, followed by ultraviolet exposure radiation through a mask containing an array of circular patterns 10 μm in diameter and subsequent developing and drying procedures according to manufacturer’s instructions. The master template thus fabricated contained cylindrical wells 10 μm in diameter and 10 μm in height. Next, the master template was coated with Sylgard 184 elastomer (Dow Corning, Elizabethtown, KY) consisting of approximately 50 g of pre-polymer and 5 g curing agent in a flat-bottomed ceramic dish and cured at 70 °C for 2 hours. The polydimethylsiloxane (PDMS) template was removed carefully from the silicon wafer master template and flushed with ethanol, followed by drying with a nitrogen stream. This intermediate PDMS template was used repeatedly in subsequent experiments to produce templates for making drug-loaded PLGA microparticles. PVA was dissolved at a concentration of 4% (w/v) in a mixture of deionized water and ethanol (40:60 v/v) with constant stirring and heating. The resulting solution was used to evenly coat the surface of PDMS intermediate template. After solvent evaporation and template solidification, the PVA template was gently peeled off the PDMS template and stored in a cool, dry place until ready to use.
2.3 Preparation of polymer-drug solutions
RIS, MPA and PTX were chosen as model poorly water-soluble compounds in this study. The properties of these compounds are presented in Table 1. The compounds were dissolved with the selected type of PLGA polymer in a mixture of BA and EA or DCM. The types of PLGA evaluated varied by lactide-to-glycolide ratio (L:G) and intrinsic viscosity (IV). Other parameters that varied were the concentration of PLGA in solution, drug concentration and ratio of BA to EA. A summary of the composition of drug solutions used in this study is provided in Table 2.
Table 1.
Physicochemical properties of model drugs risperidone, methylprednisolone acetate and paclitaxel
| Compound
|
|||
|---|---|---|---|
| RIS | MPA | PTX | |
| Molecular Structure |
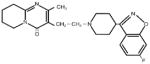
|
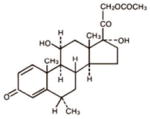
|
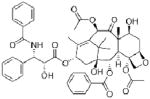
|
| Molecular Weight (g/mol) | 410.5 | 416.5 | 853.9 |
| Aqueous Solubility (μg/ml)* | 211 | 120 | 0.3 |
| LogP* | 2.5 | 1.5 | 3.0 |
| pKa* | 8.24, 3.11 | 12.58 | 10.36 |
Value obtained from DrugBank, based on experimental properties
Table 2.
Summary of formulations and particle size
| PLGA | IV (dL/g) | [PLGA]a (w/v %) | Drug | [Drug]b (w/v %) | Solventc | Average size (μm) | PDI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5050 | 0.26~0.54 | 12.5 | RIS | 7.0 | BA, EA | 10.8 ± 5.0 | 0.12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.55~0.75 | 12.5 | 9.8 ± 0.9 | 0.13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.95~1.20 | 12.5 | 8.1 ± 4.3 | 0.24 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6535 | 0.55~0.75 | 12.5 | RIS | 7.0 | BA, EA | 9.3 ± 1.2 | 0.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7525 | 0.55~0.75 | 12.5 | RIS | 7.0 | BA, EA | 9.5 ± 1.2 | 0.12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8515 | 0.55~0.75 | 12.5 | RIS | 7.0 | BA, EA | 9.2 ± 1.8 | 0.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 12.5 | BA, EA | 7.4 ± 3.4 | 0.29 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 4.3 | BA, EA | 9.4 ± 1.3 | 0.11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 7.0 | BA, EA (30:70 v/v) | 9.3 ± 0.9 | 0.11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 7.0 | BA, EA (40:60 v/v) | 9.4 ± 1.9 | 0.14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 7.0 | BA, EA (60:40 v/v) | 9.6 ± 2.2 | 0.19 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6.2 | 7.0 | BA, EA | 7.7 ± 3.1 | 0.21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25.0 | 7.0 | BA, EA | 9.2 ± 1.1 | 0.13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | 7.0 | BA, DCM | 9.1 ± 1.8 | 0.14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | MPA | 7.0 | BA, DCM | 9.6 ± 0.8 | 0.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12.5 | PTX | 7.0 | BA, EA | 9.7 ± 0.9 | 0.11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
[PLGA]: PLGA concentration; calculated as weight of PLGA per volume of solvent
[Drug]: drug concentration; calculated as weight of drug per total volume of solvent
Volume ratio of solvent system is 50:50 unless otherwise specified
2.4 Preparation of drug-loaded microparticles
Approximately 60 μl of polymer-drug solution was deposited along one edge of each PVA template and spread evenly over its surface at room temperature. Excess solution was carefully scraped away. This process was repeated several times with time in between to allow template to dry. A batch of 20 filled PVA templates were dissolved in a beaker containing 250 ml deionized water and gently stirred by magnetic stir bar at room temperature. The suspension was transferred into conical tubes (45 ml) and centrifuged for 5 minutes (Eppendorf Centrifuge 5804, Eppendorf, Hauppauge, NY) at 5,000 rpm. The pellet was re-suspended and washed at least 3 times, each time followed by centrifuging and re-suspension. The pellet obtained upon final centrifugation was freeze-dried and stored at 4 °C until further use.
2.5 Preparation of physical mixtures
Physical mixtures of drug and PLGA were prepared by weighing out each component and gently mixing for at least ten minutes using mortar and pestle. Prior to mixing with drug, PLGA was grounded into powder form using a ball mill. The amount of drug in the drug-PLGA mixture was approximately 28%, 31% and 32% respectively for RIS, MPA and PTX. These values were equal to the experimentally determined drug loading of microparticles.
2.6 Preparation of emulsions
A measured amount of RIS was dissolved in 5 ml of solvent consisting of 2.5 ml BA and 2.5 ml of EA along with 8515 PLGA. The concentration of polymer and drug (w/v) was 25 % and 7.0 % respectively. The solution (organic phase) was emulsified with 250 ml of 0.5 % (w/v) PVA solution in water (aqueous phase) at 5,000 rpm for 10 minutes. The resulting emulsion was stirred at room temperature by magnetic stirrer for 3 hours to allow droplets to harden by solvent extraction and evaporation. Thus-obtained microparticles were separated by filtration and repeated washings followed by centrifugation at 5,000 rpm for 5 minutes to collect. The collected microparticles were freeze-dried and stored at 4 °C until further use.
2.7 Characterization of microparticles
2.7.1 Particle size analysis
The mean size (volume average particle diameter) and size distribution of drug-loaded PLGA microparticles were determined by a dynamic light scattering analyzer (Microtrac S3500, Microtrac Inc., Largo, FL) equipped with appropriate analysis software (Microtrac Flex Version 10.3.3). Size measurements were performed in triplicate following suspension of microparticles in redistilled water at 25 °C. To prepare the samples for analysis, approximately 2 mg of freeze-dried microparticles were suspended in 5 ml of deionized water and vortexed for 5 minutes followed by sonication for 30 seconds.
2.7.2 Shape and morphology
The shape and surface morphology of microparticles were characterized by fluorescence microscopy and scanning electron microscopy (SEM) respectively. For fluorescence microscopy, a small amount of freeze-dried microparticles were re-suspended in deionized water and deposited onto the surface of a clean glass slide using a pipette. The sample was air-dried prior to viewing on the fluorescence microscope (Olympus BX51, Olympus, Center Valley, PA). For surface morphology, FEI Nova™ NanoSEM (FEI, Hillsboro, OR) was used. Samples for characterization were prepared by carefully depositing a small amount of freeze-dried microparticle powder onto the surface of aluminum stubs and coating with platinum under vacuum conditions.
2.7.3 Physical stability
Solid form of drug, polymer, physical mixtures, microparticles from emulsion and hydrogel template method were characterized with a powder X-ray diffraction (PXRD) apparatus (Siemens Bruker D5000, Bruker AXS, Madison, WI) using CuK radiation at 30 mA and 45 kV (scanning rate 0.4°/min), and diffraction angles (2θ) of 3–40°. Samples for PXRD were prepared by crushing a desired amount of drug, polymers, physical mixture or microparticles with mortar and pestle before adding to the sample holder. Excess powder sample was scraped away and the surface of the powder sample was leveled with a glass slide. The procedure was repeated for samples that were stored for 1 – 2 months at 4 °C for physical stability assessment.
2.7.4 Drug loading
An accurately measured amount (5 mg) of freeze-dried drug-loaded microparticles was dissolved in 1 ml of DCM and diluted with 9 ml of methanol. The solution was vortexed to ensure thorough mixing. Following centrifugation for 5 minutes at 5,000 rpm, the supernatant containing dissolved drug was collected. The pellet containing precipitated PLGA was washed with methanol several times and the washings were combined with supernatant. This solution was rotary evaporated then re-dissolved by 10 ml mobile phase for analysis by HPLC. The HPLC system (Agilent 1100 series, Agilent, Santa Clara, CA) was equipped with autosampler, in-line degasser and UV absorbance detector. The separation method for each drug is outlined below. Drug content was calculated using the external standard method. Drug loading and encapsulation efficiency were calculated by the following equations:
| (1) |
| (2) |
2.7.4.1 Risperidone
Separation was achieved using a X-Terra C-18 (250 mm × 4.6 mm) analytical column from Waters (Milford, MA) at a flow rate of 1 ml/min, a detection wavelength of 278 nm and injection volume of 65 μl. The mobile phase consists of methanol: ammonium acetate (90:10 (v/v), pH adjusted to 7 with glacial acetic acid). Samples and mobile phase were filtered through a 0.22 μm syringe filter and a 0.48 μm membrane filter respectively prior to use.
2.7.4.2 Methylprednisolone acetate
Separation was achieved using a C-12 Sinergy MAX RP (150 mm × 2.0 mm) analytical column (Chemtek Analytica, Italy) protected by a C-12 RP guard column at a flow rate of 0.2 ml/min, a detection wavelength of 278 nm and injection volume of 5 μl. The mobile phase consists of 50% water containing 0.01% formic acid (A) and 50% acetonitrile (B). The A:B ratio was maintained for 6 minutes, then the concentration of B was linearly increased to 100% B in 7 minutes, followed by another 15 minutes of isocratic elution (washing). The composition of the eluent was then restored to the original condition of A:B = 50:50 (v/v) and re-equilibrated for 10 minutes before the following injection. The column compartment was maintained at 30 °C.
2.7.4.3 Paclitaxel
Separation was achieved using a Phenomenex C-18 (250 mm × 4.6 mm) analytical column (Torrance, CA) at a flow rate of 1 ml/min, a detection wavelength of 228 nm and injection volume of 20 μl. The mobile phase consists of water, acetonitrile and methanol (40:30:30 v/v). Samples and mobile phase were filtered through a 0.22 μm syringe filter and a 0.48 μm membrane filter respectively prior to use.
2.7.5 Thermal analysis
Differential scanning calorimetry (DSC) was carried out using a DSC Q10 calorimeter (Mettler Toledo, Greifensee, Switzerland). Approximately 2 mg of sample was weighed into aluminum pans and hermetically sealed. As a reference, an empty aluminum pan was used. Samples were cooled down to −10 °C and then heated up to 180 °C at a rate of 10 °C/min. For Tg determination, the data were analyzed using STAR software (Mettler Toledo, Greifensee, Switzerland) and the midpoint of the corresponding glass transition was evaluated. Tg was determined in triplicate for every sample.
2.8 In vitro release characterization
For RIS- and MPA-loaded microparticles, approximately 10 mg of microparticles was accurately weighed into glass vials and re-suspended in 2 ml of PBS/Tween-20 (0.05%) (pH 7.4). The suspension was transferred carefully into a membrane cassette (Slide-A-Lyzer G2, Thermo Scientific, Rockford, IL) with molecular weight cut off of 3,500. The cassettes were incubated with 500 ml of PBS/Tween-20 (pH 7.4) at 37 °C in an orbital shaker set at 100 rpm throughout the duration of the release experiment. At predetermined intervals, 5 ml samples were withdrawn from the release media and replaced with 5 ml of fresh buffer to maintain sink conditions. The samples were collected in individual glass vials and stored at 4 °C till further analysis. Concentrations of RIS or MPA in the samples were determined by HPLC analysis using the method outlined previously. For each formulation, the experiment was performed in triplicate.
For PTX-loaded microparticles, due to extremely low aqueous solubility of the drug and concentration detection limits of the HPLC method, the total volume of buffer used in release studies was reduced. Approximately 2.5 mg of microparticles was accurately weighed into glass vials and re-suspended in 1 ml of PBS/Tween 20 (pH 7.4). The suspension was transferred carefully into a small-volume membrane cassette. The cassette was incubated with 10 ml of PBS/Tween 20 at 37 °C in an orbital shaker set at 100 rpm throughout the duration of the release experiment. At predetermined intervals, 5 ml samples were withdrawn from the release media and replaced with 5 ml of fresh buffer. The samples were collected in individual glass vials and stored at 4 °C till further analysis. Concentration of PTX in the samples was determined by HPLC analysis using the method outlined previously. The experiment was performed in triplicate.
2.9 Solubility determination
The saturation solubility of RIS, MPA and PTX were determined in BA, EA and DCM as follows: an excess amount of each drug was transferred into glass vials containing 2 ml of a specific solvent. The glass vials were equipped with screw-on caps to prevent solvent evaporation. The vials containing the drug in the solvent were left on an orbital shaker at room temperature under agitation. After 1 day, the solvent was removed and filtered through 0.22 μm syringe filter. The concentration of the drug in solution was determined by HPLC according to the method outlined in section 2.7.4.
2.10 Statistical analysis
Particle size measurements, drug loading and in vitro release experiments were performed in triplicate. The results are expressed as mean ± standard deviation. Statistical differences among groups were evaluated by unpaired t-test (between two groups) or one-way ANOVA (between multiple groups) with Tukey’s multiple comparison test. When p<0.05, statistical significance was considered to be achieved.
3. Results and Discussion
3.1 Development of the hydrogel template method
The ability to precisely control and manipulate microparticle geometry is highly valuable as the shape and size of drug carriers have been shown to have an impact on biological processes such as vasculature, circulation time, targeting efficiency, cellular uptake and subsequent intracellular transport for therapeutic delivery (21). While the hydrogel template approach produces microparticles of pre-defined size and shape, it requires further development for practical applications, such as scale-up production of microparticles. In the original method, gelatin was used as a hydrogel template material, because at a certain concentration it possesses a mechanical strength sufficiently high to withstand physical manipulation during template preparation and template filling. At the same time gelatin has a gel-to-sol transition temperature mild enough not to adversely affect a broad range of loaded drug. However, these gelatin templates cannot be stored for prolonged periods of time without losing shape of the delicate patterns and encountering contamination issues, which increases the difficulty associated with streamlining this process for scale-up production. To overcome this issue, PVA was used in place of gelatin as a template material. This water-soluble polymer has been used extensively as a stabilizer in nano- and micro-particle preparations, has excellent film-forming properties due to its high tensile strength and flexibility, and is resistant to commonly used organic solvents for filling templates (22, 23). In addition, PVA templates are advantageous compared with gelatin templates, since the former can be stored for an extended period of time in a dry place without losing shape of the patterns or encountering contamination issues. A large number of PVA templates can be fabricated and stored until ready to be filled by polymer-drug solutions.
In the current study, different types of PVA, as defined by the percent hydrolyzed and molecular weights, were explored for use as a template material for the hydrogel template method. The type of PVA used in the final process was chosen based the duration of template preparation time and stability of produced microparticles, as determined by retention of designed particle shape. During this process, the PVA molecules with 87~89% hydrolyzed, 96% hydrolyzed, 98~99% hydrolyzed and 99+% hydrolyzed were chosen for evaluations because they represent the range of commercially available categories of PVA. A mixture of water and ethanol was used to dissolve the PVA in preparation of template solution because a solution based on water alone was found to shrink on the surface of master PDMS template, which affected quality of templates produced. From preliminary evaluations, 87~89% hydrolyzed PVA with average molecular weight of 146,000~186,000 Da (viscosity = 40~50 cP) was found to be the most suitable type of PVA with a preparation time of 12 hours and good microparticle stability. Based on our previously defined selection criteria, 98~99% and 99+% hydrolyzed PVA with average molecular weight of 146,000~186,000 Da were not selected due to long solubilization time (>3 days at 80 °C). This is possibly due to high viscosity of the two polymers (55~65 cP), which is known to increase difficulty with powder dispersal. This in turn leads to formation of lumps that cannot be readily broken down by agitation, a factor that directly results in long solubilization times. When PVA with lower average molecular weight (13,000~23,000 Da, 31,000~50,000 Da) was used, solubilization time decreased dramatically to 5~6 hours at 80 °C due to the decrease in viscosity of polymer. However, from SEM images we can see that microparticles produced from these templates showed shape changes from the designed patterns on template (Figure 1). This result can be rationalized based on decreased ability of PVA to stabilize microparticles. Therefore, in all subsequent studies, we chose 87~89% hydrolyzed PVA with average molecular weight of 146,000~186,000 Da as the optimal template polymer.
Figure 1.

Scanning electron microscope images of microparticles prepared using 87~89% hydrolyzed PVA templates of various average molecular weights: (A) 13,000~23,000 Da; (B) 31,000~50,000 Da; and (C) 146,000~186,000 Da.
3.2 Effect of formulation and process parameters on microparticle size and morphology
Results from particle size characterization are presented in Table 2. With the exception of 3 out of the total of 16 formulations tested, the average particle size measured did not deviate more than 10% (1 μm) from the target size of 10 μm predefined by the pattern on the template. In addition, polydispersity indexes (PDI) for these groups were well below 0.5, a fact that indicates low variance in particle size distribution. In contrast, commonly used emulsion methods have been known to produce microparticles with very wide size distribution. Keeping formulation parameters the same, we used a emulsion method to prepare the microparticles. Although we were able to obtain microparticles with an average particle size of 10.5 μm, the size distribution ranged from 2 to over 30 μm, and PDI was significantly (p < 0.05) higher than the PDI of microparticles produced using the hydrogel template method. Particle size is one of the most important characteristics of microparticle-based drug delivery systems. It is a well-studied fact that particle size has important effects on microparticle degradation rate, drug loading, and drug release properties (20). Therefore, it is important to be able to control average particle size as well as size distribution of a microparticle-based drug delivery system.
Average particle size and size distribution of microparticles were not dependent on the type of PLGA (as defined by L:G ratio) or the drug type. The average particle size and PDI of samples made from 5050, 6535, 7525 and 8515 PLGA of the same intrinsic viscosity did not differ significantly (p<0.05) from each other. Similarly, the same conclusion can be reached when comparing microparticles encapsulating RIS, MPA and PTX. A robust method is advantageous because it broadens the scope of application, i.e., the method can be applied towards a larger chemical design space. However, results from Table 2 also show that size was dependent on intrinsic viscosity of polymer and polymer/drug concentration in solution. As we can see from Table 2, PLGA with intrinsic viscosity of 0.95~1.20 dL/g, a polymer-drug solution with 12.5% drug concentration and a solution with 6.2% PLGA concentration produced microparticles with −19%, −26% and −23% size deviations from target 10 μm, respectively. Observations from subsequent SEM images of these samples showed fragmented, irregularly shaped and/or microparticles with cup-like structures containing hollow centers (Figure 2). Possible causes include reduced ability of polymer-drug solution to spread evenly on the surface of template (in the case of high viscosity PLGA or high drug concentration) and inward collapse of microparticles with hollow centers (in the case of low PLGA concentration). The formation of microparticles with cup-like structures when the polymer concentration is low can be attributed to the capillary flow induced by non-uniform evaporation of the polymer-drug solution during template filling process (24). This so-called ‘coffee ring effect’ has been shown to depend on factors such as solute concentration (25). A possible method to be explored for overcoming this limitation in the hydrogel template method is to increase the number of times template filling is carried out, such that the hollow center is gradually filled.
Figure 2.

Scanning electron microscope images of RIS-containing microparticles prepared using 12.5% PLGA with intrinsic viscosity of 0.95~1.20 dL/g (A), PLGA-RIS (12.5%–12.5%) solution (B), and PLGA-RIS (6.2%–7.0%) solution (C).
The shape and morphology of microparticles prepared using the hydrogel template method as characterized by fluorescent microscopy and SEM is presented in Figure 3. From Figure 3-A showing microparticles prior to dissolving the PVA template in water we can see that the microparticles showed good conformity to the shape designed by the hydrogel template. However, the ability of microparticles to maintain original cylindrical shape as designed by the template was found to depend on the intrinsic viscosity of PLGA. SEM images show that PLGA with intrinsic viscosity of 0.55~0.75 dL/g was able to form cylindrical microparticles with good conformity to the original template design after dissolving templates in water and harvesting free microparticles (Figure 3 B–D). The same formulation produced spherical microparticles by the emulsion method. In contrast, microparticles prepared from PLGA with intrinsic viscosity of 0.24~0.54 dL/g was not able to maintain their cylindrical shapes. SEM images show the formation of more rounded microparticles that do not resemble that of the original template design (image not shown). This result can be attributed to the fact that PLGA with lower intrinsic viscosity has shorter length polymer chains and therefore more space between each polymer chain. When the templates were dissolved to collect free microparticles, it is easier for water molecules to enter the polymeric matrix and cause the microparticles to swell. This hypothesis is supported by the slightly larger measured size of microparticles prepared using lower intrinsic viscosity compared with those prepared using a higher intrinsic viscosity, even though the difference was not statistically significant (p > 0.05). The cylindrical shape microparticles may have slightly different drug release properties from the spherical microparticles, but it would not really matter as long as the drug release rate can be controlled.
Figure 3.

Fluorescent (A and B) and scanning electron microscope (C and D) images of microparticles prior to (A) dissolving PVA templates and after collecting and drying (B–D).
Another possible factor that may have led to changes in microparticle shape is the glass transition temperature (Tg) of polymer. Studies have shown that during microparticle formation the structure of polymeric microparticles is strongly affected by the Tg of the polymer, which marks the change between the rigid, glassy and the more flexible, rubbery state (26). It can be assumed that the shape and morphology of the microparticle is only formed in the rubbery state, providing that Tg is below the processing temperature. Moreover, Tg of polymer has been shown to decrease with increasing levels of residual solvent in the microparticle system (27). In our case the shape difference between microparticles prepared using 0.24~0.54 dL/g PLGA and 0.55~0.75 dL/g PLGA was not thought to be caused by differences in Tg of PLGA. The Tg of the two types of PLGA measured by DSC were 48.8 °C and 51.2 °C, respectively, which were within the range specified by the manufacturer of PLGA. The Tg of PLGA is expected to depreciate the most from the measured Tg immediately after template filling and prior to air-drying. However, the extent of Tg change is expected to be similar for both types of PLGA regardless of intrinsic viscosity, as has been shown in other studies that Tg depression as a result of residual solvent is not dependent on PLGA molecular weight, which has direct correlations with intrinsic viscosity (27). Therefore, we do not expect significant differences between Tg of 0.24~0.54 dL/g and 0.55~0.75 dL/g PLGA during production and processing of microparticles using the hydrogel template method. The measured Tg of microparticles after collecting and drying was approximately 45±1 °C. The slight depression in Tg before and after processing may be due to low levels of residual solvent in the microparticles. The microparticles were freeze-dried before use, and thus, the residual solvent, if it exists, may be very low. The potential effect of the residual solvent on drug release, however, needs to be studied in detail in the future.
In general, the hydrogel template method enabled us to prepare microparticles with relatively smooth, non-porous surfaces. The presence of pores on microparticle surfaces has been shown to increase the rate of drug release from microparticles by acting as transport pathways for drug and water molecules (28). Furthermore, pores at the surface of microparticles have been shown to correlate with initial burst release of drugs from microparticle drug delivery systems, and those with more pores tend to show a more rapid drug release (29). Therefore, from a controlled drug delivery perspective, it is often desirable to control pore size if not eliminate the presence of pores entirely. The surface characteristic of microparticles is strongly dependent on the solvent removal process, i.e., solvent evaporation and extraction kinetics (3). In the hydrogel template method, solvent removal occurs primarily through solvent evaporation during the template filling and drying stage, with limited supplementary solvent extraction when the template is exposed to water and microparticles become briefly suspended in the aqueous phase. The ability of the hydrogel template method to prepare microparticles with relatively smooth, non-porous surfaces is most likely due to slow solvent evaporation in the preparation process as well as minimized contact with water during the particle formation process. To control the rate of solvent evaporation, we used a binary co-solvent system consisting of BA (boiling point: 205 °C) and EA (boiling point: 77.1 °C). These solvents were selected based on the criteria that the solvents should be able to dissolve both the polymer and the drug at the concentrations used while having no detrimental effect on the integrity of template material. Slow solidification allows microparticles to remain soft for a longer period, which leads to particle compaction within the template. To further investigate, DCM (boiling point: 39.6 °C) instead of EA was used in combination with BA to prepare the polymer-drug solution. This binary co-solvent system is expected to have a lower boiling point than the BA and EA system, which promotes more rapid solvent evaporation. SEM study of the microparticles prepared using this new solvent system showed large pores forming on the surface of microparticles (Figure 4), which is consistent with the prediction based on faster solvent evaporation caused by boiling point depression of the new solvent system.
Figure 4.

SEM image showing large pores formed on the surface of microparticles prepared by using a co-solvent containing DCM.
3.3 Effect of formulation and process parameters on drug loading and encapsulation efficiency
Using the hydrogel template method, we successfully encapsulated model drugs, RIS, MPA and PTX, into a PLGA matrix. X-ray diffractograms of drug-loaded microparticles prepared by the hydrogel template method in comparison with drug alone, polymer alone or physical mixtures are shown in Figure 5. The X-ray diffractogram suggests that the process of encapsulating the drug in microparticles using the hydrogel template method resulted in the loss of crystallinity of the drug. The emulsion method also produced microparticles that encapsulated drug in amorphous form. For RIS, dominant peaks at 2θ angles between 13° and 15° and between 18° and 22° were observed for pure drug. The polymer PLGA is predominantly amorphous as indicated by the slight shift above baseline and lack of any dominant peaks. The peaks characteristic of RIS at 2xetas; angles of 14° and 21.5° were still observable for the physical mixture of RIS and PLGA at a concentration of 10% (w/w) RIS in the physical mixture. However, the peaks were lost in the X-ray diffractogram of microparticles prepared by the hydrogel template method, indicating amorphization of RIS. The same general trend can be observed for model drugs MPA and PTX (Figure 5-B and 4-C), indicating that all three model drugs were encapsulated in the amorphous form within the PLGA matrix. Conversion of the drug from crystalline into an amorphous form is also commonly observed in other microparticle preparation methods such as spray-drying and emulsion-based methods. Potential advantages are enhanced dissolution and saturation solubility of the drug, properties that are particularly important for delivery of poorly water soluble drugs such as the model drugs studied here. However, stability is a major concern since amorphous active pharmaceutical ingredients may undergo recrystallization during further processing and/or storage (30). The physical state of the encapsulated drug after storage at 4 °C for over 2 months was characterized by XRD experiments and comparing collected diffractograms with X-ray diffractograms obtained immediately after collecting and drying the microparticles. The results show overlap of the diffractogram patterns, which suggests that devitrification did not take place (Figure 5-A).
Figure 5.

X-ray diffractograms comparing pure drug and encapsulated form of RIS (A), MPA (B), and PTX (C).
A comparison of drug loading of RIS is presented in Figure 6 As was expected, the drug loading was found to depend on drug, polymer and solvent interactions. Figure 6-A shows that RIS drug loading increased when lactide content of PLGA increased. Drug loadings were 7.09±2.6% and 26.2±5.4%, respectively, for 5050 PLGA and 8515 PLGA, corresponding to the encapsulation efficiencies of 20.3% and 74.6%, respectively. Increasing the lactide content of PLGA most likely enhanced hydrophobic interactions between polymer and RIS, thus, resulting in higher drug loading and encapsulation efficiency. This is predicted by the interaction parameter, which can be calculated based on the following equation:
Figure 6.

Comparison of drug loading trends for RIS formulations A) effect of PLGA type based on L:G ratio; B) effect of PLGA concentration; C) effect of drug concentration; D) effect of solvent ratio. *: p<0.05; **: p<0.001
Where χ is the interaction parameter, δd and δp are solubility parameters for drug and polymer, respectively, and Vd is the molar volume of drug. With the δd value of 17.5 for RIS, 21.7 for 8515 PLGA and 22.3 for 5050 PLGA (31), the calculated χ values for RIS-8515 PLGA and RIS-5050PLGA are approximately 2.27 and 2.97, respectively. The lower value of RIS-8515 PLGA indicates better polymer-drug compatibility. Increasing PLGA concentration in the polymer-drug solution resulted in higher drug loading as well as encapsulation efficiency (Figure 6-B). At a polymer concentration of 25.0% in the polymer-drug solution, the drug loading achieved was 28.4±2.7%. This trend has also been demonstrated for emulsion-based methods (32) and can be attributed to increased viscosity of the solution such that drug diffusion through the polymer matrix is limited (33). Furthermore it has been shown that a low polymer concentration may result in polymer microparticles with drug crystals penetrating the polymer shell, which leads to drug loss during washing and further processing (34). Increasing the concentration of drug in polymer-drug solution did not result in higher drug loading or encapsulation efficiency beyond a RIS concentration of 7.0%. As we can see in Figure 6-C, from a drug concentration of 4.3% to 7.0% a corresponding increase in drug loading from 19.3±3.5% to 26.2±2.1% was seen, but further increasing the drug concentration to 12.5% did not lead to a significant change in drug loading. There were no significant differences between encapsulation efficiency at 4.3% and 7.0% drug concentration (77.2% and 74.6%, respectively), but encapsulation efficiency at 12.5% drug concentration was significantly lower at 49.8%. This observation again emphasizes the importance of polymer-drug compatibility in amount of drug loading and encapsulation efficiency.
Drug loading and encapsulation efficiency were also found to be affected by the solvent ratio (Figure 6-D). In the current study we found that increasing volume ratio of EA increased drug loading and encapsulation efficiency, though the increase was not significant beyond BA:EA volume ratio of 50:50. The higher drug loading and encapsulation efficiency seen when BA:EA volume ratio decreased is likely due to less surface associated drug. When solubility experiments were carried out, we found that the solubility of RIS is much lower in EA than in BA. Typically, migration of the drug during drying and storage steps can lead to heterogeneous distribution of drug molecules within the polymeric matrix (35). It is reasonable to assume that as BA is removed from the microparticles, it carries with it a certain amount of drug which may easily be lost to the aqueous environment or during washing steps. Lowering the BA:EA ratio decreases the amount of RIS able to diffuse along with BA to the external surface of microparticles, thereby increasing the drug loading and encapsulation efficiency. In addition, EA evaporates faster than BA at room temperature due to a much lower boiling point. The rate of solvent removal affects microparticle solidification; that is, fast solidification (fast solvent removal) may impede drug diffusion to the surface by fast formation of a dense polymer matrix. This may reduce drug diffusion to the surface and increase drug loading and encapsulation efficiency.
It is interesting to note that faster solvent removal does not automatically equate higher drug loading and encapsulation efficiency. For RIS, using EA-BA solvent system resulted in higher drug loading and encapsulation efficiency than DCM-BA solvent system (Figure 7). Due to the lower boiling point of DCM compared to EA, it is expected to be removed at a more rapid rate compared to EA. In this case, solvent removal was so rapid that it facilitated the formation of numerous surface pores, which was confirmed by SEM imaging. The presence of surface pores may have increased drug loss to the aqueous environment during washing.
Figure 7.
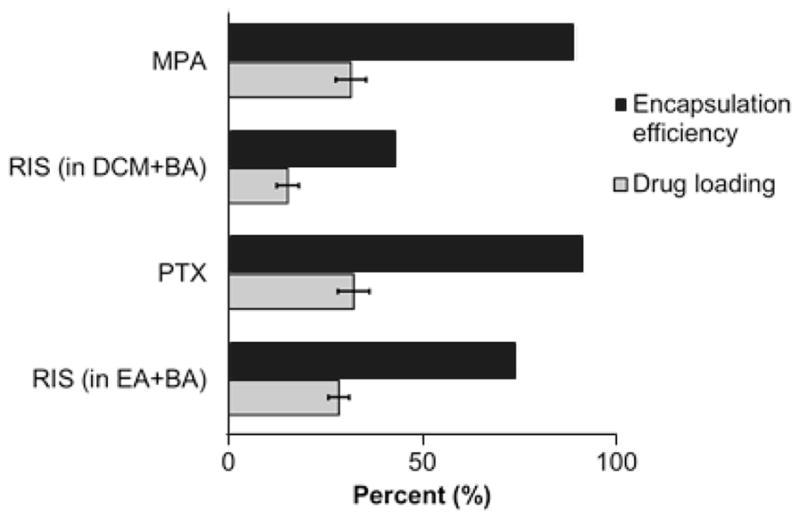
Comparison of drug loading and encapsulation efficiency of model compounds. MPA microparticles were prepared from BA and DCM solvent mix (v/v=50:50) due to low solubility in EA
Drug loading and encapsulation efficiencies are also dependent on the properties of the encapsulated drug. Using 8515 PLGA at 25.0% w/v, we achieved 31.5±3.9% and 32.2±4.1% drug loading for MPA and PTX respectively, corresponding to encapsulation efficiencies of 90.0% and 92.0% PTX, respectively. These values were significantly (p<0.05) higher than that of RIS (Figure 7). One possible explanation is that MPA and PTX have higher compatibility with the polymer 8515 PLGA, which led to enhanced interaction between polymer and drug. Using the Hildebrand-modified approach, the interaction parameter χ can be calculated based on the following equation:
where δd and δp are partial solubility parameters due to dispersion, polarity and hydrogel bonding forces for drug and polymer respectively. The values for partial solubility parameters were obtained from literature (31). Based on the above equation, the interaction parameters between 8515 PLGA and RIS, MPA and PTX are 2.74, 0.309 and 0.811, respectively. Clearly MPA and PTX have higher compatibility with 8515 PLGA compared to RIS. Another possible cause for the difference in drug loading and encapsulation efficiency can be attributed to the difference in solubilities of the drugs in the solvents. Drug with higher solubility in a particular organic solvent (e.g., RIS in BA versus MPA in BA) may diffuse towards the surface of the microparticles in greater amount than one with a lower solubility in the said solvent. A third factor that may contribute towards the drug loading trends we see here is the aqueous solubility of the drug. While PTX has a higher solubility in BA compared to RIS, the loss of surface associated PTX from microparticles may have been hindered by the extremely low aqueous solubility of the drug compared to RIS. The amount of drug loading is therefore determined by a combination of factors that involve drug-polymer compatibility, drug-solvent interaction and drug properties.
3.4 Effect of formulation and process parameters on in vitro release
The in vitro release profiles of RIS formulations are presented in Figure 8. Previously, we showed that the following formulation parameters produced fragmented microparticles or microparticles irregular in shape: low IV PLGA (IV = 0.26~0.54 dL/g), high IV PLGA (IV = 0.95~1.2 dL/g), low PLGA concentration in organic solution (6.2% w/v) and increased drug content (12.5% w/v). Because size and shape of the microparticles greatly affect drug release (36), these formulations were eliminated from direct comparison in in vitro studies with microparticles that were able to maintain original template size and shape.
Figure 8.
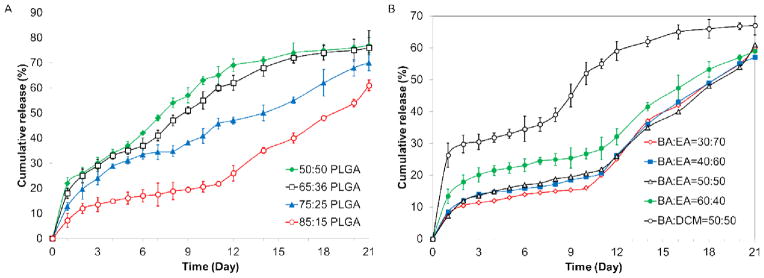
Comparison of RIS drug release from formulation in phosphate buffer (pH 7.4, 37 °C) A) comparison by PLGA type (L:G); B) comparison by different solvent combinations
Among the formulation and process-related parameters studied, initial burst release and subsequent release of RIS from microparticles produced by the hydrogel template method were found to be influenced by L:G ratio of PLGA. Figure 8-A compares release profiles of RIS-loaded microparticles made using 5050, 6535, 7525 and 8515 PLGA. Initial burst release percentages were 22.1±2.3%, 18.5±2.0%, 12.8±1.5% and 7.2±2.8% for the four formulations, respectively. The limited amount of burst release seen in microparticles made from 8515 PLGA is particularly remarkable considering the relatively high drug loading we were able to obtain from this formulation. In many cases, burst release increases with increasing drug loading (37, 38) due to the elution of surface associated drugs and a high concentration gradient between microparticle and surrounding release medium (23). As previously discussed, diffusion of drug to the surface of the microparticles in the hydrogel template method is limited by drug solubility in the solvent used and drug-polymer interaction. Increasing the lactide content presumably contributed to the latter by enhancing hydrophobic interaction between drug and polymer. In addition, decreasing the hydrophilicity of PLGA inhibited water uptake from the release medium, which in turn resulted in lower initial burst release (39). The L:G ratio of PLGA also exerted an effect on subsequent release. As is shown in Figure 8-A, the onset of more rapid release was slower for PLGA with higher lactide content. The rate of drug release over the next 21 days decreased with increasing L:G ratio. Microparticles made from 5050 PLGA showed a cumulative 77.6±3.1% release while those produced using 8515 PLGA showed 61.5±2.2% cumulative release by the end of the three week period. Following initial burst release, drug is mainly released when the polymer matrix degrades and the drug diffuses from the eroded matrix (40). It is well-established that the rate of PLGA degradation decreases with increasing the lactide content due to the more hydrophobic nature of PLGA as lactide content increases (41). Therefore, it is expected that the rate of drug released over time will be on the order of 8515 < 7525 < 6535 < 5050 PLGA. This result is in accordance with trend observed in studies using conventional preparation methods (42).
Figure 8-B compares in vitro release of RIS microparticles made using different BA and EA volume ratios or DCM instead of EA. As was expected, the initial release did not change significantly when BA content decreased from 50:50 to 30:70 (p > 0.05). When BA was increased to 60:40, the initial burst release increased significantly to 13.4±2.3% (p < 0.05). As we have previously explained, the increase in BA content likely resulted in more surface-associated RIS. In comparison, RIS microparticles made using BA and DCM showed a significantly higher initial burst release of 26.3±3.8%. The large initial release can be attributed to both surface associated drug as well as the porous structure of microparticles as seen by SEM images. The porous structure facilitated diffusion of water into the microparticle as well as drug out from the microparticle. Compared to the formulation prepared from BA and EA, the formulation prepared using BA and DCM saw a higher overall rate of release, with 67.0±3.0 % drug release over the three-week period compared to 61.5±2.2% drug release in BA-EA formulation. The higher cumulative release rate can be attributed to the formation of less dense microparticles due to the faster removal of DCM compared to EA. Typically, a more slow solvent removal process provides more time for microparticle to condense and form denser polymeric matrices. This may be a barrier to water diffusion, which may partially slow down the rate of polymer degradation and subsequent drug release.
The in vitro release profiles for RIS, MPA and PTX were compared in Figure 9. Interestingly, MPA microparticles showed very low levels of burst release (<1 %) and a distinct lag phase of approximately 10 days. Over the following 4 days, MPA was rapidly released and over 80% of drug content was released by the 16th day. The suppression of burst release in MPA-loaded microparticles may be attributed to differences in drug distribution in the polymeric matrix compared to RIS-loaded microparticles. As discussed in previous section, MPA is less soluble than RIS in both BA and DCM. During the air-drying process, it is less likely to migrate towards the surface of microparticles than RIS. On the other hand, PTX-loaded microparticles showed increased levels of burst release compared to RIS microparticles with almost no lag phase before steady release. This is in accordance with our previous observation that higher solubility of drug in organic solvent leads to higher initial burst release due to the migration of drug molecules to surface of microparticles. It is noted that the MPA was released at an accelerated rate after Day 10 of the secondary release phase. This may be due to multiple reasons, and one possible explanation is the drug release through the microchannels formed by the diffusing water. At this stage, the drug release depends on the interactions among the drug, PLGA, and water. Like MPA-loaded microparticles, more than 80 % of total drug loaded was released by day 16.
Figure 9.
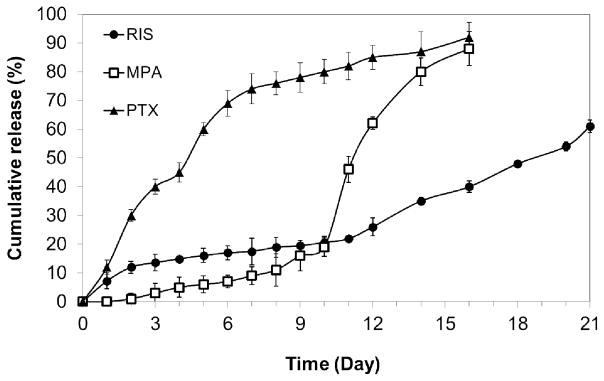
Comparison of release profiles of RIS, MPA and PTX from 8515 PLGA microparticles.
3.5 Comparison of the hydrogel template method and emulsion method
As previously discussed, the emulsion-based method is one of the most commonly used ways to prepare drug-loaded polymeric microparticles. However, three major limitations of the emulsion method are wide particle size distribution, use of heat (during solvent extraction and evaporation process) and relatively low drug loading. From the particle size data we obtained in this study, we found that microparticles produced using the emulsion method showed a polydispersity of 0.39 which is significantly higher than the polydispersity value of 0.15 for the microparticles prepared using the hydrogel template method. The microparticles were mostly spherical in shape with smooth surfaces and few pores. In commercial preparation of microparticles using emulsion-based methods, a secondary processing step is almost always required to further reduce particle size distribution. This increases time and cost of production, and is less desirable than a single process that allows us to achieve the desirable target size range.
Similar to the hydrogel template method, drug encapsulated within microparticles made by the emulsion method were amorphous in form as characterized by XRD. Drug loading and encapsulation efficiency were 28.3±0.7% and 80.8% respectively, values that were comparable to drug loading and encapsulation efficiency of the hydrogel template microparticles prepared using the same formulation (8515 PLGA, 12.5% w/v PLGA, 7.0% w/v drug, BA:EA=50:50 v/v). In vitro release profiles of RIS-loaded microparticles prepared using the two different methods are presented in Figure 10. Microparticles prepared from the hydrogel template and emulsion method showed initial burst release of 7.2±1.1% and 10.1±3.0%, respectively. The burst release from hydrogel template method was slightly lower compared to microparticle prepared from the emulsion method. Both types of microparticles displayed a period of slow release until day 10, followed by more rapid release. The cumulative release over a period of three weeks was also comparable, reaching 61±2.8% for the hydrogel template and 63.2±8.0% for emulsion. These results demonstrate that microparticles prepared using the hydrogel template method were at least comparable in in vitro performance to microparticles prepared from the conventional emulsion method. The major advantages associated with using the hydrogel template over emulsion seem to be particle size homogeneity, which is reflected in the smaller variations in subsequent in vitro release profiles. Furthermore, the hydrogel template method allows additional manipulation of PLGA composition and use of different types of polymers.
Figure 10.

Comparison of release profiles of RIS-loaded microparticles prepared using hydrogel template and emulsion methods
4. Conclusion
Drug-loaded PLGA microparticles prepared using the hydrogel template method were characterized and evaluated using three poorly-water soluble, model drugs: RIS, MPA and PTX. The microparticles were characterized based on size, shape, morphology, drug loading, encapsulation efficiency as well as in vitro release. Among the formulation and process-related parameters studied, it was found that the ability of produced microparticles to retain the designed shape was dependent on molecular weight of template material as well as viscosity of filling solution. When these two parameters were optimized, the microparticles formed showed good conformity to the original template design for a wide range of formulation conditions, which demonstrates the robustness as well as the broad applicability of the hydrogel template method. In addition, the microparticles produced showed small size distribution, which provided an advantage compared to the conventional emulsion-based method. Drug loading and encapsulation efficiency of microparticles prepared using the hydrogel template method were found to increase with lactide content of PLGA, concentration of PLGA in solution and decreasing BA content. The trends observed were expected and can be explained using well-established microencapsulation principles. Using this method, we were able to suppress initial burst release of two model compounds to below 10 % and extend the release for at least 21 days. The low initial burst release is particularly remarkable considering the relatively high drug loading we were able to achieve for these microparticles. The hydrogel template method has been demonstrated to produce microparticles that perform at least comparably in vitro to microparticles from the emulsion-based method. Efforts are underway to characterize and understand how these microparticles will perform in vivo. Applications of homogeneous micdroparticles are likely to extend to coating of drug to various biomedical devices, such as drug-eluting vascular stents and angioplasty balloons (43)
Acknowledgments
This work was supported by the Showalter Research Trust Fund and National Institute of Health through CA129287, HL062552, and GM095879.
References
- 1.Tran VT, Benoit JP, Venier-Julienne MC. Why and how to prepare biodegradable, monodispersed, polymeric microparticles in the field of pharmacy? International Journal of Pharmaceutics. 2011;407:1–11. doi: 10.1016/j.ijpharm.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 2.Wei Y, Wang YX, Kang AJ, Wang W, Ho SV, Gao JF, Ma GH, Su ZG. A Novel Sustained-Release Formulation of Recombinant Human Growth Hormone and Its Pharmacokinetic, Pharmacodynamic and Safety Profiles. Molecular pharmaceutics. 2012;9:2039–2048. doi: 10.1021/mp300126t. [DOI] [PubMed] [Google Scholar]
- 3.Yeoand Y, Park KN. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Archives of Pharmacal Research. 2004;27:1–12. doi: 10.1007/BF02980037. [DOI] [PubMed] [Google Scholar]
- 4.Su ZX, Shi YN, Teng LS, Li X, Wang LX, Meng QF, Teng LR, Li YX. Biodegradable poly(D, L-lactide-co-glycolide) (PLGA) microspheres for sustained release of risperidone: Zero-order release formulation. Pharmaceutical Development and Technology. 2011;16:377–384. doi: 10.3109/10837451003739297. [DOI] [PubMed] [Google Scholar]
- 5.Amann LC, Gandal MJ, Lin R, Liang YL, Siegel SJ. In Vitro-In Vivo Correlations of Scalable PLGA-Risperidone Implants for the Treatment of Schizophrenia. Pharmaceutical Research. 2010;27:1730–1737. doi: 10.1007/s11095-010-0152-4. [DOI] [PubMed] [Google Scholar]
- 6.Doan TVP, Couet W, Olivier JC. Formulation and in vitro characterization of inhalable rifampicin-loaded PLGA microspheres for sustained lung delivery. International Journal of Pharmaceutics. 2011;414:112–117. doi: 10.1016/j.ijpharm.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Cho SW, Song SH, Choi YW. Effects of solvent selection and fabrication method on the characteristics of biodegradable poly(lactide-co-glycolide) microspheres containing ovalbumin. Archives of Pharmacal Research. 2000;23:385–390. doi: 10.1007/BF02975452. [DOI] [PubMed] [Google Scholar]
- 8.Maaand YF, Prestrelski SJ. Biopharmaceutical Powders Particle Formation and Formulation Considerations. Current Pharmaceutical Biotechnology. 2000;1:283–302. doi: 10.2174/1389201003378898. [DOI] [PubMed] [Google Scholar]
- 9.Tran VT, Benoit JP, Venier-Julienne MC. Why and how to prepare biodegradable, monodispersed, polymeric microparticles in the field of pharmacy? International Journal of Pharmaceutics. 2011;407:1–11. doi: 10.1016/j.ijpharm.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 10.Gaumet M, Gurny R, Delie F. Localization and quantification of biodegradable particles in an intestinal cell model: The influence of particle size. European Journal of Pharmaceutical Sciences. 2009;36:465–473. doi: 10.1016/j.ejps.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Mohamedand F, van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. Journal of Pharmaceutical Sciences. 2008;97:71–87. doi: 10.1002/jps.21082. [DOI] [PubMed] [Google Scholar]
- 12.Thomas C, Gupta V, Ahsan F. Particle Size Influences the Immune Response Produced by Hepatitis B Vaccine Formulated in Inhalable Particles. Pharmaceutical Research. 27:905–919. doi: 10.1007/s11095-010-0094-x. [DOI] [PubMed] [Google Scholar]
- 13.Berkland C, Kipper MJ, Narasimhan B, Kim KK, Pack DW. Microsphere size, precipitation kinetics and drug distribution control drug release from biodegradable polyanhydride microspheres. Journal of Controlled Release. 2004;94:129–141. doi: 10.1016/j.jconrel.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Siepmann J, Faisant N, Akiki J, Richard J, Benoit JP. Effect of the size of biodegradable microparticles on drug release: experiment and theory. Journal of Controlled Release. 2004;96:123–134. doi: 10.1016/j.jconrel.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Berkland C, Kim K, Pack DW. PLG microsphere size controls drug release rate through several competing factors. Pharmaceutical Research. 2003;20:1055–1062. doi: 10.1023/a:1024466407849. [DOI] [PubMed] [Google Scholar]
- 16.Gaspar MM, Blanco D, Cruz MEM, Alonso MJ. Formulation of L-asparaginase-loaded poly(lactide-co-glycolide) nanoparticles: influence of polymer properties on enzyme loading, activity and in vitro release. Journal of Controlled Release. 1998;52:53–62. doi: 10.1016/s0168-3659(97)00196-x. [DOI] [PubMed] [Google Scholar]
- 17.Kauffman KJ, Kanthamneni N, Meenach SA, Pierson BC, Bachelder EM, Ainslie KM. Optimization of rapamycin-loaded acetalated dextran microparticles for immunosuppression. International Journal of Pharmaceutics. 2012;422:356–363. doi: 10.1016/j.ijpharm.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 18.Le Ray AM, Chiffoleau S, Iooss P, Grimandi G, Gouyette A, Daculsi G, Merle C. Vancomycin encapsulation in biodegradable poly(epsilon-caprolactone) microparticles for bone implantation. Influence of the formulation process on size, drug loading, in vitro release and cytocompatibility. Biomaterials. 2003;24:443–449. doi: 10.1016/s0142-9612(02)00357-5. [DOI] [PubMed] [Google Scholar]
- 19.Acharya G, Shin C, Park K. The hydrogel template method for fabrication of homogeneous nano/microparticles. Journal of Controlled Release. 2010;141:314– 319. doi: 10.1016/j.jconrel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 20.Acharya G, Shin CS, Vedantham K, McDermott M, Rish T, Hansen K, Fu YR, Park K. A study of drug release from homogeneous PLGA microstructures. Journal of Controlled Release. 2010;146:201–206. doi: 10.1016/j.jconrel.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 21.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141:320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Huang M, Cai DD, Liu YH, Sun J, Wang JJ, Qin CX, Dai LX, Kazuo Y. Investigation of a-PVA/s-PVA hydrogels prepared by freezing-thawing method. Fibers and Polymers. 2012;13:955–962. [Google Scholar]
- 23.Yang YY, Chung TS, Ng NP. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–241. doi: 10.1016/s0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 24.Gorr HM, Zueger JM, Barnard JA. Characteristic Size for Onset of Coffee-Ring Effect in Evaporating Lysozyme-Water Solution Droplets. Journal of Physical Chemistry B. 2012;116:12213–12220. doi: 10.1021/jp307933a. [DOI] [PubMed] [Google Scholar]
- 25.Okuzono T, Kobayashi M, Doi M. Final shape of a drying thin film. Physical Review E. 2009;80 doi: 10.1103/PhysRevE.80.021603. [DOI] [PubMed] [Google Scholar]
- 26.da Silva AA, de Matos JR, Formariz TP, Rossanezi G, Scarpa MV, do Egito EST, de Oliveira AG. Thermal behavior and stability of biodegradable spray-dried microparticles containing triamcinolone. International Journal of Pharmaceutics. 2009;368:45–55. doi: 10.1016/j.ijpharm.2008.09.054. [DOI] [PubMed] [Google Scholar]
- 27.Vay K, Friess W, Scheler S. A detailed view of microparticle formation by in-process monitoring of the glass transition temperature. European Journal of Pharmaceutics and Biopharmaceutics. 2012;81:399–408. doi: 10.1016/j.ejpb.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Jiang GQ, Ding FX. Effects of polymer degradation on drug release from PLGA-mPEG microparticles: A dynamic study of microparticle morphological and physicochemical properties. Journal of Applied Polymer Science. 2008;108:2458–2466. [Google Scholar]
- 29.Lee J, Oh YJ, Lee SK, Lee KY. Facile control of porous structures of polymer microspheres using an osmotic agent for pulmonary delivery. Journal of Controlled Release. 2010;146:61–67. doi: 10.1016/j.jconrel.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Sinclair W, Leane M, Clarke G, Dennis A, Tobyn M, Timmins P. Physical Stability and Recrystallization Kinetics of Amorphous Ibipinabant Drug Product by Fourier Transform Raman Spectroscopy. Journal of Pharmaceutical Sciences. 2011;100:4687–4699. doi: 10.1002/jps.22658. [DOI] [PubMed] [Google Scholar]
- 31.Schenderlein S, Luck M, Muller BW. Partial solubility parameters of poly(D,L-lactide-co-glycolide) International Journal of Pharmaceutics. 2004;286:19–26. doi: 10.1016/j.ijpharm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 32.Ozalp Y, Ozdemir N, Kocagoz S, Hasirci V. Controlled release of vancomycin from biodegradable microcapsules. Journal of Microencapsulation. 2001;18:89–110. doi: 10.1080/026520401750038638. [DOI] [PubMed] [Google Scholar]
- 33.Bodmeier R, McGinity JW. SOLVENT SELECTION IN THE PREPARATION OF POLY(DL-LACTIDE) MICROSPHERES PREPARED BY THE SOLVENT EVAPORATION METHOD. International Journal of Pharmaceutics. 1988;43:179–186. [Google Scholar]
- 34.Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. International Journal of Pharmaceutics. 2008;364:298– 327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 35.Huangand X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. Journal of Controlled Release. 2001;73:121–136. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 36.Lee BK, Yun YH, Choi JS, Choi YC, Kim JD, Cho YW. Fabrication of drug-loaded polymer microparticles with arbitrary geometries using a piezoelectric inkjet printing system. International Journal of Pharmaceutics. 2012;427:305–310. doi: 10.1016/j.ijpharm.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Sah H. Microencapsulation techniques using ethyl acetate as a dispersed solvent: Effects of its extraction rate on the characteristics of PLGA microspheres. Journal of Controlled Release. 1997;47:233–245. [Google Scholar]
- 38.Hora MS, Rana RK, Nunberg JH, Tice TR, Gilley RM, Hudson ME. Release of human serum-albumin from poly(lactide-co-glycolide) microspheres. Pharm Res. 1990;7:1190–1194. doi: 10.1023/a:1015948829632. [DOI] [PubMed] [Google Scholar]
- 39.Ohagan DT, Jeffery H, Davis SS. The preparation and characterization of poly(lactide-co-glycolide) microparticles .3. microparticle/polymer degradation rates and the in-vitro release of a model protein. Int J Pharm. 1994;103:37–45. [Google Scholar]
- 40.Matsumoto A, Matsukawa Y, Suzuki T, Yoshino H. Drug release characteristics of multi-reservoir type microspheres with poly(DL-lactide-co-glycolide) and poly(DL-lactide) Journal of Controlled Release. 2005;106:172–180. doi: 10.1016/j.jconrel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 41.Hong KH, Woo SH, Kang TJ. In vitro degradation and drug-release behavior of electrospun, fibrous webs of poly(lactic-co-glycolic acid) Journal of Applied Polymer Science. 2012;124:209–214. [Google Scholar]
- 42.Engineer C, Parikh J, Raval A. Effect of copolymer ratio on hydrolytic degradation of poly(lactide-co-glycolide) from drug eluting coronary stents. Chemical Engineering Research & Design. 2011;89:328–334. [Google Scholar]
- 43.Kang E, Vedantham LK, Dadarat XM, Kwon IK, Sturek M, Park K. A drug-eluting stent for delivery of signal pathway-specific 1,3-dipropyl-8-cyclopentyl xanthine (DPCPX) Mol Pharm. 2009;6:1110–1117. doi: 10.1021/mp8002623. [DOI] [PubMed] [Google Scholar]