

Abstract
This account describes a strategy for directly forming three of the six rings found in the polyketide natural product hirsutellone B via a novel cyclization cascade. The key step in our approach comprises two transformations: a large-ring forming, nucleophilic capture of a transient acyl ketene and an intramolecular Diels–Alder reaction, both of which occur in tandem through thermolyses of appropriately functionalized, polyunsaturated dioxinones. These thermally induced “double cyclization” cascades generate three new bonds, four contiguous stereocenters, and a significant fraction of the polycyclic architecture of hirsutellone B. The advanced macrolactam and macrolactone intermediates that were synthesized by this process possess key features of the hirsutellone framework, including the stereochemically dense decahydrofluorene core and the strained para-cyclophane ring. However, attempts to complete the carbon skeleton of hirsutellone B via transannular carbon-carbon bond formation were undermined by competitive O-alkylation reactions. This account also documents how we adapted to this undesired outcome through an evaluation of several distinct strategies for synthesis, as well as our eventual achievement of a formal total synthesis of hirsutellone B.
Introduction
Hirsutellone B (1) is one of five structurally related natural products isolated from the insect pathogenic fungus Hirsutella nivea BCC 2594 in 2005 by Isaka and coworkers (Figure 1).1 The stereochemically complex decahydrofluorene core (i.e. the fused 6-5-6 ring system) and 13-membered para-cyclophane ether that distinguish these natural products are also found in the related GKK1032s2 (2), pyrrocidines3 (3) and pyrrospirones4 (4). This intriguing polycyclic architectural motif is thus the hallmark of an expansive group of biologically active polyketide natural products.
Figure 1.
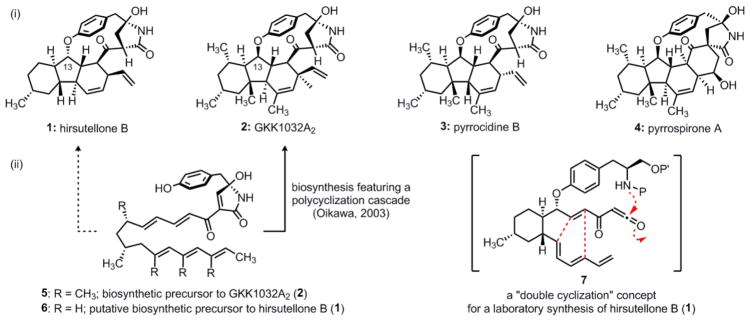
(i) Structures of hirsutellone B and related natural products and (ii) concepts for evolving these polycyclic architectures from polyunsaturated precursors.
The isotope incorporation studies of Oikawa demonstrated that the impressive molecular structure of GKK1032A2 (2) originates from one molecule of tyrosine, nine molecules of acetic acid, and five molecules of L-methionine.5 Based on these observations, they reasoned that four of its six rings arise by a remarkable tetracyclization of the polyunsaturated nonaketide 5 (Figure 1). In relation to GKK1032A2 (2), the hirsutellones possess four fewer methyl groups as well as the opposite configuration at the C-13 stereocenter. Presumably, an analogous tetracyclization of the monomethylated, tyrosine nonaketide 6 affords the related structure of hirsutellone B (1), although this seemingly plausible biogenetic connection remains unproven.
Isaka and coworkers also reported that the hirsutellones inhibit Mycobacterium tuberculosis H37Ra with minimum inhibitory concentration values in the range of 0.78–3.125 μg/mL.1 The hirsutellones thus offer a new chemotype with promising antitubercular activity, and they have emerged as compelling targets for research efforts in organic synthesis.6–11 As of this writing, three total syntheses of hirsutellone B (1) have been described. The first synthesis of this natural product was reported by Nicolaou in 2009,12 and their group later developed a second bio-inspired route to access hirsutellones A, B, and C.13 The Nicolaou approach offers an incisive, direct construction of the tricyclic decahydrofluorene framework of 1 from a polyunsaturated acyclic precursor and a late-stage Ramberg–Bäcklund ring contraction to form the strained para-cyclophane ether. The third total synthesis of hirsutellone B was achieved in 2011 by Uchiro14 and features an intramolecular Ullmann reaction to annulate the para-cyclophane onto a functionalized decahydrofluorene core.
Our laboratory was intrigued by the possibility of directly forming much of the structure of hirsutellone B from a trisubstituted cyclohexane by capitalizing on the intrinsic reactivity of acylketene 7 (Figure 1).15 In the course of this “double cyclization” event, three new rings would arise by an internal nucleophilic capture of a transient acylketene and a concomitant intramolecular Diels–Alder (IMDA) reaction. Herein, we provide the full account of our development of this concept for synthesis as well as our investigations of several distinct strategies for addressing the challenging para-cyclophane ether substructural element of hirsutellone B. We also describe the preparation of an advanced intermediate that intercepts Nicolaou’s pioneering route to hirsutellone B, constituting a formal total synthesis of this natural product.
Results and Discussion
We envisioned that much of the complexity of the hirsutellone framework could be generated in a single step through a tandem reaction sequence, as shown in Scheme 1. Inspired by Boeckman’s elegant tetronolide synthesis,16 we hoped to make use of an intramolecular acylketene capture/IMDA cascade, which, to the best of our knowledge, was unprecedented in the chemical literature. Thus, thermolysis of vinyldioxinone 8 would induce a cycloreversion, expelling acetone to give transient acylketene 7.15 This reactive intermediate could then be trapped intramolecularly by the pendent amine to form the macrocyclic β-keto amide 9. However, the thermal fragmentation of the dioxinone heterocycle in 8 would also generate an α,β-unsaturated ketone, which, in the presence of the conjugated triene, could undergo an intramolecular Diels–Alder reaction to complete the decahydrofluorene framework (9 → 10). We were willing to accept the risk associated with this concept for synthesis because of its potential to deal directly with the complex cyclic connectivity of hirsutellone B. We also anticipated that the setup costs for synthesizing the required trisubstituted cyclohexane 8 would not be excessively high.
Scheme 1.

An acylketene trapping/IMDA cascade to access the hirsutellone framework. DMB = 2,4-dimethoxybenzyl; P = TBDPS = t-BuPh2Si.
If we could generate compound 10 by a thermolysis of dioxinone 8, we believed that the γ-lactam ring could be formed via a transannular alkylation (SNi) reaction to give 11, completing the full carbon skeleton of the natural product. At this stage, a final C–H oxidation would install the hemiaminal functionality, and subsequent cleavage of the nitrogen protecting group would provide rapid access to hirsutellone B.
Having formulated an attractive strategy for contending with the polycyclic structure of hirsutellone B, we turned our attention to the synthesis of key intermediate 8 (Figure 2). Our approach focused on three strategic bond disconnections, ultimately leading to α-hydroxy aldehyde 16.7 This trisubstituted cyclohexane intermediate would then serve as a central scaffold onto which the remaining three fragments could be appended. We proposed that the desired aryl ether bond could be formed via a palladium-catalyzed Tsuji–Trost reaction with phenol 14.17 Our design also makes use of the Wittig reaction to form two key carbon-carbon bonds, utilizing phosphonium salt 1318 and the known phosphorane 15.19 In both cases, the use of a stabilized or semi-stabilized phosphorus ylide would establish the desired trans olefin geometry.20
Figure 2.

Strategic bond disconnections for the synthesis of key intermediate 8.
We began our synthesis of phenol coupling partner 14 by protecting known tyrosinol derivative 1721 as the corresponding bis-TBDPS (t-BuPh2Si) ether, as shown in Scheme 2. When this compound was treated with one equivalent of tetrabutylammonium fluoride at 0 °C, selective deprotection of the phenolic silyl ether occurred to give free phenol 18 in 81% yield over the two steps. Subsequent removal of the Boc group proceeded cleanly with two equivalents of trifluoroacetic acid to form ammonium salt 19. Based on precedent from Pfaltz and Suzuki’s synthesis of macrocidin A,22 we anticipated that transannular alkylation to form the γ-lactam ring (i.e. 10 → 11) would only be possible if the macrolactam were protected as a tertiary amide. Therefore, we decided to utilize the 2,4-dimethoxybenzyl (DMB) protecting group, which could be readily cleaved at a late stage in the synthesis under acidic conditions.23 We found that this group was easily installed via reductive amination of 19 with 2,4-dimethoxybenzaldehyde and sodium triacetoxyborohydride to give the desired DMB-protected coupling partner 14 in 77% yield over the two steps.
Scheme 2.
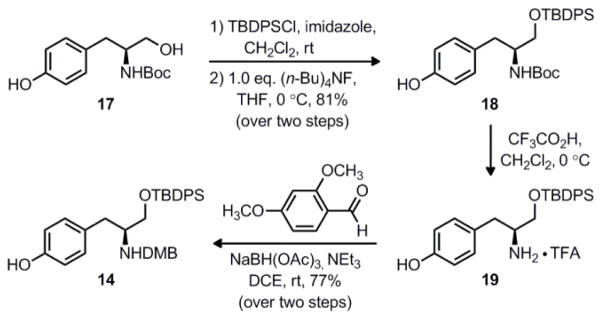
Synthesis of phenol coupling partner 14.
Having accessed phenol 14, our next objective was to investigate formation of the key aryl ether linkage via a palladium-catalyzed Tsuji–Trost reaction. Our initial efforts towards the decahydrofluorene core of the hirsutellones led us to develop a synthesis of enantioenriched allylic alcohol 12,7 which is the product of a Wittig reaction between α-hydroxy aldehyde 16 and phosphorane 15 (see Figure 2). Although we originally converted secondary alcohol 12 to the corresponding allylic carbonate, all attempts to engage this substrate in a Tsuji–Trost reaction with the sodium phenolate of 14 were unsuccessful. Similarly, no reaction was observed using the corresponding allylic acetate or trifluoroacetate esters, despite our efforts to screen many different palladium sources and ligands. In an attempt to enhance the reactivity of the substrate, we next explored preparation of the related allylic phosphate (Scheme 3).24 Thus, deprotonation of alcohol 12 at low temperature with one equivalent of methyllithium followed by the dropwise addition of diethyl chlorophosphate cleanly gave allylic phosphate 20 in 82% yield. When this substrate was treated with the sodium phenolate of 14 in the presence of 10 mol% Pd(PPh3)4 as catalyst, we were pleased to observe the rapid formation of 21, which contained the desired aryl ether linkage. Despite two potential sites of nucleophilic attack on the intermediate palladium π-allyl complex, only the regioisomer that maintains conjugation of the olefin with the dioxinone ring was observed. Furthermore, this reaction proceeds with overall retention of configuration, as is well documented for the Tsuji–Trost reaction.25
Scheme 3.

Synthesis of key triene intermediate 8 and execution of the cyclization cascade to form 10. TPAP = tetrapropylammonium perruthenate; KHMDS = potassium bis(trimethylsilyl)amide.
With the key aryl ether in place, we turned our attention to the construction of the triene side chain. Acid-induced desilylation of 21 gave primary alcohol 22 in 82% yield. However, oxidation of this alcohol to the corresponding aldehyde in the presence of the secondary benzylic amine proved to be difficult. After screening several different oxidation methods (e.g. Dess–Martin periodinane26, PCC, MnO2, Swern27), we found that the Ley oxidation28 gave the best results, producing amino aldehyde 23 in 62% yield. Subsequent Wittig olefination with the phosphorus ylide derived from 13 proceeded smoothly to give triene 8 in 76% yield as a 7:1 mixture of inseparable E/Z isomers.
With key intermediate 8 in hand, we were in a position to test the feasibility of the tandem ketene trapping/IMDA sequence portrayed in Scheme 1. Our initial attempts to bring about this transformation in refluxing toluene or xylenes were unsuccessful. Despite our best efforts to run the reaction under scrupulously anhydrous conditions (azeotropic drying of the starting material, flame-dried glassware, and freshly distilled solvent), we were only able to obtain products derived from trapping of the putative acylketene intermediate with adventitious water. However, when a 0.3 mM solution of 8 was heated in refluxing benzene under Dean–Stark conditions29 for fourteen hours, we were pleased to observe formation of the desired macrocycle 10 as a single diastereomer in 39–50% yield (Scheme 3). We suggest that the reactive acylketene 7 may well be a fleeting intermediate on the path leading from compound 8 to polycycle 10. Although the precise ordering of the new bond constructions is unclear, the production of compound 10 from unsaturated dioxinone 8 is consistent with an endo-selective IMDA reaction to form the decahydrofluorene core and an intramolecular trapping of the acylketene intermediate by the pendent secondary amine to form the macrolactam ring. Notably, this cascade process rapidly generates much of the complex topology of hirsutellone B: three new rings, including a strained cyclophane, three new bonds, and four contiguous stereocenters are formed in a single laboratory operation.
Having realized our key tandem reaction sequence, we eagerly turned our attention to the transannular carbon-carbon bond formation that would form the γ-lactam ring and complete the carbon framework of hirsutellone B. Desilylation of 10 proceeded smoothly with tetrabutylammonium triphenyldifluorosilicate (TBAT), giving primary alcohol 24 in 98% yield (Scheme 4). At this point, all that remained was to activate the primary alcohol and induce the desired SNi reaction upon treatment with a suitable base. In the event, treatment of alcohol 24 with methanesulfonyl chloride in the presence of triethylamine gave the corresponding primary mesylate 25, which proved to be somewhat unstable. To our surprise, when 25 was treated with the amidine base DBU, the desired product 11 was not formed; instead, we were only able to isolate the isomeric O-alkylated compound 26, which features an unusual acylketene hemiaminal ether. Indeed, we were never able to isolate the desired C-alkylated product 11, despite extensive experimentation with different leaving groups, bases, and Lewis acid additives.
Scheme 4.

Unexpected formation of acylketene hemiaminal ether 26.
Since our efforts to form the γ-lactam ring via an intramolecular nucleophilic displacement were unsuccessful, we decided to explore an intramolecular Knoevenagel condensation as an alternative way to make the carbon-carbon bond (Scheme 5). Importantly, we anticipated that conjugate reduction of the expected condensation product 28 would lead to the same γ-lactam 11 targeted in our previous alkylation strategy. To this end, alcohol 24 was oxidized to the corresponding aldehyde 27 in good yield using Dess–Martin periodinane (DMP).26 However, the intramolecular condensation product 28 was not observed under basic or acidic cyclization conditions, and we were unable to obtain any evidence for carbon-carbon bond formation. In fact, the only reaction that occurred under basic conditions was slow epimerization at the α-position of aldehyde 27. An attempted intramolecular Claisen condensation using the corresponding methyl ester was similarly unsuccessful, leading only to recovered starting material.
Scheme 5.

Attempted synthesis of 28 via an intramolecular Knoevenagel condensation.
Based on these unsuccessful attempts at carbon-carbon bond formation, we believe that there are several factors that could make this type of transannular reaction quite challenging. First, the preferred conformations of macrocyclic rings are difficult to predict, and it is possible that the reactive centers are too far apart to enable facile ring closure.30 In addition, restricted rotation about the amide C–N bond may further bias the conformation of the macrocycle, preventing correct alignment for nucleophilic attack of the enolate at carbon rather than at oxygen. Finally, and perhaps most significantly, there is a considerable difference in strain between the starting 14-membered cyclophane and the 13-membered cyclophane present in the natural product. Therefore, the desired transannular ring closure may be disfavored due to the buildup of strain in the transition state leading to carbon-carbon bond formation.
In light of the unexpected difficulty of transannular carbon-carbon bond formation, we chose to modify our strategy. More specifically, we reasoned that targeting a macrolactone instead of a macrolactam could impart more conformational flexibility for the desired alkylation reaction. Moreover, the carbonyl oxygen of the ester group should be less nucleophilic than that of an amide, disfavoring the undesired O-alkylation product. Finally, examination of handheld molecular models suggested that a more favorable trajectory for transannular C-alkylation might be achieved using a substrate with the alternative configuration at the carbon bearing the side chain with the leaving group.
With these points in mind, we devised a new approach to hirsutellone B, as shown in Scheme 6. Thus, thermolysis of dioxinone 29 would initiate a similar tandem acylketene capture/IMDA sequence to give macrolactone 30. At this stage, it was our hope that transannular γ-lactone formation could be achieved through intramolecular nucleophilic displacement to form 31, completing the carbon framework of hirsutellone B. Another attractive feature of this strategy is the straightforward endgame which would not require a late-stage C–H functionalization. Thus, nucleophilic opening of γ-lactone 31 with ammonia would form primary amide 32. Finally, oxidation of the resulting secondary alcohol would give ketone 33, which would yield hirsutellone B (1) directly via intramolecular hemiaminal formation. With our new strategy established, we turned our attention to the synthesis of the required triene substrate 29.
Scheme 6.

Revised strategy to hirsutellone B featuring a macrolactone intermediate.
Our first goal was preparing the new phenol coupling partner 37, as shown in Scheme 7. For this route, we selected a primary tosylate ester as a protecting group that could also serve as a leaving group for γ-lactone formation after the macrolactonization step. Thus, the known enantioenriched diol 3431 was monoprotected with p-toluenesulfonyl chloride to give the desired primary tosylate 35 in 75% yield. Under the basic conditions of the Tsuji–Trost reaction, we found that intramolecular nucleophilic displacement of tosylate to give the corresponding epoxide was a significant side reaction. Therefore, the secondary alcohol of 35 was protected as the corresponding TBS ether to give 36 in 98% yield. Finally, hydrogenolysis of the benzyl ether proceeded smoothly to give the desired phenol 37 in high yield.
Scheme 7.

Synthesis of enantioenriched phenol coupling partner 37.
We were pleased to find that the Tsuji–Trost reaction of allylic phosphate 20 and the sodium phenolate derived from 37 proceeded under our standard coupling conditions to give 38 in 65% yield (Scheme 8). Notably, intermolecular phenolate displacement of tosylate was not observed, likely due to the steric bulk of the nearby TBS group and the rapid rate of aryl ether bond formation. Simultaneous deprotection of both TBS groups could be accomplished under mild conditions using excess triethylamine trihydrofluoride to give diol 39 in 88% yield. At this point, we hoped to achieve a chemoselective oxidation of the primary alcohol in the presence of the free secondary alcohol. We found that this could be achieved using 10 mol% TEMPO and stoichiometric iodobenzene diacetate32 to give aldehyde 40 in 95% yield without any detectable ketone byproduct or undesired epimerization at the α-position of the aldehyde.
Scheme 8.

Synthesis of key triene intermediate 29 and its unexpected dimerization.
Having obtained aldehyde 40, we turned our attention to installation of the triene. In our previous routes, the triene had been introduced using a Wittig reaction with the ylide derived from phosphonium salt 13. However, this reaction had several disadvantages. First, the triene product was typically formed as a 7:1 mixture of E/Z isomers that were inseparable by silica gel chromatography. In addition, some material was always lost due to competitive nucleophilic opening of the dioxinone ring by excess ylide. Moreover, α-hydroxy tosylate 40 was unstable under basic conditions, and the corresponding epoxide was formed as a significant byproduct. To overcome these difficulties, we decided to use an alternative two-step procedure to introduce the triene, as reported by Liu.8 Thus, Takai olefination33 of aldehyde 40 gave exclusively E vinyl iodide 41 in 74% yield, avoiding the mixture of olefin isomers obtained in the Wittig reaction. A subsequent Stille coupling34 of 41 with known dienyl stannane 4235 proceeded under mild conditions at room temperature to give the desired triene 29 in 85% yield.
Having secured our key intermediate, the stage was set for the tandem intramolecular acylketene capture/IMDA reaction. Interestingly, we found that this macrolactonization was much more sensitive to concentration than our previous acylketene-trapping cascade. As with any macrocyclization, dilute conditions are necessary to disfavor competing intermolecular oligomerization reactions. Typically, we had been able to achieve good yields of our intramolecular acylketene-trapping products at a concentration of 0.3 mM. However, when the cyclization of compound 29 was attempted at this concentration, the desired macrolactone 30 was obtained only in low yield; to our surprise, the major product was the 28-membered dimer 43. The formation of 43 suggests that the acylketene derived from 29 may be relatively long-lived; that is, trapping of this intermediate by the pendent secondary alcohol is slow. We attribute this phenomenon to the reduced nucleophilicity of the secondary alcohol due to the inductive effect of the nearby tosylate group. Indeed, it has been experimentally observed that the rate of alcohol addition to acylketenes correlates with the relative nucleophilicity of the attacking species.36
Fortunately, we were able to minimize the formation of 43 by running the reaction at a lower concentration of 0.1 mM; this afforded a single diastereomer of the desired macrolactone 30 in 50% yield, as shown in Scheme 9. At this stage, we began screening conditions to induce γ-lactone formation via a transannular SNi reaction (see 30 → 31, Scheme 6). Unfortunately, treatment of 30 with a wide variety of bases resulted either in no reaction or slow decomposition of the starting material. Closer examination of a handheld model suggested that a more favorable trajectory for alkylation might be achieved if the two carbonyl groups were held in a syn orientation. Thus, several chelating Lewis acids were also screened in combination with either triethylamine or DBU as a base. However, none of the desired γ-lactone product 31 was observed under any of these conditions.
Scheme 9.

Attempted γ-lactone synthesis and formation of acylketene acetal 45.
In an effort to enhance the reactivity of the substrate, we decided to convert the primary tosylate into a different leaving group. We found that the Finkelstein reaction of 30 with sodium iodide in acetone was relatively slow and required heating to 45 °C to give primary iodide 44 in 54% yield. However, when this compound was treated with DBU at 45 °C in toluene, we were surprised to isolate the relatively unstable acylketene acetal 45. As was observed in our previous macrolactam route, compound 45 is the product of undesired O-alkylation of the ester carbonyl group.
Similarly, when the alkylation reaction was run in the presence of chelating Lewis acids, acylketene acetal 45 was still the only isolable product. Indeed, controlling the selectivity of C vs. O alkylation is a general problem in the reaction of electrophiles with enolates derived from 1,3-dicarbonyl compounds.37 For intermolecular alkylation reactions, C-alkylation can often be favored by choosing an appropriate solvent, counterion, or leaving group. However, the corresponding intramolecular alkylation reactions are often controlled by stereoelectronic effects and strongly favor O-alkylation regardless of the reaction conditions.38
One of the few reliable methods to favor C-alkylation of 1,3-dicarbonyl compounds involves the use of thallium enolates.39 This methodology was pioneered by Taylor, who postulated that the general insolubility of thallium enolates accounted for the observed selectivity. Although thallium enolates of β-keto esters are typically generated using thallium (I) ethoxide, we found that tosylate 30 decomposed under these conditions. However, the combination of thallium (I) carbonate and DBU cleanly converted 30 to a new compound, which was identified as 46 (Scheme 10). Notably, compound 46 is the geometrical isomer of acylketene acetal 45, the product obtained upon exposure of 44 to DBU alone (see Scheme 9). The origin of this remarkable switch in stereoselectivity is unclear. One possibility is that the thallium salt forms a strong chelate with the 1,3-dicarbonyl that enforces a specific geometry for the alkylation step. Notably, the thallium (I) cation is a soft electrophile and known to be highly enophilic;39 therefore, coordination with the nearby vinyl group could lead to the formation of a π-complex that influences the observed enolate geometry.
Scheme 10.

Formation of acylketene acetal 46 and its subsequent ring expansion to 49.
Interestingly, acylketene acetal 46 was unstable in solution and slowly isomerized to its geometrical isomer 45 at a rate of approximately 1% per hour, as determined by 1H-NMR. As mentioned previously, 45 is a somewhat sensitive compound, complicating isolation and purification. We found that upon exposure to silica gel, acylketene acetals 45 and 46 were converted to macrolactone 49 in quantitative yield. We propose that 45 is protonated by the mildly acidic silica gel to give a resonance-stabilized oxonium ion 47 that could be trapped by adventitious water to give 48. This intermediate could then open to give macrolactone 49; presumably, this transformation is driven by release of strain upon ring-expansion to the larger 15-membered cyclophane.
Despite extensive experimentation, we were never able to observe formation of the desired γ-lactone 31. Having obtained the same undesired O-alkylation product in both of our transannular SNi substrates, we decided to abandon this particular strategy towards hirsutellone B. It is likely that the conformation of the macrocycle does not allow the two reactive centers to come close enough for carbon-carbon bond formation to occur. Undoubtedly, this conformational restriction is due to the strained nature of the cyclophane ring, which remains the most challenging feature of this natural product.
In developing a new route towards hirsutellone B, we wished to retain the intramolecular acylketene trapping/IMDA cascade to simultaneously construct the tricyclic core and the cyclophane ring. However, instead of using a polar SNi mechanism to form the transannular carbon-carbon bond, we sought to explore pericyclic and radical reactivity. To this end, we targeted macrolactone 50 as a potentially attractive substrate, as shown in Scheme 11. One possibility for ring contraction of 50 is the Carroll rearrangement, which transforms allyl β-keto esters into γ,δ-unsaturated ketones.40 This reaction would proceed through enol tautomer 51, which undergoes a [3,3]-sigmatropic rearrangement to give β-keto acid 52. Under the thermal reaction conditions, 52 would spontaneously decarboxylate to give ring-contracted ketone 53. Alternatively, macrolactone 50 might be directly converted to 53 under more mild conditions using a metal-catalyzed decarboxylative allylation, constituting a formal Carroll rearrangement.41–42 Next, diastereoselective acylation of 53 could be achieved using LiHMDS and Mander’s reagent43 to give β-keto ester 54. Finally, chemoselective oxidative cleavage of the exocyclic alkene would give ketone 55, intercepting the penultimate intermediate in Nicolaou’s total synthesis of hirsutellone B.12
Scheme 11.

Proposed Carroll rearrangement for the formal synthesis of hirsutellone B. LiHMDS = lithium bis(trimethylsilyl)amide.
The synthesis of the phenol coupling partner required for this route began with TBS-protected 4-bromophenol 5644, which was converted to the corresponding Grignard reagent 57 under standard conditions (Scheme 12). After titration, a solution of 57 was added to known allylic bromide 5845 to form the coupled product 59. Upon workup, the crude product was treated with lithium hydroxide to chemoselectively cleave the phenolic TBS group, giving the desired phenol 60 in 77% yield over the three steps.
Scheme 12.
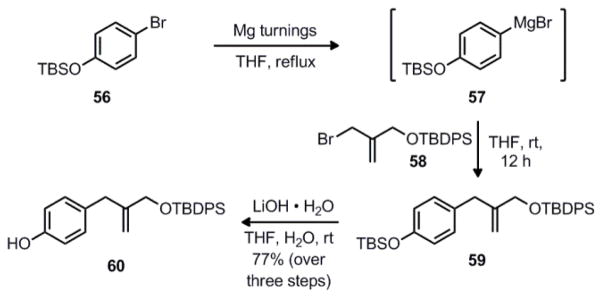
Synthesis of phenol coupling partner 60.
The Tsuji–Trost reaction of allylic phosphate 20 and the sodium phenolate derived from 60 proceeded under our standard coupling conditions to give aryl ether 61 (Scheme 13). After chemoselective desilylation of the primary TBS group, alcohol 62 was isolated in 60% yield over the two steps. Oxidation of 62 with DMP cleanly gave aldehyde 63 in 90% yield, and subsequent Takai olefination proceeded in 86% yield to give exclusively E vinyl iodide 64. At this stage, we found that the TBDPS protecting group could be conveniently removed using TBAT to give allylic alcohol 65 in good yield.
Scheme 13.
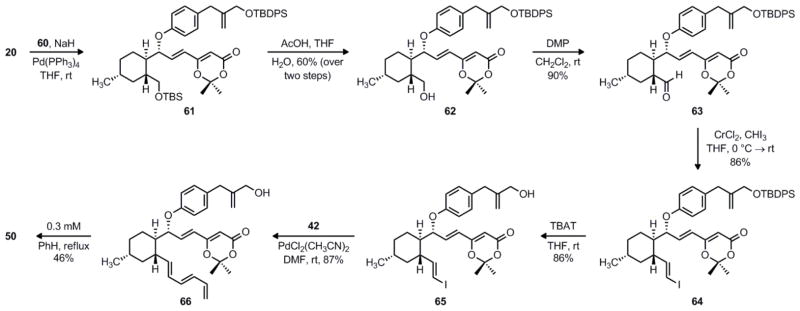
Synthesis of macrolactone 50.
Next, vinyl iodide 65 was coupled with dienyl stannane 42 as before to give triene 66 as a single geometrical isomer in 87% yield. With cascade precursor 66 in hand, we were again in a position to test our tandem intramolecular acylketene trapping/IMDA cascade. Thus, we were pleased to observe that heating a dilute solution of 66 in refluxing benzene gave the desired macrolactone 50 in 46% yield as a single diastereomer. Unfortunately, all attempts to induce Carroll rearrangement of 50 under thermal conditions were unsuccessful. At the elevated temperatures required for rearrangement (175 °C in 1,2-dichlorobenzene or 200 °C in 1,2,4 trichlorobenzene), macrolactone 50 likely underwent the retro-acylketene trapping, leading to decomposition products.15 Efforts to induce a more mild transition metal-catalyzed decarboxylative allylation using a range of palladium41 and ruthenium42 catalysts were similarly unsuccessful.
Having investigated pericyclic and transition metal-catalyzed processes to achieve the ring contraction of macrolactone 50, we next explored a radical reaction to form the key transannular carbon-carbon bond. For this strategy, we decided to use manganese (III) acetate which readily oxidizes enolizable β-ketoesters to give carbon-centered radicals.46–47 However, treatment of 50 with manganese (III) acetate and copper (II) acetate in acetic acid did not result in the desired transannular bond formation; instead, the only observable product was cyclized compound 67 (Scheme 14). We rationalize the formation of this byproduct in the following way. First, hydrogen atom abstraction from 50 by the action of Mn(OAc)3 gives the stabilized radical 68. However, instead of reacting with the exocyclic alkene, this radical engages the proximal vinyl group in an alternative 6-endo-trig cyclization to form secondary radical 69. Reaction of this intermediate with Cu(OAc)2 then generates an organocopper species which, in turn, undergoes β-hydride elimination to deliver 67. Although the radical cyclization did not form the desired carbon-carbon bond, we note that this reactivity could prove useful for the synthesis of the tetracyclic framework of the related pyrrospirone (4) natural products.4
Scheme 14.

The construction of polycycle 67 via a radical cyclization reaction.
At this stage, we began to investigate an alternative strategy for the synthesis of hirsutellone B that would not require a late-stage ring contraction. In particular, we were attracted to the possibility of directly forming the 13-membered cyclophane using a palladium-catalyzed coupling reaction (Scheme 15). After examining handheld molecular models, it seemed critical that the synthesis of the decahydrofluorene core precede the macrocyclization step; otherwise, the presence of the trans alkene required for the Diels–Alder reaction would introduce even more strain into the 13-membered cyclophane. Therefore, our initial target was decahydrofluorene 70, which features a functionalized dioxinone.
Scheme 15.

An approach to macrocyclic ketone 72 using a palladium-catalyzed coupling reaction.
Strategically, we envisioned that 70 could be elaborated to a compound such as 71, setting the stage for a pivotal palladium-catalyzed macrocyclization reaction to give the elusive cyclophane 72. We found the intramolecular Stille coupling34,48 to be especially appealing since the reaction would proceed through a larger 14-membered palladacycle intermediate. In this way, ring contraction to the more highly strained 13-membered paracyclophane accompanies the irreversible reductive elimination step. Another attractive feature of this route is the straightforward endgame; thus, thermolysis of dioxinone 72 would again expel acetone in a retro-hetero-Diels–Alder reaction to give reactive acylketene 73. If this reaction were conducted in the presence of ammonia, nucleophilic capture of the reactive acylketene would occur to form the same primary amide 33 that appears in Nicolaou’s route.12 Finally, spontaneous hemiaminal formation would complete the total synthesis of hirsutellone B in a single cascade reaction sequence.
Our first goal in pursuing this new route was to gain access to the substituted dioxinone fragment, as shown in Scheme 16. Our synthesis began with tert-butyl acetoacetate 74, which was alkylated with methyl bromoacetate to give diester 75. This crude material was then cyclized under acidic conditions in a mixture of acetone and acetic anhydride to afford substituted dioxinone 76 in 63% yield over the two steps. When 76 was brominated under Wohl–Ziegler conditions,49 primary bromide 77 was formed as the major product along with a small amount of the regioisomeric α-bromo ester. Next, treatment of this mixture with triphenylphosphine led to formation of phosphonium salt 78, and subsequent deprotonation with aqueous sodium carbonate under biphasic conditions gave the desired ylide 79 in 24% yield over the three steps.
Scheme 16.

Synthesis of dioxinone ylide 79.
Having secured functionalized dioxinone ylide 79, we returned to the tandem dihydroxylation/Wittig sequence from our original synthesis of the decahydrofluorene core of the hirsutellones.7 Thus, diastereoselective dihydroxylation of enol ether 80 (Scheme 17) under Upjohn conditions50 gave α-hydroxyaldehyde 16, which was subsequently treated with dioxinone ylide 79 following an aqueous workup. This sequence gave the expected Wittig product as a 3:1 mixture of separable alcohol epimers, favoring the desired allylic alcohol 81. As before, deprotonation of 81 with methyllithium followed by the addition of diethyl chlorophosphate gave allylic phosphate 82 in 77% yield.
Scheme 17.
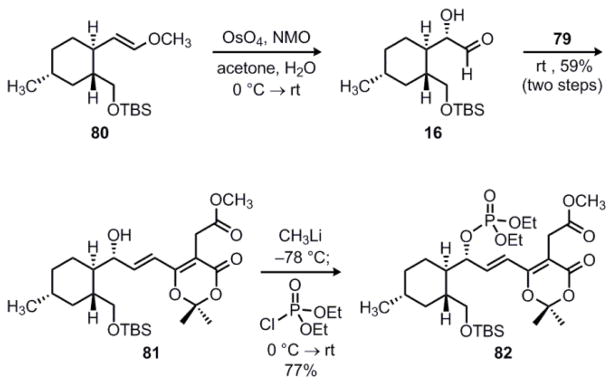
Synthesis of allylic phosphate 82.
Next, treatment of 82 with the sodium phenolate of 8351 under our standard Tsuji–Trost conditions produced aryl ether 84 (Scheme 18). Subsequent chemoselective cleavage of the TBS silyl ether of 84 using acetic acid gave primary alcohol 85 in 50% yield over the two steps. Next, oxidation of primary alcohol 85 with DMP afforded aldehyde 86 in 97% yield. Proceeding as in our previous route, Takai olefination of aldehyde 86 gave exclusively E vinyl iodide 87 in 94% yield. The Stille coupling of vinyl iodide 87 and dienyl stannane 42 occurred under our usual conditions to give the desired triene 88 in 88% yield.
Scheme 18.
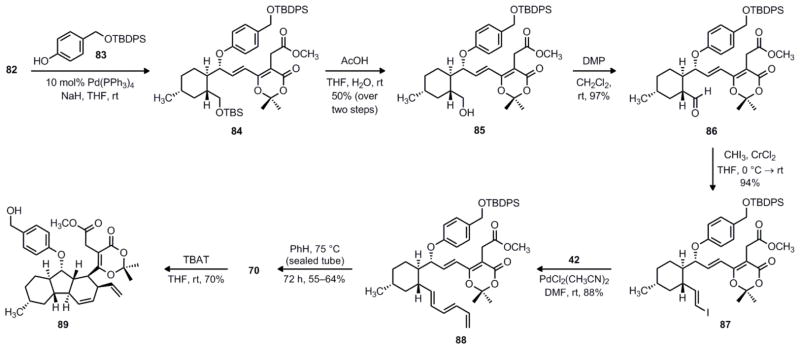
Synthesis of decahydrofluorene 89 via an IMDA reaction.
At this stage, we were ready to test whether we could achieve the intramolecular Diels–Alder reaction while retaining the dioxinone group. In the event, we were pleased to observe that the IMDA reaction occurred, albeit slowly, when a solution of 88 was heated to 75 °C in benzene. It is interesting to note that 70 was obtained as an inseparable 3:1 mixture of endo/exo isomers, whereas all of our tandem acylketene trapping/IMDA reactions occurred with complete diastereoselectivity. This observation provides some evidence that acylketene formation precedes the Diels–Alder reaction in the tandem sequences. Continuing on toward the precursor for the Stille reaction, deprotection of the TBDPS silyl ether of 70 using TBAT proceeded smoothly to give the corresponding benzylic alcohol. Conveniently, the endo/exo diastereomers were separable by chromatography at this stage, and the major product 89 was isolated in 70% yield.
At this stage, we needed to activate the benzylic alcohol as the corresponding halide as a prelude to forming the required stannane (Scheme 19). Although we were able to transform 89 into the corresponding benzylic bromide using the Appel reaction,52 this intermediate was unstable to chromatography. Therefore, we decided to form the less reactive chloride by treating 89 with methanesulfonyl chloride, which gave the isolable compound 90 in 86% yield.
Scheme 19.

Synthesis of key intermediate 71 and attempted intramolecular Stille coupling.
The conversion of benzylic chloride 90 to the corresponding stannane proved to be a challenging transformation. Ultimately, we were able to synthesize 91 via nucleophilic displacement of chloride with trimethylstannyllithium. After screening several conditions, this reagent was most conveniently prepared in situ from the reaction of hexamethylditin and methyllithium.53 Nevertheless, the displacement reaction itself suffered from low yields and reproducibility issues. We found that conducting the reaction at −40 °C was critical; at higher temperatures, the strongly nucleophilic stannyllithium reacts with the methyl ester or opens the dioxinone. Using our optimized conditions, we could consistently achieve yields in the 50% range, although this required the addition of several aliquots of the stannyllithium reagent. Interestingly, the reaction of 90 with the less toxic reagent tributylstannyllithium led to complete decomposition of the starting material.
Having secured benzylic stannane 91, we next focused on synthesizing the acid chloride in order to test the intramolecular Stille reaction. Hydrolysis of the methyl ester of 91 using lithium hydroxide in aqueous THF proceeded slowly at room temperature to give carboxylic acid 92 in 79% yield. Notably, the substituted dioxinone heterocycle survives the action of excess aqueous lithium hydroxide whereas the corresponding unsubstituted dioxinones are highly susceptible to ring openings under basic conditions. An exceptionally mild method for converting carboxylic acids to acid chlorides under neutral conditions utilizes chloroenamine 93, also known as the Ghosez reagent.54 We were pleased to observe that the reaction of carboxylic acid 92 with 93 proceeded almost instantaneously at room temperature to give the desired acid chloride 71 in essentially quantitative yield. The identity of 71 was confirmed when an aliquot was quenched with methanol, producing methyl ester 91; alternatively, the formation of 71 could be monitored by 1H-NMR. Since the only byproduct of this reaction is an innocuous amide, the crude solution of 71 in chloroform was used directly without purification. Conveniently, Stille’s original coupling protocol also used chloroform as the reaction solvent along with 94 as the palladium pre-catalyst.48 In order to minimize possible decarbonylation, we ran all reactions under an atmosphere of carbon monoxide. Unfortunately, we were unable to observe formation of the desired cyclophane 72 when acid chloride 71 was exposed to these reaction conditions. No reaction occurred at room temperature, and only decomposition of 71 was observed upon heating.
Notably, we found that acid chloride 71 was a competent substrate for an intermolecular Stille coupling with phenyltrimethylstannane under the conditions shown in Scheme 19. Unfortunately, we were never able to isolate the desired cyclophane 72 that would arise from an intramolecular Stille coupling, despite screening a wide variety of palladium catalysts, ligands, and solvents. One possible explanation is that the transfer of benzyl groups in the transmetallation step of the Stille reaction is often extremely slow. Indeed, benzyl groups are only slightly more reactive than alkyl groups, which are typically used as “inert” dummy ligands in the stannane component.34
Having developed an efficient route to compounds featuring a substituted dioxinone, we decide to pursue a formal total synthesis of hirsutellone B. We returned to triene 88 and found that heating this compound to 160 °C in a biphasic mixture of m-xylene and water gave decahydrofluorene 96 in 72% yield as a single diastereomer (Scheme 20). Presumably, the acylketene is trapped by water to give an intermediate β-keto acid 95 that undergoes in situ decarboxylation to form 96. Interestingly, thermolysis of substituted dioxinone 88 required a much higher temperature than was needed for any of the unsubstituted dioxinone substrates. For example, when 88 was heated to 120 °C in a mixture of toluene/water for three hours, Diels–Alder adduct 70 was again isolated as an inseparable 3:1 mixture of endo/exo isomers. In contrast, 96 was formed as a single diastereomer under the conditions shown in Scheme 20. These observations suggest that proceeding through the acylketene intermediate is critical in order to achieve high levels of diastereoselectivity in the IMDA reaction.
Scheme 20.

Synthesis of diol 97.
At this stage, exhaustive reduction of 96 with diisobutylaluminum hydride gave diol 97, which was formed as an inconsequential 1:1 mixture of diastereomers. The primary alcohol could then be selectively protected as the corresponding TBDPS ether in 72% yield to give secondary alcohol 98 (Scheme 21). Next, oxidation of 98 with DMP proceeded smoothly in 97% yield to provide ketone 99. Finally, removal of both silyl ether protecting groups could be achieved with buffered TBAF to give diol 100 in 97% yield. This constitutes a formal total synthesis since Nicolaou has previously shown that 100 can be converted to hirsutellone B in a ten-step sequence.12
Scheme 21.

Formal synthesis of hirsutellone B.
Conclusion
In summary, we have described the evolution of a strategy for a total synthesis of hirsutellone B featuring a novel “double cyclization” cascade. This key transformation takes advantage of the intrinsic reactivity of an acylketene to trigger macrocyclization and IMDA events that rapidly construct both the decahydrofluorene core and the strained para-cyclophane ring in a single step. We have demonstrated that the intramolecular acylketene trapping can be achieved using either amines or alcohols (both primary and secondary) as the nucleophile to form 14-membered macrolactams and macrolactones, respectively. Notably, the IMDA reaction is highly diastereoselective, establishing the correct configuration at four contiguous stereocenters along the decahydrofluorene core. Although we were unable to achieve the necessary ring-contraction to form the complete carbon framework of hirsutellone B, our attempts at transannular carbon-carbon bond formation did produce a series of unexpected cyclization products with interesting structures. This surprising reactivity demonstrates how the subtle interplay of stereoelectronic effects, conformation, and proximity can still be difficult to predict, especially in complex molecular settings.
Experimental Section
All reactions were carried out in oven or flame-dried glassware (unless water was present in the reaction mixture) with magnetic stirring under a positive pressure of argon unless otherwise indicated. Reactions were monitored by thin layer chromatography (TLC) carried out on 0.25 mm Merck silica gel plates (60 F254) containing a fluorescent indicator (254 nm). TLC plates were visualized under a UV lamp before treatment with the indicated stain and development with heat. Flash column chromatography was performed using Silicycle SiliaFlash P60 silica gel (60 Å pore size, 40–63 μm particle size, 230–400 mesh) and ACS reagent grade solvents. Preparative TLC was performed using Analtech silica gel GF glass-backed UNIPLATES (250 μm, 500 μm, 1000 μm, or 2000 μm thickness). All Grignard and alkyllithium reagents were titrated with salicylaldehyde phenylhydrazone before use.55 Anhydrous methanol, 1,2-dichloroethane, and acetone were purchased and used without further purification. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), toluene, diethyl ether, benzene, acetonitrile (CH3CN), dimethyl sulfoxide (DMSO), triethylamine (NEt3), and pyridine were dried by passing previously degassed solvents through activated alumina columns. High resolution mass spectral (HRMS) data was obtained on an electrospray ionization LC/MS equipped with a time-of-flight mass analyzer.
tert-butyl (S)-(1-((tert-butyldiphenylsilyl)oxy)-3-(4-hydroxyphenyl)propan-2-yl)carbamate (18)
To a solution of 1721 (223 mg, 0.83 mmol, 1.00 eq.) in 6 mL of CH2Cl2 was added imidazole (382 mg, 5.62 mmol, 6.75 eq.) followed by the dropwise addition of neat tert-butyldiphenylchlorosilane (487 μL, 1.87 mmol, 2.25 eq.). After 30 minutes at room temperature, TLC (10:1 hexanes/EtOAc, UV/ninhydrin) showed complete consumption of the starting material and formation of the product of Rf = 0.33. The reaction was quenched with water, the layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2. The combined organics were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure to give 598 mg of the crude bis-TBDPS protected tyrosinol as a colorless oil.
A solution of this crude product in 11 mL of THF was cooled to 0 °C in an ice bath, and a 1.0 M solution of TBAF in THF (804 μL, 0.80 mmol, 1.00 eq.) was added dropwise. The resulting light yellow solution was stirred at 0 °C for 30 minutes, at which point TLC (3:1 hexanes/EtOAc, ninhydrin) showed complete consumption of the starting material (Rf = 0.70) and formation of the product (Rf = 0.37). The reaction was quenched with saturated NH4Cl and diluted with water and EtOAc. The layers were separated, and the organic phase was washed with brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residual oil was purified by column chromatography (3:1 hexanes/EtOAc) to afford 18 as a colorless, viscous oil (340 mg, 81% over two steps).
[α]20D = +18.7 (c 1.04, CH2Cl2); IR (neat) ν 3050, 2870, 2857, 1617, 1542, 1522, 1438, 1294, 1237, 1148, 1022, 847 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.10 (9H, s), 1.43 (9H, s), 2.81 (2H, m), 3.56 (1H, dd, J = 3.08 Hz, 10.16 Hz), 3.61 (1H, m), 3.84 (1H, m), 4.87 (1H, d, J = 8.96 Hz), 6.24 (1H, br s), 6.68 (2H, d, J = 8.08 Hz), 6.97 (2H, d, J = 7.89 Hz), 7.38-7.63 (10H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 27.0, 28.5, 36.9, 53.5, 64.0, 79.6, 115.3, 127.8, 129.7, 129.8, 130.4, 133.2, 135.60, 135.63, 154.6, 155.7; HRMS (ESI+) calculated for C30H40NO4Si ([M+H]+): 506.2727, found 506.2730.
(S)-4-(3-((tert-butyldiphenylsilyl)oxy)-2-((3,4-dimethylbenzyl)amino)propyl)phenol (14)
A solution of 18 (1.35 g, 2.67 mmol, 1.00 eq.) in 35 mL of dry CH2Cl2 was cooled to 0 °C in an ice bath, and neat trifluoroacetic acid (17.5 mL, 236 mmol, 89 eq.) was added dropwise. After 15 minutes, TLC (100% EtOAc, UV/ninhydrin) showed complete consumption of the starting material and formation of the product of Rf = 0.70. The solvent was removed under reduced pressure, and the residue was dried under high vacuum to give crude TFA salt 19 as a white amorphous solid (1.20 g, 87%).
To a portion of this crude material (150 mg, 0.29 mmol, 1.00 eq.) and 2,4-dimethoxybenzaldehyde (46 mg, 0.27 mmol, 0.95 eq.) in 2.5 mL of 1,2-dichloroethane was added triethylamine (80.4 μL, 0.58 mmol, 2.00 eq.) and solid sodium triacetoxyborohydride (128 mg, 0.61 mmol, 2.10 eq.). The resulting cloudy reaction mixture was stirred at room temperature for 15 hours at which point TLC (2:1 EtOAc/hexanes, UV/KMnO4) showed clean conversion to the product of Rf = 0.31. The reaction mixture was diluted with CH2Cl2 and quenched with pH 7 phosphate buffer. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2. The combined organics were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure to give a yellow oil. The crude product was then purified by column chromatography (1:1 hexanes/EtOAc → 100% EtOAc) to give phenol 14 as a white foam (142 mg, 77% over two steps).
[α]20D = −6.3 (c 1.45, CH2Cl2); IR (neat) ν 3071, 2999, 2931, 2857, 1614, 1589, 1508, 1463, 1428, 1289, 1260, 1208, 1157, 1137, 1112, 1037, 823, 702 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.05 (9H, s), 2.66 (1H, dd, J = 7.11 Hz, 13.86 Hz), 2.76 (1H, dd, J = 6.43 Hz, 13.87 Hz), 2.88 (1H, q, J = 6.29 Hz), 3.57 (1H, m), 3.59 (3H, s), 3.67 (1H, d, J = 13.46 Hz), 3.68 (1H, m), 3.71 (1H, d, J = 13.23 Hz), 3.80 (3H, s), 6.37-6.39 (2H, m), 6.65 (2H, d, J = 8.28 Hz), 6.90 (2H, d, J = 8.29 Hz), 6.98 (1H, d, J = 7.77 Hz), 7.33-7.38 (4H, m), 7.40-7.44 (2H, m), 7.61 (2H, d, J = 7.53 Hz), 7.64 (2H, d, J = 7.53 Hz); 13C NMR (125 MHz, CDCl3): δ 19.3, 26.9, 36.6, 46.6, 55.1, 55.5, 59.4, 65.5, 98.5, 103.6, 115.3, 127.74, 127.77, 129.71, 129.74, 130.3, 130.7, 133.44, 133.49, 135.62, 135.65, 154.4, 158.6, 160.3 [Note: one fully substituted carbon from the tyrosine ring was not observed]; HRMS (ESI+) calculated for C34H42NO4Si ([M+H]+): 556.2883, found 556.2874.
Allylic phosphate 20
A solution of 12 (1.04 g, 2.46 mmol, 1.00 eq.) in 25 mL of THF was cooled to −78 °C, and a 1.6 M solution of methyllithium in ether (1.69 mL, 2.70 mmol, 1.10 eq.) was added dropwise to give a dark yellow solution. After five minutes, neat diethyl chlorophosphate (530 μL, 3.69 mmol, 1.50 eq.) was added, and the flask was transferred to an ice bath. After 10 minutes, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material (Rf = 0.66) and formation of the product of Rf = 0.36. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc. The combined organics were dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 hexanes/EtOAc) to give 20 as a colorless oil (1.13 g, 82%).
[α]20D = −18.8 (c 2.66, CH2Cl2); IR (neat) ν 2951, 2928, 2857, 1732, 1659, 1597, 1391, 1271, 1034, 986, 837 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.04 (3H, s), 0.05 (3H, s), 0.83 (1H, m), 0.89 (9H, s), 1.01 (1H, q, J = 12.09 Hz), 1.23-1.37 (8H, m), 1.47-1.68 (4H, m), 1.72 (6H, s), 3.58 (1H, d, J = 10.24 Hz), 3.84 (1H, dd, J = 4.73 Hz, 10.23 Hz), 4.10 (4H, m), 5.18 (1H, m), 5.32 (1H, s), 6.12 (1H, d, J = 15.58 Hz), 6.48 (1H, dd, J = 5.06 Hz, 15.59 Hz); 13C NMR (125 MHz, CDCl3): δ −5.5, −5.4, 16.2 (d, JC-P = 6.96 Hz), 18.3, 22.6, 25.0, 25.06, 25.09, 25.9, 32.2, 34.5, 38.9, 39.8, 42.6 (d, JC-P = 6.02 Hz), 63.87 (d, JC-P = 5.80 Hz), 63.89 (d, JC-P = 5.80 Hz), 64.2, 77.7 (d, JC-P = 6.24 Hz), 95.2, 106.6, 123.0, 138.9 (d, JC-P = 1.95 Hz), 161.8, 162.3; HRMS (ESI+) calculated for C27H49NaO8PSi ([M+Na]+): 583.2832, found 583.2807.
Aryl ether 21
A 0.015 M stock solution of tetrakis(triphenylphosphine)palladium(0) was prepared by dissolving 52.5 mg of the solid catalyst in 3.0 mL of THF. A 0.125 M solution of phenolate was prepared by adding a solution of phenol 14 (223 mg, 0.40 mmol, 1.05 eq.) in 3.21 mL of THF to 16 mg of 60% sodium hydride dispersion in mineral oil. Vigorous bubbling was observed, and a colorless phenolate solution was obtained after 10 minutes. To a flask containing neat allylic phosphate 20 (214 mg, 0.38 mmol, 1.00 eq.) was added a portion of the bright yellow catalyst solution (2.54 mL, 38.2 μmol, 0.10 eq.) followed by dropwise addition of the phenolate solution. After each drop, the reaction mixture turned dark orange before returning to light yellow after several seconds. After five minutes, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the phosphate and formation of the product of Rf = 0.54. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 3:1 hexanes/EtOAc → 1:1 hexanes/EtOAc) to give aryl ether 21 as a colorless oil (236 mg, 64%).
[α]20D = −9.6 (c 1.11, CH2Cl2); IR (neat) ν 2928, 2856, 1730, 1655, 1612, 1590, 1508, 1463, 1390, 1375, 1258, 1235, 1208, 1113 cm−1; 1H NMR (500 MHz, CDCl3): δ −0.06 (3H, s), −0.04 (3H, s), 0.86 (9H, s), 0.89 (1H, m), 0.91 (3H, d, J = 6.31 Hz), 0.97 (1H, m), 1.04 (9H, s), 1.43 (1H, m), 1.47 (1H, m), 1.59 (1H, m), 1.62 (1H, m), 1.64 (1H, m), 1.67 (1H, m), 1.71 (3H, s), 1.72 (3H, s), 1.74 (1H, m), 2.67 (1H, dd, J = 6.57 Hz, 1.83 Hz), 2.74 (1H, dd, J = 6.18 Hz, 13.85 Hz), 2.88 (1H, m), 3.48 (2H, m), 3.56 (1H, m), 3.62 (3H, s), 3.65 (3H, m), 3.81 (3H, s), 4.97 (1H, m), 5.24 (1H, s), 6.02 (1H, d, J = 15.67 Hz), 6.38 (2H, m), 6.56 (1H, dd, J = 4.01 Hz, 15.72 Hz), 6.68 (2H, d, J = 8.50 Hz), 6.97 (2H, d, J = 8.45 Hz), 7.00 (1H, m), 7.33 (4H, m), 7.36 (2H, m), 7.62 (4H, m); 13C NMR (125 MHz, CDCl3): δ −5.43, −5.41, 18.4, 19.3, 22.7, 25.1, 25.2, 25.8, 26.0, 26.9, 32.3, 34.9, 36.9, 39.1, 40.3, 43.1, 46.6, 55.1, 55.4, 59.6, 65.1, 65.6, 77.2, 94.7, 98.4, 103.5, 106.5, 115.0, 121.0, 122.8, 127.68, 127.71, 129.64, 129.67, 130.3, 131.8, 133.63, 133.68, 135.64, 135.65, 140.8, 156.7, 158.6, 160.0, 161.9, 162.5; HRMS (ESI+) calculated for C57H80NO8Si2 ([M+H]+): 962.5422, found 962.5431.
Primary alcohol 22
To a solution of aryl ether 21 (416 mg, 0.43 mmol, 1.00 eq.) in 40 mL of 1:1 THF/water was added glacial acetic acid (17.32 mL, 303 mmol, 700 eq.), and the reaction mixture was stirred at room temperature for 14 hours. After this time, TLC (100% EtOAc, UV/anisaldehyde) showed complete conversion to the product of Rf = 0.47. The reaction mixture was diluted with water and EtOAc and quenched by the dropwise addition of saturated aqueous K2CO3 solution. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc. The combined organics were dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (1:1 hexanes/EtOAc → 100% EtOAc) to give primary alcohol 22 as a colorless oil (302 mg, 82%).
[α]20D = −12.6 (c 1.64, CH2Cl2); IR (neat) ν 2928, 2858, 1724, 1654, 1612, 1590, 1508, 1463, 1390, 1375, 1289, 1236, 1208, 1112 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.91 (1H, m), 0.92 (3H, d, J = 6.53 Hz), 0.98 (1H, m), 1.03 (9H, s), 1.42 (1H, m), 1.47 (1H, m), 1.60 (1H, m), 1.68 (1H, m), 1.69 (1H, m), 1.70 (3H, s), 1.71 (3H, s), 1.72 (1H, m), 1.78 (1H, m), 2.66 (1H, dd, J = 6.65 Hz, 13.87 Hz), 2.74 (1H, dd, J = 6.45 Hz, 13.89 Hz), 2.85 (1H, m), 3.53 (2H, m), 3.62 (3H, s), 3.64 (4H, m), 3.81 (3H, s), 4.97 (1H, m), 5.26 (1H, s), 6.06 (1H, dd, J = 1.41 Hz, 15.68 Hz), 6.38 (2H, m), 6.60 (1H, dd, J = 4.49 Hz, 15.67 Hz), 6.70 (2H, d, J = 8.57 Hz), 6.96 (2H, d, J = 8.64 Hz), 6.98 (1H, m), 7.34 (4H, m), 7.40 (2H, m), 7.60 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.3, 22.6, 25.1, 25.2, 26.9, 27.1, 32.3, 34.8, 36.9, 38.9, 40.5, 43.5, 46.6, 55.1, 55.5, 59.5, 65.6, 65.9, 79.0, 95.0, 98.5, 103.5, 106.6, 115.1, 120.9, 123.4, 127.69, 127.72, 129.65, 129.68, 130.4, 132.3, 133.60, 133.66, 135.65, 135.66, 139.3, 156.3, 158.6, 160.0, 161.8, 162.3; HRMS (ESI+) calculated for C51H66NO8Si ([M+H]+): 848.4558, found 848.4547.
Aldehyde 23
To a solution of alcohol 22 (187 mg, 0.22 mmol, 1.00 eq.) and NMO (30.9 mg, 0.26 mmol, 1.20 eq.) in 6.0 mL of anhydrous CH2Cl2 was added 500 mg of 4Å molecular sieves. A 0.05 M solution of TPAP was prepared by dissolving 17.6 mg of the solid catalyst in 1.0 mL of CH2Cl2. A portion of the TPAP solution (352 μL, 0.18 μmol, 0.08 eq.) was added to the reaction mixture, and the dark black solution was stirred at room temperature for 1.5 hours. After this time, TLC (100% EtOAc, UV/anisaldehyde) showed complete conversion of the starting material (Rf = 0.55) to the product of Rf = 0.68. The reaction was filtered through a plug of Celite/Florisil, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 1:1 → 100% EtOAc) to give aldehyde 23 as a white foam (116 mg, 62%).
[α]20D = −12.0 (c 1.40, CH2Cl2); IR (neat) ν 2928, 2856, 1724, 1656, 1611, 1590, 1508, 1462, 1391, 1375, 1273, 1233, 1208, 1112, 1038 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.89 (1H, m), 0.94 (1H, m), 0.95 (3H, d, J = 6.49 Hz), 1.02 (9H, s), 1.39 (1H, m), 1.48 (1H, m), 1.75 (1H, m), 1.77 (1H, m), 1.84 (1H, m), 2.07 (1H, m), 2.47 (1H, m), 2.65 (1H, dd, J = 6.77 Hz, 13.96 Hz), 2.73 (1H, dd, J = 6.48 Hz, 13.93 Hz), 2.85 (1H, m), 3.54 (1H, dd, J = 5.30 Hz, 10.12 Hz), 3.59 (3H, s), 3.62 (2H, m), 3.69 (1H, d, J = 13.19 Hz), 3.80 (3H, s), 4.91 (1H, m), 5.28 (1H, s), 6.07 (1H, dd, J = 1.38 Hz, 15.67 Hz), 6.38 (2H, m), 6.55 (1H, dd, J = 4.79 Hz, 15.67 Hz), 6.65 (2H, d, J = 8.60 Hz), 6.94 (2H, d, J = 8.56 Hz), 6.97 (1H, d, J = 8.64 Hz), 7.34 (4H, m), 7.41 (2H, m), 7.61 (4H, m), 9.52 (1H, d, J = 3.00 Hz); 13C NMR (125 MHz, CDCl3): δ 19.3, 22.4, 25.1, 25.2, 25.4, 26.9, 31.7, 34.0, 34.5, 36.9, 41.5, 46.6, 51.3, 55.1, 55.5, 59.4, 65.6, 78.2, 95.3, 98.4, 103.5, 106.6, 115.2, 120.9, 124.0, 127.69, 127.72, 129.65, 129.69, 130.4, 132.3, 133.59, 133.65, 135.65, 135.65, 138.3, 155.9, 158.6, 160.0, 161.7, 162.1, 203.5; HRMS (ESI+) calculated for C51H64NO8Si ([M+H]+): 846.4401, found 846.4397.
Triene 8
A suspension of phosphonium salt 13 (449 mg, 1.10 mmol, 8.00 eq.) in 3.5 mL of dry THF was cooled to −78 °C, and a 0.5 M solution of KHMDS in toluene (1.92 mL, 9.60 mmol, 7.00 eq.) was added dropwise. The resulting dark red ylide solution was warmed to 0 °C for 30 minutes before the dropwise addition of a solution of aldehyde 23 (116 mg, 0.14 mmol, 1.00 eq.) in 3.0 mL of dry THF. After 30 minutes at 0 °C, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the aldehyde and formation of the product of Rf = 0.49. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with one additional portion of EtOAc. The combined organic phases were dried over anhydrous Na2SO4 before removal of the solvent under reduced pressure. The residue was purified by column chromatography to give triene 8 as a colorless oil (93.8 mg, 76%). 1H-NMR showed that the triene was formed as an 7:1 mixture of E/Z isomers.
IR (neat) ν 2928, 2857, 1726, 1654, 1612, 1590, 1507, 1463, 1390, 1375, 1267, 1233, 1207, 1156, 1112, 1008 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.89 (1H, m), 0.90 (3H, d, J = 6.46 Hz), 0.96 (1H, m), 1.03 (9H, s), 1.38 (1H, m), 1.48 (1H, m), 1.76 (1H, m), 1.78 (1H, m), 1.82 (1H, m), 2.08 (1H, m), 2.25 (1H, m), 2.72 (2H, m), 2.88 (1H, m), 3.56 (1H, dd, J = 5.25 Hz, 10.30 Hz), 3.61 (3H, s), 3.63-3.67 (2H, m), 3.70 (1H, d, J = 13.26 Hz), 3.80 (3H, s), 4.90 (1H, m), 4.99 (1H, d, J = 10.29 Hz), 5.05 (1H, d, J = 16.93 Hz), 5.22 (1H, s), 5.43 (1H, dd, J = 9.30 Hz, 15.17 Hz), 5.63 (1H, dd, J = 10.59 Hz, 15.19 Hz), 5.75 (1H, dd, J = 10.71 Hz, 14.80 Hz), 6.01 (1H, dd, J = 1.46 Hz, 15.64 Hz), 6.07 (1H, dd, J = 10.63 Hz, 15.05 Hz), 6.25 (1H, dt, J = 10.37 Hz, 16.92 Hz), 6.38 (2H, m), 6.54 (1H, dd, J = 4.05 Hz, 15.61 Hz), 6.66 (2H, d, J = 8.52 Hz), 6.94 (2H, d, J = 8.45 Hz), 6.98 (1H, d, J = 8.73 Hz), 7.34 (4H, m), 7.40 (2H, m), 7.61 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.3, 22.5, 24.8, 25.1, 25.2, 26.9, 32.0, 34.6, 36.7, 42.0, 43.2, 46.5, 47.3, 55.1, 55.5, 59.5, 65.4, 77.6, 94.7, 98.4, 103.6, 106.5, 115.3, 117.0, 120.5, 122.7, 127.70, 127.74, 129.67, 129.69, 130.2, 130.5, 131.7, 132.0, 132.7, 132.9, 133.55, 133.59, 135.63, 135.65, 136.9, 138.3, 140.9, 156.7, 158.6, 160.1, 161.9, 162.5; HRMS (ESI+) calculated for C56H70NO7Si ([M+H]+): 896.4922, found 896.4920.
Macrolactam 10
A 2-neck 500 mL flask equipped with a magnetic stirring bar, Dean–Stark trap, and a reflux condenser was flame-dried under high vacuum and cooled to room temperature under argon. A solution of triene 8 (93.8 mg, 0.11 mmol, 1.00 eq.) in 350 mL of freshly distilled benzene (sodium/benzophenone) was added, and the reaction mixture was heated to reflux in a 105 °C oil bath overnight. After cooling to room temperature, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.63. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 hexanes/EtOAc → 1:1 hexanes/EtOAc) to give macrolactam 10 (34.1 mg, 39%) as a colorless oil. NMR showed that this compound exists as a 3:1 mixture of enol/keto tautomers in CDCl3 solution but almost exclusively as the enol tautomer in C6D6 solution.
[α]20D = +17.7 (c 1.10, CH2Cl2); IR (neat) ν 2923, 2854, 1613, 1588, 1505, 1462, 1427, 1343, 1287, 1257, 1207, 1187, 1156, 1111, 1037, 1005, 963, 822, 738, 702, 504 cm−1; 1H NMR (500 MHz, C6D6): δ 0.86 (1H,m ), 0.90 (3H, d, J = 7.03 Hz), 0.96 (1H, m), 0.97 (1H, m), 1.17 (9H, s), 1.25 (1H, m), 1.29 (1H, m), 1.67 (1H, m), 1.74 (1H, m), 1.85 (1H, m), 2.06 (1H, m), 2.39 (1H, m), 2.52 (1H, m), 2.59 (1H, d, J = 11.98 Hz), 2.71 (1H, dd, J = 12.0 Hz, 6.8 Hz), 2.81 (1H, d, J = 12.69 Hz), 2.93 (1H, m), 3.20 (3H, s), 3.34 (3H, s), 3.43 (1H, m), 3.59 (1H, m), 3.84 (1H, s), 3.86 (1H, m), 4.49 (1H, d, J = 15.87 Hz), 4.82 (1H, m), 5.05 (2H, m), 5.28 (1H, d, J = 16.10 Hz), 5.62 (1H, m), 6.03 (1H, d, J = 9.96 Hz), 6.10 (1H, m), 6.25 (1H, dd, J = 8.48 Hz, 2.30 Hz), 6.39 (1H, d, J = 2.26 Hz), 6.56 (1H, dd, J = 8.34 Hz, 1.92 Hz), 6.71 (1H, dd, J = 8.34 Hz, 1.92 Hz), 6.79 (1H, dd, J = 8.29 Hz, 2.34 Hz), 6.88 (1H, dd, J = 8.22 Hz, 2.41 Hz), 7.22 (6H, m), 7.51 (1H, d, J = 8.40 Hz), 7.59 (2H, m), 7.64 (2H, m), 15.18 (1H, s); 13C NMR (125 MHz, C6D6): δ 19.4, 22.8, 27.1, 29.6, 33.4, 36.1, 37.0, 38.7, 39.1, 42.9, 44.8, 46.8, 47.4, 50.4, 54.6, 54.9, 56.4, 64.7, 65.1, 82.8, 87.6, 98.6, 104.2, 115.6, 118.8, 120.0, 120.7, 128.3, 128.4, 128.5, 129.3, 129.5, 130.0, 130.1, 130.7, 130.8, 132.9, 133.59, 133.63, 136.06, 136.08, 139.1, 157.4, 159.5, 160.2, 174.9, 175.1; HRMS (ESI+) calculated for C53H64NO6Si ([M+H]+): 838.4503, found 838.4506.
Macrolactam alcohol 24
To a solution of macrolactam 10 (34.1 mg, 40.7 μmol, 1.00 eq.) in 3 mL of THF was added solid tetrabutylammonium triphenyldifluorosilicate (TBAT, 132 mg, 0.24 mmol, 6.00 eq.), and the reaction mixture was stirred at room temperature. After six hours, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde), showed complete consumption of the starting material and formation of the product of Rf = 0.45. The reaction mixture was diluted with EtOAc and quenched with pH 7 phosphate buffer. The layers were separated, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 1:1 hexanes/EtOAc) to give primary alcohol 24 as a colorless oil (24 mg, 98%). NMR showed that this compound exists as a 6:1 mixture of enol/keto tautomers in C6D6 solution.
[α]20D = +24.7 (c 0.97, CH2Cl2); IR (neat) ν 3496, 2922, 2853, 1616, 1589, 1506, 1463, 1291, 1258, 1237, 1208, 1182, 1157, 1121, 1038 cm−1; 1H NMR (500 MHz, C6D6): δ 0.88 (1H, m), 0.91 (3H, d, J = 6.60 Hz), 0.93 (1H, m), 1.01 (1H, td, J = 12.4 Hz, 3.9 Hz), 1.21 (1H, m), 1.27 (1H, m), 1.70 (1H, m), 1.85 (1H, ddd, J = 13.1 Hz, 8.0 Hz, 2.7 Hz), 1.92 (1H, m), 2.05 (1H, m), 2.06 (1H, ddd, J = 11.7 Hz, 9.1 Hz, 4.5 Hz), 2.28 (1H, m), 2.33 (1H, m), 2.38 (1H, m), 2.57 (1H, dd, J = 12.0 Hz, 6.7 Hz), 2.89 (1H, m), 3.28 (3H, s), 3.29 (3H, s), 3.37 (1H, m), 3.65 (1H, m), 3.45 (1H, m), 3.93 (1H, s), 4.31 (1H, d, J = 15.9 Hz), 4.83 (1H, t, J = 3.6 Hz), 5.06 (2H, m), 5.30 (1H, d, J = 15.8 Hz), 5.58 (1H, m), 6.01 (1H, d, J = 10.0 Hz), 6.12 (1H, m), 6.15 (1H, dd, J = 8.3 Hz, 2.5 Hz), 6.41 (1H, d, J = 2.4 Hz), 6.53 (1H, dd, J = 8.3 Hz, 2.2 Hz), 6.63 (1H, dd, J = 8.3 Hz, 2.2 Hz), 6.80 (1H, dd, J = 8.3 Hz, 2.5 Hz), 6.87 (1H, dd, J = 8.2 Hz, 2.6 Hz), 7.51 (1H, d, J = 8.4 Hz), 15.10 (1H, s); 13C NMR (125 MHz, C6D6): δ 22.8, 29.6, 33.4, 35.4, 37.0, 37.7, 38.4, 42.9, 44.8, 46.7, 47.3, 50.5, 54.87, 54.89, 56.4, 63.6, 65.5, 82.9, 87.6, 99.2, 104.6, 115.7, 118.8, 119.7, 123.1, 128.1, 129.4, 129.9, 130.5, 130.7, 132.5, 139.1, 157.4, 159.5, 160.6, 175.3, 176.2; HRMS (ESI+) calculated for C37H46NO6 ([M+H]+): 600.3325, found 600.3314.
Acylketene hemiaminal ether 26
A solution of primary alcohol 24 (4.5 mg, 7.50 μmol, 1.0 eq.) in 1 mL of CH2Cl2 was cooled to 0 °C, and triethylamine (10 μL, 71.7 μmol, 9.6 eq.) was added followed by neat methanesulfonyl chloride (6.0 μL, 77.5 μmol, 10.3 eq.). The reaction mixture was stirred at 0 °C for 30 minutes before the addition of neat DBU (10 μL, 66.9 μmol, 8.9 eq.). The cooling bath was removed, and the reaction mixture was stirred at room temperature for 15 minutes before the addition of water and EtOAc. The layers were separated, and the organic phase was washed with water and brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (eluting with 100% EtOAc) to give 26 as a colorless oil (1.2 mg, 28%).
[α]20D = −16.0 (c 0.10, CH2Cl2); IR (neat) ν 2919, 2850, 1729, 1612, 1536, 1508, 1456, 1292, 1240, 1209, 1173, 1158, 1114, 1032 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.88 (1H, m), 0.93 (1H, m), 0.95 (3H, d, J = 6.6 Hz), 1.07 (1H, qd, J = 12.5 Hz, 4.1 Hz), 1.40 (1H, m), 1.42 (1H, m), 1.56 (1H, m), 1.80 (1H, m), 1.83 (1H, m), 1.99 (1H, m), 2.04 (1H, m), 2.06 (1H, m), 2.66 (1H, d, J = 14.1 Hz), 2.85 (1H, dd, J = 11.7 Hz, 7.0 Hz), 2.91 (1H, dd, J = 14.1 Hz, 4.7 Hz), 2.96 (1H, m), 3.76 (1H, s), 3.81 (3H, s), 3.82 (1H, m), 3.85 (3H, s), 3.99 (1H, d, J = 14.8 Hz), 4.24 (1H, d, J = 14.9 Hz), 4.26 (1H, m), 4.38 (1H, d, J = 8.9 Hz), 4.78 (1H, d, J = 17.0 Hz), 4.86 (1H, m), 4.87 (1H, m), 5.41 (1H, dt, J = 9.8 Hz, 3.4 Hz), 5.66 (1H, ddd, J = 17.6 Hz, 9.8 Hz, 8.4 Hz), 5.90 (1H, d, J = 9.8 Hz), 6.43 (1H, dd, J = 8.3 Hz, 2.3 Hz), 6.47 (1H, d, J = 2.2 Hz), 6.80 (1H, dd, J = 8.3 Hz, 2.6 Hz), 6.91 (1H, dd, J = 8.6 Hz, 2.5 Hz), 6.94 (1H, dd, J = 8.3 Hz, 2.0 Hz), 7.06 (1H, d, J = 8.0 Hz), 7.16 (1H, dd, J = 8.6 Hz, 2.0 Hz); 13C NMR (125 MHz, CDCl3): δ 22.7, 29.2, 33.1, 36.6, 37.6, 38.2, 42.0, 43.1, 43.3, 46.6, 49.2, 50.4, 55.52, 55.46, 56.2, 58.6, 71.3, 79.6, 81.6, 98.6, 104.1, 114.7, 116.0, 117.1, 119.0, 126.3, 127.8, 129.7, 130.0, 130.3, 131.1, 139.1, 158.3, 159.8, 160.8, 163.1, 192.5; HRMS (ESI+) calculated for C37H44NO5 ([M+H]+): 582.3219, found 582.3215.
Aldehyde 27
To a solution of macrolactam alcohol 24 (3.0 mg, 5.0 μmol, 1.00 eq.) in 500 μL of CH2Cl2 was added solid Dess–Martin periodinane (2.75 mg, 6.50 μmol, 1.30 eq.). After 15 minutes at room temperature, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the product of Rf = 0.62. The reaction was diluted with CH2Cl2 and quenched with saturated aqueous sodium thiosulfate solution and saturated aqueous NaHCO3 solution. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2. The combined organics were dried over anhydrous Na2SO4 before removal of the solvent under reduced pressure to give essentially pure aldehyde 27 (2.5 mg, 84%) which was used without further purification.
(S)-4-(3-((tert-butyldiphenylsilyl)oxy)-2-((3,4-dimethylbenzyl)amino)propyl)phenol (35)
To a solution of enantioenriched diol 3431 (400 mg, 1.55 mmol, 1.00 eq.) in 8 mL of CH2Cl2 was added dibutyltin oxide (7.71 mg, 31 μmol, 0.02 eq.), p-toluenesulfonyl chloride (295 mg, 1.55 mmol, 1.00 eq.) and neat triethylamine (216 μL, 1.55 mmol, 1.00 eq.). Over the course of 30 minutes, the reaction mixture turned cloudy and the dibutyltin oxide dissolved. After four hours at room temperature, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed formation of the primary tosylate at Rf = 0.26 and the secondary tosylate byproduct of Rf = 0.19. The reaction mixture was quenched with water and diluted with CH2Cl2. The layers were separated, and the aqueous phase was extracted with two additional portions of CH2Cl2. The combined organics were dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 hexanes/EtOAc) to give primary tosylate 35 as a colorless oil (476 mg, 75%) along with the corresponding secondary tosylate byproduct (72.4 mg, 11%).
[α]20D = −1.9 (c 1.03, CH2Cl2); IR (neat) ν 3523, 3034, 2922, 1611, 1511, 1454, 1358, 1241, 1189, 1175, 1096, 978 cm−1; 1 H NMR (500 MHz, CDCl3): δ 2.02 (1H, d, J = 4.66 Hz), 2.46 (3H, s), 2.72 (2H, m), 3.93 (1H, dd, J = 5.97 Hz, 9.06 Hz), 4.03 (2H, m), 5.04 (2H, s), 6.89 (2H, d, J = 8.05 Hz), 7.06 (2H, d, J = 8.02 Hz), 7.32-7.43 (7H, m), 7.79 (2H, d, J = 7.84 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 38.5, 70.1, 70.5, 72.7, 115.1, 127.5, 128.1, 128.7, 128.8, 130.0, 130.4, 132.6, 137.0, 145.2, 157.8; HRMS (ESI+) calculated for C23H25O5S ([M+H]+): 413.1423, found 413.1430.
(R)-3-(4-(benzyloxy)phenyl)-2-((tert-butyldimethylsilyl)oxy)propyl 4-methylbenzenesulfonate (36)
A solution of 35 (238 mg, 0.58 mmol, 1.00 eq.) in 4 mL of CH2Cl2 was cooled to −78 °C, and neat 2,6-lutidine (100 μL, 0.87 mol, 1.50 eq.) was added followed by the dropwise addition of neat TBSOTf (159 μL, 0.69 mmol, 1.20 eq.). After 1.5 hours at −78 °C, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.68. The reaction was diluted with CH2Cl2 and quenched with water at −78 °C, at which point a bright yellow color appeared. After warming to room temperature, the organic layer was isolated, and the aqueous phase was extracted with one additional portion of CH2Cl2. The combined organics were dried over anhydrous Na2SO4 before removal of the solvent under reduced pressure and purification of the residue by column chromatography (3:1 hexanes/EtOAc) to give 36 as a colorless oil (298 mg, 98%).
[α]20D = +4.3 (c 2.05, CH2Cl2); IR (neat) ν 2952, 2928, 2856, 1612, 1511, 1471, 1454, 1362, 1244, 1190, 1176, 1118, 1097, 983, 834, 813, 777 cm−1; 1H NMR (500 MHz, CDCl3): δ −0.26 (3H, s), −0.10 (3H, s), 0.78 (9H, s), 2.45 (3H, s), 2.58 (1H, dd, J = 7.10 Hz, 13.73 Hz), 2.73 (1H, dd, J = 5.23 Hz, 13.74 Hz), 3.85 (2H, d, J = 5.25 Hz), 3.95 (1H, quintet, J = 5.59 Hz), 5.04 (2H, s), 6.85 (2H, d, J = 7.78 Hz), 7.00 (2H, d, J = 7.90 Hz), 7.33 (3H, m), 7.38 (2H, t, J = 7.31 Hz), 7.43 (2H, d, J = 7.57 Hz), 7.77 (2H, d, J = 7.68 Hz); 13C NMR (125 MHz, CDCl3): δ −5.2, −4.9, 18.0, 21.8, 25.8, 39.8, 70.0, 71.3, 72.5, 114.8, 127.5, 128.0, 128.1, 128.7, 129.6, 129.9, 130.8, 132.9, 137.1, 144.9, 157.5; HRMS (ESI+) calculated for C29H39O5SSi ([M+H]+): 527.2287, found 527.2290.
(R)-2-((tert-butyldimethylsilyl)oxy)-3-(4-hydroxyphenyl)propyl 4-methylbenzenesulfonate (37)
To a flask containing solid 10% palladium on carbon (150 mg, 0.14 mmol, 0.25 eq.) was added a solution of protected diol 36 (298 mg, 0.57 mmol, 1.00 eq.) in 10 mL of anhydrous methanol and 5 mL of EtOAc. The flask was evacuated, put under an atmosphere of hydrogen gas (balloon), and allowed to stir at room temperature. After three hours, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.46. The crude reaction mixture was filtered through Celite topped with a thin layer of silica gel to remove the solids, and the pad was washed with additional EtOAc. The solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc) to afford phenol 37 as a colorless oil (242 mg, 98%).
[α]20D = +0.59 (c 1.41, CH2Cl2); IR (neat) ν 3480, 2953, 2929, 2857, 1614, 1598, 1516, 1472, 1359, 1258, 1189, 1175, 1118, 983, 835, 778 cm−1; 1H NMR (500 MHz, CDCl3): δ −0.25 (3H, s), −0.10 (3H, s), 0.78 (9H, s), 2.45 (3H, s), 2.57 (1H, dd, J = 7.11 Hz, 13.79 Hz), 2.72 (1H, dd, J = 5.32 Hz, 13.79 Hz), 3.84 (2H, d, J = 5.35 Hz), 3.94 (1H, td, J = 5.39 Hz, 10.80 Hz), 4.75 (2H, s), 6.71 (2H, d, J = 8.48 Hz), 6.96 (2H, d, J = 8.45 Hz), 7.34 (2H, d, J = 8.03 Hz), 7.77 (2H, d, J = 8.28 Hz); 13C NMR (125 MHz, CDCl3): δ −5.2, −4.9, 18.0, 21.8, 25.8, 39.7, 71.3, 72.5, 115.2, 128.0, 129.4, 129.9, 130.9, 132.8, 145.0, 154.2; HRMS (ESI+) calculated for C22H33O5SSi ([M+H]+): 437.1818, found 437.1813.
Aryl ether 38
To a solution of phenol 37 (345 mg, 0.46 mmol, 1.10 eq.) in 6 mL of THF was added solid 60% sodium hydride dispersion in mineral oil (18 mg, 0.0.46 mmol, 1.10 eq.). Vigorous bubbling was observed, and the resulting colorless phenolate solution was used immediately. A 15 mM solution of Pd(PPh3)4 was also prepared by dissolving 47.8 mg of the solid catalyst in 2.75 mL of THF. The palladium catalyst solution was then added to a flask containing neat allylic phosphate 20 (232 mg, 0.41 mmol, 1.00 eq.), and the phenolate solution was added dropwise. The reaction remained yellow throughout the addition and only turned light orange after the addition was complete. Over five minutes, the color gradually faded to yellow again. After this time, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting phosphate and formation of the product of Rf = 0.47. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 4:1) to give 38 as a pale yellow oil (227 mg, 65%).
[α]20D = −13.3 (c 0.74, CH2Cl2); IR (neat) ν 2950, 2927, 2856, 1726, 1655, 1596, 1509, 1471, 1462, 1390, 1366, 1237, 1206, 1189, 1176, 1117, 1097, 972, 834, 812, 776 cm−1; 1H NMR (500 MHz, CDCl3): δ −0.35 (3H, s), −0.14 (3H, s), −0.04 (3H, s), −0.03 (3H, s), 0.76 (9H, s), 0.85 (9H, s), 0.86 (1H, m), 0.90 (3H, d, J = 6.66 Hz), 0.97 (1H, m), 1.43 (1H, m), 1.46 (1H, m), 1.61 (1H, m), 1.64 (3H, m), 1.71 (3H, s), 1.72 (3H, s), 1.74 (1H, m), 2.45 (3H, s), 2.52 (1H, dd, J = 7.65 Hz, 13.73 Hz), 2.75 (1H, dd, J = 4.31 Hz, 13.61 Hz), 3.48 (2H, m), 3.86 (2H, m), 3.93 (1H, m), 5.00 (1H, m), 5.21 (1H, s), 5.97 (1H, d, J = 15.68 Hz), 6.53 (1H, dd, J = 3.50 Hz, 15.74 Hz), 6.72 (2H, d, J = 7.65 Hz), 6.99 (2H, d, J = 7.79 Hz), 7.34 (2H, d, J = 7.71 Hz), 7.78 (2H, d, J = 7.68 Hz); 13C NMR (125 MHz, CDCl3): δ −5.4, −5.36, −5.3, −4.9, 18.0, 18.4, 21.8, 22.7, 25.1, 25.2, 25.72, 25.74, 26.0, 32.4, 35.0, 39.1, 39.8, 40.2, 42.9, 65.0, 71.4, 72.6, 76.9, 94.7, 106.5, 115.0, 122.9, 128.1, 129.93, 129.95, 130.9, 132.9, 140.4, 145.0, 157.1, 161.9, 162.5; HRMS (ESI+) calculated for C45H71O9SSi2 ([M+H]+): 843.4357, found 843.4365.
Diol 39
To a solution of 38 (227 mg, 0.27 mmol, 1.00 eq.) in 10 mL of dry THF was added neat triethylamine trihydrofluoride (951 μL, 0.27 mmol, 22 eq.), and the reaction mixture was stirred at room temperature for 36 hours. After this time, TLC (100% EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.57. The reaction was quenched with water and diluted with EtOAc. The layers were separated, and the organic phase was washed with an additional portion of water before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 1:1 hexanes/EtOAc → 100% EtOAc) to give diol 39 as a colorless oil (146 mg, 88%).
[α]20D = −24.7 (c 0.55, CH2Cl2); IR (neat) ν 3434, 2923, 1709, 1654, 1595, 1510, 1453, 1392, 1375, 1276, 1235, 1205, 1189, 1176, 1097, 1021, 976 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.89 (1H, m), 0.91 (3H, d, J = 6.37 Hz), 0.95 (1H, m), 1.42 (1H, m), 1.45 (1H, m), 1.60 (1H, m), 1.68 (1H, m), 1.69 (1H, m), 1.70 (1H, m), 1.71 (6H, s), 1.77 (1H, m), 2.07 (1H, d, J = 3.75 Hz), 2.46 (3H, s), 2.68 (1H, dd, J = 7.48 Hz, 13.86 Hz), 2.74 (1H, dd, J = 5.05 Hz, 13.90 Hz), 3.52 (1H, m), 3.64 (1H, m), 3.93 (1H, dd, J = 5.82 Hz, 9.43 Hz), 4.04 (2H, m), 5.02 (1H, m), 5.26 (1H, s), 6.05 (1H, d, J = 15.71 Hz), 6.59 (1H, dd, J = 4.06 Hz, 15.57 Hz), 6.78 (2H, d, J = 7.78 Hz), 7.06 (2H, d, J = 7.84 Hz), 7.36 (2H, d, J = 7.74 Hz), 7.79 (2H, d, J = 7.58 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.6, 25.13, 25.16, 26.9, 32.3, 34.7, 38.4, 38.8, 40.5, 43.5, 65.9, 70.5, 72.7, 78.8, 95.1, 106.6, 115.6, 123.4, 128.1, 129.4, 130.1, 130.6, 132.6, 139.1, 145.3, 157.0, 161.9, 162.2; HRMS (ESI+) calculated for C33H43O9S ([M+H]+): 615.2628, found 615.2621.
Aldehyde 40
To a solution of diol 39 (509 mg, 0.83 mmol, 1.00 eq.) in 20 mL of CH2Cl2 was added iodobenzene diacetate (280 mg, 0.87 mmol, 1.05 eq.) and TEMPO (12.9 mg, 82.7 μmol, 0.10 eq.). After 12 hours at room temperature, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and clean formation of the product of Rf = 0.26. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 hexanes/EtOAc → 1:1 hexanes/EtOAc) to afford aldehyde 40 as a colorless oil (483 mg, 95%).
[α]20D = −24.5 (c 0.57, CH2Cl2); IR (neat) ν 3430, 2924, 1720, 1655, 1596, 1509, 1454, 1392, 1361, 1275, 1234, 1205, 1189, 1176, 1097, 1019, 976 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.88 (1H, m), 0.92 (1H, m), 0.96 (3H, d, J = 6.46 Hz), 1.40 (1H, ddd, J = 3.07 Hz, 12.86 Hz, 16.44 Hz), 1.48 (1H, m), 1.71 (6H, s), 1.75 (1H, m), 1.79 (1H, m), 1.86 (1H, m), 2.07 (1H, m), 2.46 (3H, s), 2.49 (1H, m), 2.67 (1H, dd, J = 7.45 Hz, 13.95 Hz), 2.72 (1H, dd, J = 5.44 Hz, 13.95 Hz), 3.93 (1H, dd, J = 5.84 Hz, 9.51 Hz), 4.02 (2H, m), 4.94 (1H, m), 5.28 (1H, s), 6.05 (1H, dd, J = 1.27 Hz, 15.68 Hz), 6.54 (1H, dd, J = 4.70 Hz, 15.67 Hz), 6.73 (2H, d, J = 8.56 Hz), 7.04 (2H, d, J = 8.55 Hz), 7.36 (2H, d, J = 8.13 Hz), 7.79 (2H, d, J = 8.26 Hz), 9.53 (1H, d, J = 2.93 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.3, 25.13, 25.14, 25.4, 31.7, 34.0, 34.5, 38.4, 41.5, 51.2, 70.5, 72.7, 78.1, 95.4, 106.7, 115.6, 124.1, 128.1, 129.5, 130.1, 130.6, 132.6, 138.0, 145.3, 156.6, 161.8, 162.0, 203.6; HRMS (ESI+) calculated for C33H41O9S ([M+H]+): 613.2471, found 613.2444.
Vinyl iodide 41
To a flame-dried 50 mL flask under argon equipped with a magnetic stirring bar was added solid chromium (II) chloride (774 mg, 6.30 mmol, 8.00 eq.) in the glove box. The flask was sealed with a septum, removed from the glove box, and put under a balloon of argon. 15 mL of anhydrous THF was added, and the resulting suspension was cooled to 0 °C. A solution of aldehyde 40 (483 mg, 0.79 mmol, 1.00 eq.) and iodoform (620 mg, 1.57 mmol, 2.00 eq.) in 15 mL of THF was then added dropwise. After addition was complete, the reaction mixture was allowed to warm slowly to room temperature. After 1.5 hours, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.44. The reaction was quenched with water and diluted with ether. The organic phase was isolated, and the aqueous phase was extracted with two additional portions of ether. The combined organics were washed with saturated aqueous Na2S2O3 solution and brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 3:1 → 2:1 → 1:1) to give vinyl iodide 41 as a white foam (431 mg, 74%).
[α]20D = −80.9 (c 0.75, CH2Cl2); IR (neat) ν 3487, 2922, 1718, 1654, 1596, 1508, 1454, 1391, 1362, 1271, 1233, 1204, 1189, 1175, 1097, 1020, 971 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.86 (1H, m), 0.89 (3H, d, J = 6.33 Hz), 0.91 (1H, m), 1.41 (1H, m), 1.44 (1H, m), 1.46 (1H, m), 1.63 (1H, m), 1.70 (1H, m), 1.71 (6H, s), 1.77 (1H, m), 2.25 (1H, m), 2.46 (3H, s), 2.69 (1H, dd, J = 7.37 Hz, 13.86 Hz), 2.76 (1H, dd, J = 5.23 Hz, 13.83 Hz), 3.95 (1H, dd, J = 5.54 Hz, 9.41 Hz), 4.05 (2H, m), 4.89 (1H, m), 5.25 (1H, s), 5.51 (1H, d, J = 14.34 Hz), 6.03 (1H, d, J = 15.62 Hz), 6.22 (1H, dd, J = 10.06 Hz, 14.29 Hz), 6.53 (1H, dd, J = 3.61 Hz, 15.62 Hz), 6.75 (2H, d, J = 8.00 Hz), 7.05 (2H, d, J = 7.94 Hz), 7.36 (2H, d, J = 7.86 Hz), 7.81 (2H, d, J = 7.80 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.4, 24.8, 25.1, 25.2, 31.8, 34.4, 38.5, 41.1, 46.5, 47.1, 70.5, 72.6, 76.5, 77.7, 95.0, 106.5, 115.5, 123.0, 128.1, 129.3, 130.0, 130.6, 132.6, 139.7, 145.2, 149.2, 157.4, 161.9, 162.3; HRMS (ESI+) calculated for C34H42IO8S ([M+H]+): 737.1645, found 737.1628.
Triene 29
A 0.01 M solution of dichlorobis(acetonitrile)palladium(II) was prepared by dissolving 15.2 mg of the solid catalyst in 5 mL of DMF under argon. To a solution of vinyl iodide 41 (431 mg, 0.59 mmol, 1.00 eq.) in 3 mL of DMF was added a 0.25 M solution of stannane 42 in DMF (4.68 mL, 1.17 mmol, 2.00 eq.). The palladium catalyst solution was then added dropwise, and the reaction mixture immediately turned dark brown/black. After stirring at room temperature for two hours, the reaction was diluted with water and ether. The layers were separated, and the organic phase was washed with two additional portions of water before drying over anhydrous Na2SO4. TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed the triene of Rf = 0.51, which was essentially copolar with the starting vinyl iodide. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 3:1 → 2:1 → 1:1) to afford triene 29 as a pale yellow oil (331 mg, 85%).
[α]20D = −18.6 (c 1.10, CH2Cl2); IR (neat) ν 3449, 2924, 1719, 1654, 1596, 1509, 1455, 1391, 1374, 1273, 1234, 1205, 1189, 1176, 1097, 1020, 976, 904, 835 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.88 (1H, m), 0.90 (3H, d, J = 6.40 Hz), 0.91 (1H, m), 1.37 (1H, m), 1.45 (1H, m), 1.51 (1H, m), 1.65 (1H, m), 1.69 (1H, m), 1.695 (3H, s), 1.701 (3H, s), 1.78 (1H, m), 2.23 (1H, m), 2.45 (3H, s), 2.68 (1H, dd, J = 7.47 Hz, 14.10 Hz), 2.74 (1H, dd, J = 5.40 Hz, 13.96 Hz), 3.94 (1H, dd, J = 5.98 Hz, 9.62 Hz), 4.02 (1H, br s), 4.05 (1H, dd, J = 3.48 Hz, 9.59 Hz), 4.91 (1H, m), 5.02 (1H, d, J = 10.03 Hz), 5.10 (1H, d, J = 16.88 Hz), 5.24 (1H, s), 5.42 (1H, dd, J = 9.39 Hz, 15.13 Hz), 5.62 (1H, dd, J = 10.62 Hz, 15.16 Hz), 5.75 (1H, dd, J = 10.71 Hz, 14.93 Hz), 6.00 (1H, d, J = 15.65 Hz), 6.06 (1H, m), 6.27 (1H, ddd, J = 10.41 Hz, 10.41 Hz, 16.98 Hz), 6.52 (1H, dd, J = 3.99 Hz, 15.63 Hz), 6.74 (2H, d, J = 8.53 Hz), 7.03 (2H, d, J = 8.52 Hz), 7.35 (2H, d, J = 8.14 Hz), 7.80 (2H, d, J = 8.23 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.5, 24.8, 25.1, 25.2, 31.9, 34.6, 38.5, 42.0, 43.4, 47.2, 70.5, 72.7, 77.8, 94.8, 106.5, 115.7, 117.0, 122.8, 128.1, 129.0, 130.1, 130.4, 131.77, 131.82, 132.6, 133.0, 137.0, 138.2, 140.5, 145.2, 157.5, 161.9, 162.4; HRMS (ESI+) calculated for C38H47O8S ([M+H]+): 663.2992, found 663.2980.
Macrolactone 30 and dimer 43
An oven-dried 2-neck 2 L flask was equipped with a glass stopper, a Dean–Stark trap with a reflux condenser, and a magnetic stirring bar. The apparatus was evacuated and put under an atmosphere of argon before the addition of 1.5 L of freshly distilled benzene (sodium/benzophenone). A solution of triene 29 (100 mg, 0.15 mmol, 1.00 eq.) in 5 mL of benzene was added, and the reaction mixture was heated to reflux in a pre-heated 110 °C oil bath. Three 25 mL aliquots of benzene were drained through the side arm of the Dean–Stark trap, and the reaction mixture was heated at reflux for 12 hours. After cooling to room temperature, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of macrolactone 30 as a spot of Rf = 0.52 along with a small amount of dimer 43 of Rf = 0.54. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (1500 μm plate, loaded in CH2Cl2, eluted in 9:1 toluene/EtOAc, silica washed with EtOAc) to give macrolactone 30 as a white foam (46 mg, 50%) along with dimer 43 (10 mg) as a colorless oil.
Macrolactone 30
[α]20D = +83.5 (c 0.70, CH2Cl2); IR (neat) ν 2916, 1759, 1714, 1610, 1508, 1454, 1365, 1237, 1190, 1177, 1148, 1097, 1067, 1035, 985, 928, 815 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.91 (1H, m), 0.92 (1H, m), 0.95 (3H, d, J = 6.5 Hz), 1.09 (1H, qd, J = 12.7 Hz, 4.1 Hz), 1.42 (1H, m), 1.44 (1H, m), 1.61 (1H, m), 1.65 (1H, m), 1.84 (1H, m), 1.99 (1H, m), 2.10 (1H, m), 2.13 (1H, m), 2.46 (3H, s), 2.69 (1H, m), 2.74 (1H, d, J = 15.7 Hz), 2.88 (1H, d, J = 15.7 Hz), 2.94 (2H, m), 3.01 (1H, dd, J = 11.4 Hz, 6.5 Hz), 4.15 (2H, m), 4.86 (1H, m), 4.93 (1H, d, J = 17.5 Hz), 4.94 (1H, d, J = 8.4 Hz), 5.14 (1H, m), 5.29 (1H, m), 5.34 (1H, m), 5.90 (1H, d, J = 9.8 Hz), 6.81 (1H, m), 6.88 (1H, m), 6.89 (1H, m), 6.91 (1H, m), 7.36 (2H, d, J = 7.9 Hz), 7.80 (2H, d, J = 8.2 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.6, 29.2, 33.1, 35.9, 36.5, 38.0, 42.3, 42.7, 45.8, 46.6, 49.5, 52.1, 56.0, 70.0, 70.5, 81.9, 117.0, 119.5, 120.0, 128.0, 128.09, 128.10, 128.8, 129.0, 130.0, 132.5, 132.6, 136.4, 145.2, 157.3, 164.4, 198.6; HRMS (ESI+) calculated for C35H41O7S ([M+H]+): 605.2573, found 605.2571.
Dimer 43
[α]20D = +67.2 (c 1.19, CH2Cl2); IR (neat) ν 2920, 2867, 1749, 1714, 1610, 1510, 1454, 1367, 1296, 1242, 1190, 1177, 1149, 1097, 1044, 991, 955, 815 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.81 (2H, q, J = 11.7 Hz), 0.89 (6H, d, J = 6.5 Hz), 0.90 (2H, m), 1.03 (2H, m), 1.29 (2H, m), 1.36 (2H, m), 1.38 (2H, m), 1.69 (2H, m), 1.93 (2H, m), 1.96 (2H, m), 1.97 (2H, m), 2.05 (2H, m), 2.35 (2H, dd, J = 14.2 Hz, 5.2 Hz), 2.47 (6H, s), 2.50 (2H, dd, J = 14.3 Hz, 7.6 Hz), 3.22 (2H, d, J = 14.1 Hz), 3.32 (2H, m), 3.41 (2H, m), 3.44 (2H, d, J = 14.2 Hz), 3.57 (2H, dd, J = 10.9 Hz, 4.4 Hz), 3.67 (2H, dd, J = 10.9 Hz, 3.2 Hz), 4.56 (2H, m), 5.07 (2H, d, J = 11.0 Hz), 5.08 (2H, d, J = 15.9 Hz), 4.65 (2H, dd, J = 6.5 Hz, 3.8 Hz), 5.47 (2H, dt, J = 9.7 Hz, 3.2 Hz), 5.60 (2H, dt, J = 17.7 Hz, 9.2 Hz), 5.98 (2H, d, J = 9.8 Hz), 6.61 (4H, d, J = 8.5 Hz), 6.77 (4H, d, J = 8.5 Hz), 7.37 (4H, d, J = 8.1 Hz), 7.78 (4H, d, J = 8.2 Hz); 13C NMR (125 MHz, CDCl3): δ 21.8, 22.5, 30.7, 32.8, 34.7, 36.1, 38.0, 43.0, 44.1, 45.3, 49.1, 49.8, 51.9, 56.5, 68.4, 72.9, 77.9, 116.0, 117.0, 127.6, 128.1, 128.8, 129.0, 130.0, 130.4, 132.8, 137.0, 145.0, 156.4, 165.7, 202.1; HRMS (ESI+) calculated for C70H81O14S2 ([M+H]+): 1209.5068, found 1209.5092.
Primary iodide 44
To a solution of macrolactone tosylate 30 (11.1 mg, 18.4 μmol, 1.00 eq.) in 500 μL of anhydrous acetone was added sodium iodide (207 mg, 1.38 mmol, 75 eq.). The flask was tightly sealed with a yellow Teflon cap, and the reaction mixture was heated to 40 °C in an oil bath for 12 hours. After this time, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the iodide of Rf = 0.65. The reaction was diluted with water and EtOAc, the layers were separated, and the organic phase was washed with one additional portion of water before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (500 μm plate, sample loaded in EtOAc, plate eluted in 3:1 hexanes/EtOAc, silica washed with EtOAc) to give primary iodide 44 as a colorless oil (5.6 mg, 54%) that was used immediately in the next step.
[α]20D = +54.3 (c 0.30, CH2Cl2); IR (neat) ν 2924, 1762, 1712, 1610, 1510, 1454, 1232, 1151, 1097, 1022, 954, 587, 562 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.91 (1H, m), 0.92 (1H, m), 0.96 (3H, d, J = 6.6 Hz), 1.09 (1H, qd, J = 12.4 Hz, 4.3 Hz), 1.42 (1H, m), 1.44 (1H, m), 1.61 (1H, m), 1.66 (1H, td, J = 11.5 Hz, 4.7 Hz), 1.85 (1H, m), 1.99 (1H, m), 2.12 (1H, m), 2.14 (1H, m), 2.65 (1H, dd, J = 13.2 Hz, 10.9 Hz), 2.84 (1H, d, J = 15.8 Hz), 2.94 (1H, m), 2.96 (1H, d, J = 15.7 Hz), 3.05 (1H, dd, J = 11.5 Hz, 6.4 Hz), 3.22 (1H, dd, J = 13.2 Hz, 5.5 Hz), 3.35-3.36 (2H, m), 4.88 (1H, m), 4.93 (1H, d, J = 10.4 Hz), 4.94 (1H, m), 4.95 (1H, d, J = 17.1 Hz), 5.31 (1H, m), 5.35 (1H, m), 5.90 (1H, d, J = 9.8 Hz), 6.84 (1H, dd, J = 8.4 Hz, 2.5 Hz), 6.90 (1H, dd, J = 8.7 Hz, 2.5 Hz), 6.95 (1H, dd, J = 8.4 Hz, 2.3 Hz), 6.97 (1H, dd, J = 8.7 Hz, 2.3 Hz); 13C NMR (125 MHz, CDCl3): δ 7.9, 22.6, 29.3, 33.1, 36.6, 38.1, 40.8, 42.4, 42.7, 46.0, 46.7, 49.5, 52.1, 56.1, 72.1, 82.0, 117.1, 119.5, 120.1, 128.11, 128.12, 128.8, 130.0, 132.5, 136.5, 157.4, 164.4, 198.7; HRMS (ESI+) calculated for C28H34IO4 ([M+H]+): 561.1502, found 561.1489.
Acylketene acetal 45
To a solution of primary iodide 44 (5.6 mg, 10 μmol, 1.00 eq.) in 500 μL of toluene was added neat DBU (25 μL, 167 μmol, 16.7 eq.), and the reaction mixture was heated to 45 °C for three hours. After this time, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.07. After cooling to room temperature, the mixture was diluted with EtOAc, and the organic phase was washed twice with water and once with brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (500 μm plate, loaded in CH2Cl2, eluted with 3:1 EtOAc/hexanes, silica washed with EtOAc) to give acylketene acetal 45. Although sufficient material was obtained for characterization, the product is very susceptible to hydrolysis and decomposes over time in chloroform, upon storage, and during purification efforts.
[α]20D = +110 (c 0.24, CH2Cl2); IR (neat) ν 2925, 1610, 1562, 1422, 1364, 1225, 1190, 1143, 1097, 1063, 1035, 928, 833 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.85 (1H, m), 0.91 (1H, m), 0.95 (3H, d, J = 6.6 Hz), 1.07 (1H, qd, J = 12.6 Hz, 4.2 Hz), 1.39 (1H, m), 1.44 (1H, m), 1.58 (1H, m), 1.71 (1H, td, J = 11.5 Hz, 6.0 Hz), 1.83 (1H, m), 1.99 (1H, m), 2.01 (1H, m), 2.12 (1H, m), 2.87 (1H, d, J = 13.9 Hz), 2.91 (1H, dd, J = 11.6 Hz, 6.6 Hz), 3.01 (1H, m), 3.38 (1H, dd, J = 13.8 Hz, 9.0 Hz), 4.31-4.34 (2H, m), 4.36 (1H, s), 4.81 (1H, d, J = 17.0 Hz), 4.86 (1H, d, J = 10.1 Hz), 4.96 (1H, t, J = 5.3 Hz), 5.11 (1H, m), 5.41 (1H, dt, J = 9.8 Hz, 3.5 Hz), 5.48 (1H, ddd, J = 17.6 Hz, 9.8 Hz, 8.5 Hz), 5.89 (1H, d, J = 9.8 Hz), 6.71 (1H, dd, J = 8.2 Hz, 2.5 Hz), 6.92 (1H, dd, J = 8.2 Hz, 1.8 Hz), 7.00 (1H, dd, J = 8.6 Hz, 2.4 Hz), 7.05 (1H, dd, J = 8.6 Hz, 1.8 Hz); 13C NMR (125 MHz, CDCl3): δ 22.7, 29.1, 32.9, 36.3, 38.2, 40.6, 43.3, 44.5, 45.7, 48.4, 51.7, 55.8, 72.5, 77.4, 79.3, 82.8, 115.6, 118.7, 119.8, 126.3, 128.0, 128.3, 129.1, 133.5, 138.0, 159.3, 166.4, 195.4; HRMS (ESI+) calculated for C28H33O4 ([M+H]+): 433.2379, found 433.2377.
Acylketene acetal 46 and ring-expanded macrolactone 49
To a solution of macrolactone tosylate 30 (8.0 mg, 13.2 μmol, 1.00 eq.) in 1 mL of THF was added thallium (I) carbonate (15 mg, 32 μmol, 2.42 eq.) After stirring at room temperature for 20 minutes, neat DBU (25 μL, 167 μmol, 12.7 eq.) was added, and the reaction was stirred for an additional 10 minutes at room temperature. After this time, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion of the starting material to the product of Rf = 0.24. The reaction was diluted with ether, and water was added. The layers were separated, and the organic phase was washed with pH 7 phosphate buffer before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was dried under high vacuum to give acylketene acetal 46, which was immediately submitted for NMR analysis. In CDCl3 solution, 46 slowly isomerized to 45 at the rate of 1% per hour; this isomerization was accelerated upon exposure to silica, eventually leading to the ring-expanded macrolactone 49.
Acylketene acetal 46
1H NMR (500 MHz, CDCl3): δ 0.94 (1H, m), 0.96 (3H, d, J = 6.62 Hz), 1.00 (1H, m), 1.11 (1H, qd, J = 12.39 Hz, 4.21 Hz), 1.41 (1H, ddd, J = 16.08 Hz, 10.53 Hz, 3.81 Hz), 1.46 (1H, m), 1.68 (1H, ddd, J = 13.35 Hz, 5.98 Hz, 2.90 Hz), 1.81 (1H, m), 1.95 (1H, dt, J = 11.5 Hz, 4.5 Hz), 2.00 (1H, m), 2.05 (1H, m), 2.07 (1H, m), 2.69 (1H, dd, J = 14.32 Hz, 1.75 Hz), 2.84 (1H, m), 2.93 (1H, dd, J = 11.03 Hz, 7.76 Hz), 3.13 (1H, dd, J = 14.30 Hz, 3.31 Hz), 4.24 (1H, dd, J = 8.33 Hz, 6.70 Hz), 4.34 (1H, dd, J = 8.71 Hz, 1.03 Hz), 4.51 (1H, s), 4.71 (1H, m), 4.83 (1H, m), 4.85 (1H, m), 4.99 (1H, m), 5.46 (1H, dt, J = 10 Hz, 3 Hz), 5.53 (1H, ddd, J = 16.82 Hz, 10.10 Hz, 8.82 Hz), 5.93 (1H, d, J = 9.68 Hz), 6.82 (1H, dd, J = 8.25 Hz, 2.55 Hz), 6.85 (1H, dd, J = 8.29 Hz, 2.57 Hz), 7.03 (1H, dd, J = 8.23 Hz, 2.10 Hz), 7.08 (1H, dd, J = 8.31 Hz, 2.14 Hz); 13C NMR (125 MHz, CDCl3): δ 22.7, 29.5, 33.3, 36.6, 38.2, 39.0, 41.9, 42.5, 45.7, 48.2, 49.6, 56.2, 70.3, 78.0, 80.6, 84.6, 114.9, 115.6, 119.9, 123.7, 128.8, 129.6, 129.7, 134.3, 138.8, 159.6, 167.7, 198.7; HRMS (ESI+) calculated for C28H33O4 ([M+H]+): 433.2379, found 433.2375.
Ring-expanded macrolactone 49
[α]20D = +19.5 (c 0.70, CH2Cl2); IR (neat) ν 3456, 2916, 2849, 1749, 1711, 1612, 1583, 1508, 1456, 1406, 1377, 1335, 1296, 1237, 1176, 1158, 1114, 1063, 1037, 1014, 964, 927, 805 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.88 (1H, q, J = 11.2 Hz), 0.94 (1H, m), 0.95 (3H, d, J = 6.5 Hz), 1.06 (1H, qd, J = 12.7 Hz, 4.2 Hz), 1.40 (1H, m), 1.43 (1H, m), 1.56 (1H, m), 1.75 (1H, m), 1.82 (1H, m), 2.00 (1H, m), 2.05 (1H, m), 2.08 (1H, m), 2.63 (1H, dd, J = 13.1 Hz, 9.3 Hz), 2.93 (1H, d, J = 16.6 Hz), 2.99 (1H, d, J = 16.6 Hz), 3.02 (1H, m), 3.04 (1H, m), 3.19 (1H, dd, J = 13.1 Hz, 4.1 Hz), 3.94 (1H, d, J = 11.0 Hz), 4.07 (1H, m), 4.21 (1H, dd, J = 11.0 Hz, 8.1 Hz), 4.91 (1H, t, J = 5.1 Hz), 4.97 (1H, d, J = 17.0 Hz), 4.99 (1H, d, J = 9.9 Hz), 5.37 (1H, m), 5.41 (1H, m), 5.93 (1H, d, J = 9.7 Hz), 6.73 (1H, dd, J = 8.3 Hz, 2.7 Hz), 6.80 (1H, dd, J = 8.4 Hz, 2.5 Hz), 7.01 (1H, dd, J = 8.2 Hz, 2.1 Hz), 7.04 (1H, dd, J = 8.5 Hz, 2.1 Hz); 13C NMR (125 MHz, CDCl3): δ 22.6, 29.1, 32.9, 36.3, 38.0, 42.5, 43.3, 43.6, 45.2, 47.6, 48.6, 51.1, 56.1, 68.7, 69.7, 79.8, 115.5, 117.1, 117.7, 128.3, 128.38, 128.44, 130.3, 130.7, 136.3, 157.4, 165.2, 201.0; HRMS (ESI+) calculated for C28H35O5 ([M+H]+): 451.2484, found 451.2480.
4-(2-(((tert-butyldiphenylsilyl)oxy)methyl)allyl)phenol (60)
To a 2-neck 250 mL flask equipped with a magnetic stirring bar, a rubber septum, and a reflux condenser were added magnesium turnings (2.37 g, 97.5 mmol, 7.00 eq.). The entire apparatus was flame-dried under high vacuum and cooled to room temperature under argon. Approximately 8 mL of THF was added to the magnesium turnings followed by 100 μL of 1,2-dibromoethane; bubbling was observed, and the solution turned grey. A solution of aryl bromide 5644 (4.00 g, 13.9 mmol, 1.00 eq.) in 50 mL of THF was added slowly, and the reaction was initiated with a heat gun. The flask was then transferred to a preheated 100 °C oil bath, and the reaction was heated to reflux for two hours. After cooling to room temperature, the dark grey/black solution of Grignard reagent 57 was titrated with salicylaldehyde phenylhydrazone,55 giving a final concentration of 0.14 M.
To an oven-dried flask under argon equipped with a magnetic stirring bar was added a solution of allylic bromide 5845 (2.39 g, 6.13 mmol, 1.00 eq.) in 50 mL of THF. The 0.14 M solution of Grignard reagent 57 (52 mL, 7.28 mmol, 1.19 eq.) was transferred to the flask via cannula, and the homogeneous grey/black reaction mixture was left to stir at room temperature overnight. After this time, TLC (10:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the allylic bromide (Rf = 0.59) and formation of the coupled product of Rf = 0.73. The reaction was quenched with saturated aqueous NH4Cl solution and diluted with ether. The layers were separated, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the mixture containing crude 59 was used directly in the next step without further purification.
Solid lithium hydroxide monohydrate (692 mg, 16.5 mmol, 2.69 eq.) was added in a single portion to a solution of crude 59 (3.74 g) in 100 mL of THF and 50 mL of water; the reaction mixture immediately turned yellow and eventually became purple. After stirring at room temperature for 30 minutes, TLC (10:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the deprotected product of Rf = 0.16. The reaction was quenched with pH 7 phosphate buffer (discharging the purple color to yellow) and diluted with EtOAc. The layers were separated, and the organic phase was dried over anhydrous Na2SO4 before the solvent was removed under reduced pressure. The residue was purified by column chromatography (9:1 hexanes/EtOAc, → 2:1 → 1:1) to afford phenol 60 as a colorless oil (1.90 g, 77% over three steps).
IR (neat) ν 3339, 3071, 2930, 2892, 2857, 1613, 1597, 1512, 1472, 1428, 1226, 1112, 824, 741, 702 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.05 (9H, s), 3.29 (2H, s), 4.07 (2H, s), 4.58 (1H, s), 4.85 (1H, s), 5.21 (1H, d, J = 1.49 Hz), 6.71 (2H, d, J = 8.49 Hz), 7.00 (2H, d, J = 8.46 Hz), 7.35 (4H, m), 7.41 (2H, m), 7.64 (4H, d, J = 6.62 Hz); 13C NMR (125 MHz, CDCl3): δ 19.3, 26.8, 38.7, 65.9, 110.7, 115.1, 127.7, 129.6, 130.0, 131.4, 133.6, 135.5, 147.8, 153.8; HRMS (ESI+) calculated for C26H31O2Si ([M+H]+): 403.2093, found 403.2080.
Primary alcohol 62
A 15 mM solution of Pd(PPh3)4 was prepared under argon by dissolving 43 mg of the solid catalyst in 2.50 mL of THF. To a 0.125 M solution of phenol 60 (3.30 mL, 0.41 mmol, 1.10 eq.) in dry THF was added solid 60% sodium hydride dispersion in mineral oil (16.5 mg, 0.41 mmol, 1.10 eq.); vigorous bubbling was observed, and the resulting phenolate solution was used immediately. The bright yellow catalyst solution was added to a flask containing neat allylic phosphate 20 (210 mg, 0.38 mmol, 1.00 eq.) followed by the dropwise addition of the phenolate solution. After each drop, the solution would turn dark orange before fading to yellow again over the course of several seconds. After addition was complete, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting phosphate and formation of the desired product of Rf = 0.60 along with the elimination byproduct of Rf = 0.54 and unreacted phenol at Rf = 0.46. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (9:1 hexanes/EtOAc → 4:1) to give a mixture of aryl ether 61 and the elimination byproduct that was used directly in the next step (248 mg total).
To a solution of mixed 61 (248 mg, 0.31 mmol, 1.00 eq.) in 16 mL of 1:1 THF/H2O was added glacial acetic acid (15 mL, 262 mmol, 845 eq.). After 12 hours at room temperature, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the product of Rf = 0.49. The acid was neutralized with saturated aqueous K2CO3 solution and diluted with water and EtOAc. The layers were separated, and the aqueous phase was extracted with three additional portions of EtOAc. The combined organics were dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 3:1 → 2:1 → 1:1) to give primary alcohol 62 as a white foam (157 mg, 60% over two steps).
[α]20D = −15.1 (c 2.80, CH2Cl2); IR (neat) ν 3482, 2927, 2857, 1717, 1654, 1592, 1507, 1472, 1428, 1390, 1375, 1273, 1234, 1205, 1174, 1111, 1020, 971, 939, 903, 823, 742, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.92 (1H, m), 0.93 (3H, d, J = 6.51 Hz), 0.97 (1H, m), 1.03 (9H, s), 1.43 (1H, m), 1.46 (1H, m), 1.59 (1H, m), 1.68 (1H, m), 1.69 (1H, m), 1.70 (3H, s), 1.71 (4H, m), 1.76 (1H, m), 3.29 (2H, s), 3.52 (1H, d, J = 11.04 Hz), 3.66 (1H, d, J = 11.02 Hz), 4.07 (2H, s), 4.84 (1H, s), 4.99 (1H, m), 5.22 (1H, s), 5.26 (1H, s), 6.07 (1H, d, J = 15.69 Hz), 6.61 (1H, dd, J = 4.44 Hz, 15.66 Hz), 6.74 (2H, d, J = 8.54 Hz), 7.03 (2H, d, J = 8.46 Hz), 7.35 (4H, m), 7.41 (2H, m), 7.63 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.3, 22.6, 25.1 25.2, 26.9, 27.1, 32.3, 34.8, 38.7, 38.9, 40.5, 43.5, 65.9, 66.0, 79.1, 95.0, 106.6, 110.9, 115.3, 123.5, 127.7, 129.7, 130.1, 132.3, 133.6, 135.6, 139.2, 147.7, 156.4, 161.8, 162.3; HRMS (ESI+) calculated for C43H54NaO6Si ([M+Na]+): 717.3587, found 717.3591.
Aldehyde 63
To a solution of primary alcohol 62 (157 mg, 0.23 mmol, 1.00 eq.) in 5 mL of CH2Cl2 was added solid Dess–Martin periodinane (115 mg, 0.27 mmol, 1.20 eq.). The reaction mixture immediately turned slightly yellow and was stirred at room temperature for 30 minutes. After this time, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the product of Rf = 0.67. The reaction was diluted with water and CH2Cl2 and quenched with saturated aqueous sodium thiosulfate solution and saturated aqueous NaHCO3 solution. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2. The combined organics were dried over anhydrous Na2SO4 before removal of the solvent under reduced pressure and purification of the residue by column chromatography (3:1 hexanes/EtOAc → 1:1) to afford aldehyde 63 as a colorless oil (141 mg, 90%).
[α]20D = −11.2 (c 1.24, CH2Cl2); IR (neat) ν 2932, 1724, 1653, 1596, 1507, 1457, 1428, 1390, 1273, 1235, 1112, 1019 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.90 (1H, m), 0.95 (1H, m), 0.96 (3H, d, J = 6.51 Hz), 1.03 (9H, s), 1.42 (1H, ddd, J = 3.89 Hz, 13.72 Hz, 16.53 Hz), 1.49 (1H, m), 1.70 (3H, s), 1.71 (3H, s), 1.76 (1H, m), 1.78 (1H, m), 1.85 (1H, m), 2.08 (1H, m), 2.49 (1H, m), 3.27 (2H, s), 4.06 (2H, s), 4.82 (1H, s), 4.92 (1H, m), 5.21 (1H, d, J = 1.38 Hz), 5.27 (1H, s), 6.06 (1H, dd, J = 1.45 Hz, 15.68 Hz), 6.55 (1H, dd, J = 4.77 Hz, 15.66 Hz), 6.69 (2H, d, J = 8.63 Hz), 7.01 (2H, d, J = 8.61 Hz), 7.35 (4H, m), 7.41 (2H, m), 7.63 (4H, m), 9.53 (1H, d, J = 3.00 Hz); 13C NMR (125 MHz, CDCl3): δ 19.3, 22.3, 25.1, 25.5, 26.9, 31.7, 34.0, 34.5, 38.7, 41.6, 51.3, 66.0, 78.3, 95.4, 106.6, 110.9, 115.3, 124.1, 127.7, 129.7, 130.1, 132.3, 133.6, 135.6, 138.2, 147.7, 16.1, 161.7, 162.1, 203.6; HRMS (ESI+) calculated for C43H53O6Si ([M+H]+): 693.3611, found 693.3588.
Vinyl iodide 64
To a flame-dried 25 mL round bottom flask equipped with a magnetic stirring bar was added solid chromium (II) chloride (200 mg, 1.63 mmol, 8.00 eq.) in the glove box. The flask was tightly sealed with a septum, removed from the glove box, and put under a balloon of argon. 3.50 mL of freshly distilled THF (sodium/benzophenone) was added, and the resulting green slurry was cooled to 0 °C. To a separate flask containing neat aldehyde 63 (141 mg, 0.20 mmol, 1.00 eq.) was added solid iodoform (160 mg, 0.41 mol, 2.00 eq.). This mixture was dissolved in 3 mL of THF and added slowly to the chromium suspension; the vial was rinsed with two additional 2.0 mL portions of THF, which were also added to the reaction mixture. The cooling bath was removed, and after 1.5 hours, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed clean formation of the product of Rf = 0.51. The reaction was quenched with water and diluted with ether. The layers were separated, and the aqueous phase was extracted with two additional portions of ether. The combined organics were washed with saturated aqueous sodium Na2S2O3 solution (yellow color discharged) and brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (9:1 hexanes/EtOAc → 4:1 → 3:1) to afford vinyl iodide 64 as a white foam (144 mg, 86%).
[α]20D = −29.5 (c 1.04, CH2Cl2); IR (neat) ν 2928, 2856, 1728, 1655, 1596, 1507, 1428, 1390, 1375, 1269, 1233, 1204, 1174, 1111, 1020, 951, 902 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.88 (1H, m), 0.90 (1H, m), 0.91 (3H, d, J = 6.44 Hz), 1.05 (9H, s), 1.40 (1H, m), 1.45 (1H, m), 1.49 (1H, m), 1.64 (1H, m), 1.69 (1H, m), 1.70 (6H, s), 1.75 (1H, m), 2.25 (1H, ddd, J = 3.55 Hz, 10.54 Hz, 10.63 Hz), 3.29 (2H, s), 4.07 (1H, d, J = 14.06 Hz), 4.11 (1H, d, J = 14.11 Hz), 4.84 (1H, s), 4.89 (1H, m), 5.23 (2H, m), 5.52 (1H, d, J = 14.35 Hz), 6.04 (1H, dd, J = 1.59 Hz, 15.61 Hz), 6.23 (1H, dd, J = 9.96 Hz, 14.34 Hz), 6.53 (1H, dd, J = 3.88 Hz, 15.61 Hz), 6.71 (2H, d, J = 8.56 Hz), 7.02 (2H, d, J = 8.52 Hz), 7.36 (4H, m), 7.41 (2H, m), 7.65 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.4, 24.7, 25.1, 25.2, 26.9, 31.8, 34.5, 38.7, 41.2, 46.5, 47.0, 66.1, 76.5, 77.5, 94.9, 106.5, 110.9, 115.1, 123.0, 127.7, 129.7, 130.1, 132.0, 133.6, 135.6, 140.1, 147.8, 149.3, 156.7, 161.9, 162.4; HRMS (ESI+) calculated for C44H53INaO5Si ([M+Na]+): 839.2605, found 839.2594.
Primary alcohol 65
To a solution of vinyl iodide 64 (129 mg, 0.16 mmol, 1.00 eq.) in 5 mL of THF was added solid TBAT (340 mg, 0.63 mmol, 4.00 eq.). The flask was then wrapped in aluminum foil to protect the reaction mixture from light. After seven hours at room temperature, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the product of Rf = 0.15. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. The layers were separated, and the organic phase was washed with water before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 → 1:1) to give primary alcohol 65 as a colorless oil (79 mg, 86%).
[α]20D = −42.3 (c 0.77, CH2Cl2); IR (neat) ν 3520, 1720, 1654, 1594, 1507, 1391, 1375, 1275, 1233, 1204, 1174, 1021, 952, 905 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.87 (1H, m), 0.88 (1H, m), 0.89 (3H, d, J = 6.53 Hz), 1.40 (1H, m), 1.45 (1H, m), 1.49 (1H, m), 1.64 (1H, m), 1.705 (3H, s), 1.709 (3H, s), 1.71 (1H, m), 1.77 (1H, m), 2.27 (1H, ddd, J = 3.58 Hz, 10.36 Hz, 10.39 Hz), 3.35 (2H, s), 4.04 (2H, d, J = 3.42 Hz), 4.90 (2H, s), 5.11 (1H, s), 5.25 (1H, s), 5.54 (1H, d, J = 14.35 Hz), 6.05 (1H, dd, J = 1.72 Hz, 15.61 Hz), 6.24 (1H, dd, J = 9.99 Hz, 14.34 Hz), 6.54 (1H, dd, J = 3.87 Hz, 15.61 Hz), 6.76 (2H, d, J = 8.60 Hz), 7.09 (2H, d, J = 8.56 Hz); 13C NMR (125 MHz, CDCl3): δ 22.4, 24.7, 25.1, 25.2, 31.8, 34.5, 39.1, 41.2, 46.5, 47.1, 65.4, 76.5, 77.7, 94.9, 106.5, 111.4, 115.3, 123.0, 130.1, 131.8, 140.0, 148.5, 149.3, 157.0, 161.9, 162.4; HRMS (ESI+) calculated for C28H36IO5 ([M+H]+): 579.1607, found 579.1596.
Triene 66
A 0.01 M solution of dichlorobis(acetonitrile)palladium (II) was prepared by dissolving 13 mg of the solid catalyst in 5 mL of dry DMF. To a separate flask containing neat vinyl iodide 65 (78.5 mg, 0.14 mmol, 1.00 eq.) was added a 0.25 M solution of stannane 42 in DMF (679 μL, 0.17 mmol, 1.25 eq.) followed by a portion of the palladium catalyst solution (1.36 mL, 13.6 μmol, 0.10 eq.). The reaction mixture immediately turned dark brown/black, and this was stirred at room temperature for 1.5 hours. After this time, the reaction was diluted with ether and water and poured into a separatory funnel. The layers were separated, and the organic phase was washed with two additional portions of water before drying over anhydrous Na2SO4. TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed the product of Rf = 0.28, which was essentially copolar with the starting vinyl iodide. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 4:1 → 3:1 → 2:1 → 1:1) to afford triene 66 as a yellow oil (59.5 mg, 87%).
[α]20D = −31.0 (c 0.57, CH2Cl2); IR (neat) ν 3436, 2923, 2866, 1717, 1654, 1594, 1507, 1455, 1391, 1375, 1271, 1232, 1204, 1175, 1111, 1060, 1020, 1008, 974, 902 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.87 (1H, m), 0.89 (1H, m), 0.90 (3H, d, J = 6.49 Hz), 1.36 (1H, m), 1.45 (1H, m), 1.50 (1H, m), 1.64 (1H, m), 1.67 (1H, m), 1.69 (3H, s), 1.70 (3H, s), 1.77 (1H, m), 2.24 (1H, ddd, J = 3.52 Hz, 12.41 Hz, 12.77 Hz), 3.34 (2H, s), 4.03 (2H, s), 4.87 (1H, s), 4.91 (1H, m), 5.03 (1H, d, J = 10.27 Hz), 5.10 (1H, s), 5.11 (1H, d, J = 16.68 Hz), 5.24 (1H, s), 5.41 (1H, dd, J = 9.40 Hz, 15.17 Hz), 5.62 (1H, dd, J = 10.60 Hz, 15.19 Hz), 5.78 (1H, dd, J = 10.71 Hz, 15.02 Hz), 6.02 (1H, dd, J = 1.74 Hz, 15.72 Hz), 6.07 (1H, dd, J = 11.02 Hz, 15.33 Hz), 6.27 (1H, dt, J = 10.45 Hz, 17.04 Hz), 6.54 (1H, dd, J = 3.95 Hz, 15.62 Hz), 6.75 (2H, d, J = 8.57 Hz), 7.06 (2H, d, J = 8.54 Hz); 13C NMR (125 MHz, CDCl3): δ 22.5, 24.8, 25.1, 25.2, 31.9, 34.7, 39.1, 42.0, 43.4, 47.3, 65.4, 77.9, 94.7, 106.5, 111.2, 115.5, 116.8, 122.8, 129.9, 131.5, 131.81, 131.83, 133.0, 137.1, 138.3, 140.8, 148.6, 157.1, 161.9, 162.5; HRMS (ESI+) calculated for C32H41O5 ([M+H]+): 505.2954, found 505.2931.
Macrolactone 50
Benzene (400 mL) was freshly distilled from sodium/benzophenone directly into a flame-dried 2-neck 500 flask equipped with a magnetic stirring bar and a 24/40 glass stopper. A solution of triene 66 (46.2 mg, 91.5 μmol, 1.00 eq.) in 5 mL of dry benzene was added, a flame-dried Dean–Stark trap/reflux condenser was attached, and the flask was heated to reflux in a pre-heated 110 °C oil bath. 3 × 10 mL of benzene was drained from the Dean–Stark side arm, and the reaction was heated at reflux for 22 hours. After this time, TLC (4:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the triene and formation of the product of Rf = 0.53. After cooling to room temperature, the solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 4:1) to give the macrolactone 50 as a white foam (18.8 mg, 46%).
[α]20D = +125.0 (c 0.77, CH2Cl2); IR (neat) ν 2947, 2915, 2851, 1748, 1715, 1647, 1610, 1507, 1456, 1402, 1326, 1238, 1173, 1153, 1000, 922 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.89 (1H, q, J = 11.4 Hz), 0.92 (1H, m), 0.95 (3H, d, J = 6.57 Hz), 1.01 (1H, qd, J = 12.44 Hz, 4.02 Hz), 1.39 (1H, m), 1.41 (1H, m), 1.54 (1H, m), 1.76 (1H, td, J = 11.2 Hz, 5.6 Hz), 1.79 (1H, m), 1.97 (1H, m), 2.03 (1H, m), 2.05 (1H, m), 2.89 (1H, d, J = 16.63 Hz), 2.94 (1H, d, J = 16.70 Hz), 3.01 (2H, m), 3.32 (1H, d, J = 14.24 Hz), 3.54 (1H, d, J = 14.23 Hz), 4.23 (1H, d, J = 11.34 Hz), 4.61 (1H, d, J = 11.27 Hz), 4.92 (1H, m), 4.97 (1H, d, J = 16.9 Hz), 4.98 (1H, d, J = 8.9 Hz), 5.11 (1H, s), 5.24 (1H, s), 5.37 (1H, m), 5.42 (1H, m), 5.92 (1H, d, J = 9.77 Hz), 6.74 (1H, dd, J = 8.64 Hz, 2.60 Hz), 6.79 (1H, dd, J = 8.87 Hz, 2.54 Hz), 7.01 (1H, m), 7.03 (1H, m); 13C NMR (125 MHz, CDCl3): δ 22.6, 29.1, 32.9, 36.3, 38.0, 42.6, 43.1, 43.4, 45.3, 47.4, 48.7, 51.0, 56.1, 68.5, 79.5, 115.6, 117.0, 117.9, 118.9, 128.3, 128.4, 129.4, 130.6, 131.3, 136.4, 143.0, 156.8, 165.1, 201.0; HRMS (ESI+) calculated for C29H34NaO4 ([M+Na]+): 469.2355, found 469.2349.
Macrolactone 67
To a solution of macrolactone 50 (2.0 mg, 4.5 μmol, 1.00 eq.) in 500 μL of acetic acid was added solid manganese (III) acetate dihydrate (3.1 mg, 11.6 μmol, 2.60 eq.) and copper (II) acetate (0.8 mg, 4.40 μmol, 0.98 eq.). The vial was then purged with argon and and heated to 45 °C in an oil bath for five hours. After cooling to room temperature, the mixture was diluted with ether and washed sequentially with saturated aqueous NaHCO3, water, and brine. The organic phase was dried over anhydrous MgSO4, the solvent was removed under reduced pressure, and the residue was purified by preparative TLC (eluted in 30% EtOAc in hexanes) to give a small amount (< 1 mg) of macrolactone 67.
[α]20D = −6.2 (c 0.10, CH2Cl2); IR (neat) ν 2917, 2849, 1746, 1723, 1644, 1507, 1456, 1378, 1352, 1313, 1235, 1175, 1107 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.87 (1H, m), 0.94 (1H, m), 0.96 (3H, d, J = 6.6 Hz), 1.07 (1H, qd, J = 12.5 Hz, 4.1 Hz), 1.40 (1H, m), 1.42 (1H, m), 1.57 (1H, m), 1.67 (1H, td, J = 11.8 Hz, 6.0 Hz), 1.81 (1H, m), 2.00 (1H, m), 2.13 (1H, m), 2.20 (1H, t, J = 11.5 Hz), 2.68 (1H, m), 2.82 (1H, m), 3.34 (1H, d, J = 14.3 Hz), 3.41 (1H, dd, J = 11.0 Hz, 8.4 Hz), 3.59 (1H, d, J = 14.4 Hz), 3.74 (1H, d, J = 11.8 Hz), 4.39 (1H, d, J = 11.2 Hz), 4.53 (1H, d, J = 11.3 Hz), 4.95 (1H, t, J = 5.3 Hz), 5.13 (1H, s), 5.25 (1H, s), 5.47 (1H, d, J = 3.3 Hz), 5.87 (1H, d, J = 9.8 Hz), 5.94 (1H, dd, J = 9.7 Hz, 2.5 Hz), 6.77 (1H, dd, J = 8.3 Hz, 2.6 Hz), 6.95 (1H, dd, J = 8.4 Hz, 2.6 Hz), 7.02 (1H, dd, J = 8.3 Hz, 2.0 Hz), 7.06 (1H, dd, J = 8.5 Hz, 1.9 Hz); 13C NMR (125 MHz, CDCl3): δ 22.6, 29.1, 32.4, 32.8, 36.2, 38.0, 42.7, 43.5, 47.5, 48.2, 49.0, 52.7, 55.7, 69.0, 81.2, 117.5, 118.6, 119.2, 122.7, 128.6, 129.6, 129.9, 130.6, 131.9, 136.7, 142.9, 157.4, 168.2, 203.7; HRMS (ESI+) calculated for C29H33O4 ([M+H]+): 445.2379, found 445.2381.
methyl 2-(2,2,6-trimethyl-4-oxo-4H-1,3-dioxin-5-yl)acetate (76)
A solution of tert-butyl acetoacetate 74 (20 mL, 121 mmol 1.00 eq.) in 900 mL of THF was cooled to 0 °C, and 60% NaH dispersion in mineral oil (4.82 g, 121 mmol, 1.00 eq.) was added in five portions over the course of 40 minutes. After the last addition, the cooling bath was removed, and the reaction mixture was stirred at room temperature for one hour to give a pale yellow solution. The reaction mixture was again cooled to 0 °C, and neat methyl bromoacetate (11.42 mL, 121 mmol, 1.00 eq.) was added. The cooling bath was removed, and the reaction was left to stir at room temperature for 14 hours. After this time, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed clean formation of the product of Rf = 0.36. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to give crude diester 75 that was used directly in the next step without further purification.
A solution of crude diester 75 in 20 mL of acetone was cooled to 0 °C, and neat acetic anhydride (38.6 mL, 409 mmol, 3.00 eq.) was added followed by the dropwise addition of 18 M sulfuric acid (7.68 mL). After addition of the acid was complete, the cooling bath was removed, and the reaction was allowed to warm to room temperature. After 10 hours, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.20. The reaction mixture was cooled in an ice bath and quenched by the slow addition of cold water to decompose the excess acetic anhydride. The reaction was diluted with CH2Cl2, the layers were separated, and the aqueous phase was extracted with two additional portions of CH2Cl2 before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc) to give dioxinone ester 76 as a tan amorphous solid (16.3 g, 63% over two steps).
IR (neat) ν 2999, 2953, 1714, 1648, 1436, 1400, 1379, 1360, 1333, 1272, 1241, 1197, 1151, 1058, 1000, 973, 921, 832, 786, 758 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.71 (6H, s), 1.98 (3H, s), 3.33 (2H, s), 3.70 (3H, s); 13C NMR (125 MHz, CDCl3): δ 17.7, 25.0, 30.4, 52.2, 99.8, 105.6, 162.1, 165.7, 171.5; HRMS (ESI+) calculated for C10H15O5 ([M+H]+): 215.0919, found 215.0914.
methyl 2-(2,2-dimethyl-4-oxo-6-((triphenyl-λ5-phosphanylidene)methyl)-4H-1,3-dioxin-5-yl)acetate (79)
A two-neck 2 L flask equipped with a reflux condenser and a magnetic stirring bar was flame-dried under high vacuum and cooled to room temperature under argon. A solution of dioxinone ester 76 (18.52 g, 86.5 mmol, 1.00 eq.) in 1050 mL of CCl4 was added followed by solid recrystallized NBS (16.17 g, 90.8 mmol, 1.05 eq.) and benzoyl peroxide (978 mg, 3.30 mmol, 0.035 eq.). The reaction mixture was then heated to reflux for five hours before cooling to room temperature. TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed almost complete consumption of the starting material and formation of the desired product of Rf = 0.45 and the regioisomeric α-bromoester byproduct at Rf = 0.37. The solvent was removed under reduced pressure, and the residue was absorbed onto silica gel for purification by column chromatography (3:1 hexanes/EtOAc → 2:1 → 1:1) to give slightly impure 77 as a colorless oil (8.48 g, 33%) that was used directly in the next step.
To a solution of 77 (8.48 g, 28.9 mmol, 1.00 eq.) in 60 mL of toluene was added a solution of triphenylphosphine (8.35 g, 31.8 mmol, 1.10 eq.) in 20 mL of toluene. A white precipitate formed immediately, and the reaction was left to stir at room temperature for 12 hours. After this time, the solid was collected by vacuum filtration and washed with cold toluene to give crude phosphonium salt 78 that was used directly in the next step.
To a solution of phosphonium salt 78 in 60 mL of CH2Cl2 was added a solution of sodium carbonate (6.00 g, 56.6 mmol, 1.96 eq.) in 150 mL of water. The resulting biphasic reaction mixture was then stirred vigorously at room temperature for one hour. After this time, the layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2 before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on neutral alumina (100% EtOAc) to give ylide 79 as a yellow microcrystalline solid (9.84 g, 24% over three steps).
IR (neat) ν 3056, 2994, 2949, 1719, 1665, 1625, 1526, 1437, 1387, 1198, 1150, 1118, 752, 721, 694, 542, 521 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.09 (6H, s), 3.14 (1H, d, 2JH-P = 17.1 Hz), 3.42 (2H, s), 3.66 (3H, s), 7.49-7.53 (6H, m), 7.59-7.63 (9H, m); 13C NMR (125 MHz, CDCl3): δ 24.7, 31.2, 40.1 (d, 1JC-P = 122 Hz), 51.7, 102.7, 110.0, 127.1 (d, 1JC-P = 91.5 Hz), 129.0 (d, 3JC-P = 12 Hz), 132.6 (d, 4JC-P = 3 Hz), 133.1 (d, 2JC-P = 10 Hz), 163 (d, 2JC-P = 3 Hz), 169.4, 173.9; HRMS (ESI+) calculated for C28H28O5P ([M+H]+): 475.1674, found 475.1666.
Allylic alcohol 81
A solution of enol ether 807 (3.11 g, 10.4 mmol, 1.00 eq.) in 60 mL of acetone and 6 mL of water was cooled to 0 °C, and solid NMO (1.28 g, 10.9 mmol, 1.05 eq.) was added followed by a 4% aqueous solution of OsO4 (663 μL, 0.10 mmol, 0.01 eq.). After stirring at 0 °C for five hours, TLC showed almost complete conversion of the starting material to α-hydroxyaldehyde 16. The reaction was quenched with saturated aqueous Na2S2O3 solution and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc. The combined organics were washed with saturated aqueous Na2S2O3 and brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure to give crude α-hydroxy aldehyde 16 that was used directly in the next step.
To a solution of the crude α-hydroxy aldehyde 16 in 60 mL of CH2Cl2 was added solid dioxinone ylide 79 (4.94 g, 10.4 mmol, 1.00 eq.), and the reaction was stirred at room temperature for 12 hours. After this time, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed formation of the desired product of Rf = 0.33 along with the mixed α-hydroxy aldehyde, α-hydroxy ketone, and their dimers at Rf = 0.62. When the mobile phase was changed to 9:1 toluene/acetone, the major allylic alcohol diastereomer 81 appeared at Rf = 0.42 and the minor diastereomer appeared at Rf = 0.37. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (19:1 toluene/acetone) to give 81 as a colorless oil (2.73 g, 59%).
[α]20D = +10.5 (c 0.74, CH2Cl2); IR (neat) ν 3469, 2950, 2927, 2856, 1742, 1722, 1650, 1601, 1461, 1390, 1376, 1338, 1253, 1202, 1149, 1118, 1093, 1004, 836 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.086 (3H, s), 0.091 (3H, s), 0.75 (1H, q, J = 12.8 Hz), 0.87 (1H, m), 0.88 (3H, d, J = 6.5 Hz), 0.90 (9H, s), 1.16 (1H, qd, J = 13.0 Hz, 3.7 Hz), 1.37-1.43 (2H, m), 1.50-1.57 (3H, m), 1.73 (1H, m), 1.745 (3H, s), 1.748 (3H, s), 3.46 (2H, s), 3.53 (1H, dd, J = 10.4 Hz, 7.2 Hz), 3.60 (1H, dd, J = 10.5 Hz, 2.5 Hz), 3.69 (3H, s), 3.83 (1H, d, J = 6.3 Hz), 4.51 (1H, m), 6.39 (1H, dd, J = 15.3 Hz, 1.9 Hz), 6.59 (1H, dd, J = 15.3 Hz, 4.5 Hz); 13C NMR (125 MHz, CDCl3): δ −5.5, −5.4, 18.3, 22.6, 25.06, 25.13, 26.0, 27.4, 29.9, 32.5, 34.8, 39.1, 41.4, 48.1, 52.3, 68.7, 73.2, 100.1, 105.5, 118.5, 143.4, 159.4, 163.1, 171.5; HRMS (ESI+) calculated for C26H45O7Si ([M+H]+): 497.2935, found 497.2930.
Allylic phosphate 82
A solution of allylic alcohol 81 (1.55 g, 3.12 mmol, 1.00 eq.) in 40 mL of THF was cooled to −78 °C, and a 1.6 M solution of methyllithium in ether (2.19 mL, 3.43 mmol, 1.10 eq.) was added dropwise. The reaction mixture immediately turned dark red; after 10 minutes, neat diethyl chlorophosphate (905 μL, 3.65 mmol, 2.00 eq.) was added, and the flask was transferred to a 0 °C ice bath. After 15 minutes, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.11. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 → 1:1) to afford allylic phosphate 82 as a yellow oil (1.497 g, 77%).
[α]20D = −20.5 (c 1.53, CH2Cl2); IR (neat) ν 2950, 2927, 2856, 1723, 1656, 1605, 1437, 1391, 1376, 1337, 1257, 1201, 1149, 1115, 1093, 1030, 984, 939, 837, 776 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.04 (3H, s), 0.05 (3H, s), 0.84 (1H, m), 0.88 (3H, d, J = 6.5 Hz), 0.89 (9H, s), 1.01 (1H, q, J = 12.3 Hz), 1.28 (1H, m), 1.29 (3H, t, J = 6.5 Hz), 1.32 (3H, t, J = 6.5 Hz), 1.36 (1H, m), 1.53-1.58 (3H, m), 1.66-1.70 (2H, m), 1.75 (6H, s), 3.41 (2H, s), 3.57 (1H, dd, J = 10.3 Hz, 2.0 Hz), 3.69 (3H, s), 3.87 (1H, dd, J = 10.3 Hz, 4.7 Hz), 4.04-1.14 (4H, m), 5.20 (1H, m), 6.35 (1H, dd, J = 15.3 Hz, 1.5 Hz), 6.50 (1H, dd, J = 15.3 Hz, 5.1 Hz); 13C NMR (125 MHz, CDCl3): δ −5.40, −5.37, 16.2 (3JC-P = 5 Hz), 16.3 (3JC-P = 5 Hz), 18.3, 22.7, 24.9, 25.0, 25.2, 26.0, 29.8, 32.3, 35.5, 39.0, 39.8, 42.7 (d, 3JC-P = 6 Hz), 52.3, 63.9 (2JC-P = 2.5 Hz), 64.0 (2JC-P = 2.5 Hz), 64.2, 77.9 (2JC-P = 6 Hz), 101.1, 105.7, 119.8, 140.1 (3JC-P = 2 Hz), 158.5, 162.7, 171.1; HRMS (ESI+) calculated for C30H53NaO10PSi ([M+Na]+): 655.3043, found 655.3041.
Aryl ether 84
To a a solution of phenol 8351 (943 mg, 2.60 mmol, 1.10 eq.) in 21 mL of THF was added solid 60% sodium hydride dispersion in mineral oil (104 mg, 2.60 mmol, 1.10 eq.); gas evolution was observed, and a colorless phenolate solution was obtained. To a separate flask containing neat allylic phosphate 82 (1.50 g, 2.36 mmol, 1.00 eq.) was added a solution of Pd(PPh3)4 (273 mg, 0.24 mmol, 0.10 eq.) in 15 mL of THF. The phenolate solution was immediately added dropwise; after each drop, the reaction mixture would turn dark orange before returning to yellow over the course of several seconds. After addition was complete, TLC (4:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the phosphate, formation of the product of Rf = 0.51, and formation of the elimination byproduct of Rf = 0.40. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (10:1 hexanes/EtOAc → 4:1 → 2:1) to give a mixture of 84 and the elimination byproduct, which was used directly in the next step (1.80 g). An analytical sample of 84 was obtained by repeating the chromatographic separation on a smaller sample of the mixture.
[α]20D = −12.0 (c 0.45, CH2Cl2); IR (neat) ν 3071, 2951, 2928, 2856, 1742, 1724, 1653, 1604, 1509, 1472, 1462, 1428, 1391, 1377, 1336, 1234, 1204, 1171, 1150, 1113, 1086, 835, 741, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ −0.05 (3H, s), −0.04 (3H, s), 0.86 (9H, s), 0.89 (1H, m), 0.91 (3H, d, J = 6.5 Hz), 0.99 (1H, m), 1.07 (9H, s), 1.45 (1H, m), 1.49 (1H, m), 1.63-1.67 (4H, m), 1.73 (1H, m), 1.75 (3H, s), 1.76 (3H, s), 3.33 (2H, s), 3.45-3.52 (2H, m), 3.58 (3H, s), 4.68-4.70 (2H, m), 5.04 (1H, m), 6.23 (1H, dd, J = 15.4 Hz, 1.7 Hz), 6.58 (1H, dd, J = 15.4 Hz, 4.2 Hz), 6.79 (2H, d, J = 8.5 Hz), 7.22 (2H, d, J = 8.5 Hz), 7.36-7.39 (4H, m), 7.41-7.44 (2H, m), 7.67-7.69 (4H, m); 13C NMR (125 MHz, CDCl3): δ −5.43, −5.42, 18.4, 19.4, 22.8, 25.0, 25.2, 25.6, 26.0, 26.9, 29.8, 32.4, 35.0, 39.1, 40.2, 43.4, 52.2, 65.17, 65.23, 77.5, 100.4, 105.7, 115.1, 119.7, 127.8, 129.8, 133.6, 133.7, 135.6, 141.9, 157.5, 158.9, 162.9, 171.2; HRMS (ESI+) calculated for C49H68NaO8Si2 ([M+Na]+): 863.4350, found 863.4343.
Primary alcohol 85
To a solution of mixed 84 (1.80 g, 2.14 mmol, 1.00 eq.) in 75 mL of THF and 75 mL of water was added glacial acetic acid (92 mL, 1.61 mol, 751 eq.). After 12 hours at room temperature, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed formation of the desired product of Rf = 0.52 and the deprotected byproduct of Rf = 0.25. The reaction was diluted with EtOAc, and the acid was quenched by the slow addition of saturated aqueous K2CO3 solution. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (9:1 hexanes/EtOAc → 4:1 → 3:1 → 2:1 → 1:1) to give primary alcohol 85 as a colorless oil (858 mg, 50% over two steps).
[α]20D = −22.7 (c 0.76, CH2Cl2); IR (neat) ν 3487, 2998, 2928, 2857, 1746, 1720, 1652, 1603, 1509, 1472, 1428, 1391, 1376, 1337, 1233, 1203, 1171, 1150, 1111, 1090, 1008, 938, 824, 796, 741, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.92 (3H, d, J = 6.6 Hz), 0.93 (1H, m), 0.97 (1H, m), 1.08 (9H, s), 1.42 (1H, m), 1.47 (1H, m), 1.62 (1H, m), 1.69-1.73 (3H, m), 1.745 (3H, s), 1.750 (3H, s), 1.78 (1H, m), 3.36 (2H, s), 3.54-3.58 (1H, m), 3.60 (3H, s), 3.62-3.67 (1H, m), 4.69 (2H, s), 5.04 (1H, m), 6.29 (1H, dd, J = 15.4 Hz, 1.7 Hz), 6.63 (1H, dd, J = 15.4 Hz, 4.4 Hz), 6.83 (2H, d, J = 8.6 Hz), 7.23 (2H, d, J = 8.6 Hz), 7.36-7.39 (4H, m), 7.41-7.44 (2H, m), 7.67-7.69 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.6, 25.05, 25.13, 26.9, 27.1, 29.8, 32.3, 34.8, 38.9, 40.6, 43.4, 52.3, 65.1, 65.8, 79.4, 100.6, 105.7, 115.3, 120.4, 127.5, 127.8, 129.8, 133.5, 134.2, 135.6, 140.2, 157.0, 158.8, 162.8, 171.3; HRMS (ESI+) calculated for C43H54NaO8Si ([M+Na]+): 749.3486, found 749.3487.
Aldehyde 86
To a solution of primary alcohol 85 (858 mg, 1.18 mmol, 1.00 eq.) in 12 mL of CH2Cl2 was added Dess–Martin periodinane (651 mg, 1.54 mmol, 1.30 eq.). After 20 minutes, TLC (3:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material (Rf = 0.11) and formation of the product of Rf = 0.26. The reaction mixture was diluted with CH2Cl2 and quenched with saturated aqueous Na2S2O3 solution and saturated aqueous NaHCO3 solution. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2 before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 → 1:1) to afford aldehyde 86 as a yellow foam (832 mg, 97%).
[α]20D = −19.1 (c 2.08, CH2Cl2); IR (neat) ν 2929, 2857, 1742, 1720, 1654, 1605, 1509, 1428, 1391, 1377, 1337, 1230, 1203, 1150, 1111, 1090, 1008, 982, 938, 824, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.91 (1H, m), 0.96 (3H, d, J = 6.5 Hz), 0.97 (1H, m), 1.07 (9H, s), 1.42 (1H, m), 1.50 (1H, m), 1.75 (6H, s), 1.79-1.87 (3H, m), 2.10 (1H, m), 2.51 (1H, m), 3.37 (2H, s), 3.60 (3H, s), 4.67 (2H, s), 4.98 (1H, m), 6.29 (1H, dd, J = 15.5 Hz, 1.5 Hz), 6.58 (1H, dd, J = 15.4 Hz, 4.6 Hz), 6.78 (2H, d, J = 8.6 Hz), 7.21 (2H, d, J = 8.3 Hz), 7.37 (4H, t, J = 7.2 Hz), 7.41-7.44 (2H, m), 7.67 (4H, m), 9.54 (1H, d, J = 3.0 Hz); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.4, 25.1, 25.5, 26.9, 29.8, 31.7, 34.0, 34.5, 41.6, 51.3, 52.3, 65.1, 78.5, 101.0, 105.8, 115.4, 121.1, 127.5, 127.8, 129.8, 133.5, 134.2, 135.6, 139.1, 156.7, 158.6, 162.8, 171.2, 203.6; HRMS (ESI+) calculated for C43H52NaO8Si ([M+Na]+): 747.3329, found 747.3320.
Vinyl iodide 87
To a flame-dried 100 mL flask equipped with a magnetic stirring bar was added solid chromium (II) chloride (1.13 g, 9.18 mmol, 8.00 eq.) in the glove box. The flask was tightly sealed with a rubber septum, removed from the glove box, and put under an atmosphere of argon. 25 mL of THF was added to give a pale green slurry, which was cooled to 0 °C. A solution of aldehyde 86 (832 mg, 1.15 mmol, 1.00 eq.) and iodoform (903 mg, 2.29 mmol, 2.00 eq.) in 25 mL of THF was added dropwise, and the reaction immediately turned dark red. The flask was removed from the cooling bath and stirred at room temperature for one hour. After this time, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material (Rf = 0.38) and formation of the product of Rf = 0.49. The reaction was diluted with water and ether, and the layers were separated. The aqueous phase was extracted with two additional portions of ether, and the combined organics were washed with saturated aqueous Na2S2O3 solution and brine before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (9:1 hexanes/EtOAc → 3:1 → 2:1) to give vinyl iodide 87 as an off-white foam (911 mg, 94%).
[α]20D = −75.8 (c 4.88, CH2Cl2); IR (neat) ν 2927, 2856, 1742, 1720, 1653, 1604, 1509, 1460, 1428, 1391, 1376, 1336, 1232, 1203, 1173, 1149, 1111, 1090, 1008, 940, 824, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.89 (1H, m), 0.90 (3H, d, J = 6.5 Hz), 0.91 (1H, m), 1.08 (9H, s), 1.44-1.50 (3H, m), 1.66 (1H, m), 1.72 (1H, m), 1.74 (3H, s), 1.75 (3H, s), 1.78 (1H, m), 2.29 (1H, td, J = 10.1 Hz, 6.6 Hz), 3.34 (2H, s), 3.58 (3H, s), 4.69 (2H, s), 4.95 (1H, m), 5.61 (1H, d, J = 14.3 Hz), 6.24-6.30 (2H, m), 6.56 (1H, dd, J = 15.4 Hz, 3.9 Hz), 6.79 (2H, d, J = 8.5 Hz), 7.22 (2H, d, J = 8.3 Hz), 7.36-7.39 (4H, m), 7.41-7.44 (2H, m), 7.68-7.70 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.4, 24.9, 25.0, 25.1, 26.9, 29.8, 31.8, 34.5, 41.2, 46.4, 47.0, 52.2, 65.2, 76.4, 77.8, 100.6, 105.7, 115.2, 120.0, 127.5, 127.8, 129.8, 133.6, 133.9, 135.6, 140.9, 149.4, 157.2, 158.7, 162.8, 171.2; HRMS (ESI+) calculated for C44H53IKO7Si ([M+K]+): 887.2242, found 887.2241.
Triene 88
To a solution of vinyl iodide 87 (911 mg, 1.07 mmol, 1.00 eq.) in 5 mL of DMF was added a solution of dichlorobis(acetonitrile)palladium(II) (28 mg, 0.11 mmol, 0.10 eq.) in 5 mL of DMF. Next, a 0.25 M solution of stannane 42 (8.61 mL, 2.15 mmol, 2.00 eq.) in DMF was added dropwise; the reaction mixture immediately turned black, and stirring was continued at room temperature for two hours. After this time, the reaction was diluted with ether and water. The layers were separated, and the organic phase was washed with two additional portions of water before drying over anhydrous Na2SO4. TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed the product of Rf = 0.56 which was essentially copolar with the vinyl iodide starting material. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (9:1 hexanes/EtOAc → 3:1) to give triene 88 as a yellow foam (735 mg, 88%).
[α]20D = −26.3 (c 0.38, CH2Cl2); IR (neat) ν 2999, 2928, 2857, 1741, 1721, 1653, 1605, 1509, 1428, 1391, 1376, 1336, 1232, 1203, 1172, 1149, 1112, 1089, 1007, 977, 824, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.90 (3H, d, J = 6.4 Hz), 0.91-0.92 (2H, m), 1.08 (9H, s), 1.38 (1H, m), 1.47 (1H, m), 1.52 (1H, m), 1.67 (1H, m), 1.71 (1H, m), 1.727 (3H, s), 1.734 (3H, s), 1.79 (1H, m), 2.27 (1H, m), 3.32 (2H, s), 3.57 (3H, s), 4.69 (2H, s), 4.96 (1H, m), 4.99 (1H, d, J = 10.3 Hz), 5.04 (1H, d, J = 16.7 Hz), 5.44 (1H, dd, J = 15.1 Hz, 9.3 Hz), 5.68 (1H, dd, J = 15.2 Hz, 10.6 Hz), 5.80 (1H, dd, J = 15.2 Hz, 10.7 Hz), 6.09 (1H, dd, J = 15.1 Hz, 10.5 Hz), 6.23 (1H, dd, J = 15.3 Hz, 1.3 Hz), 6.26 (1H, m), 6.55 (1H, dd, J = 15.4 Hz, 4.0 Hz), 6.79 (2H, d, J = 8.6 Hz), 7.21 (2H, d, J = 8.3 Hz), 7.36-7.39 (4H, m), 7.41-7.44 (2H, m), 7.68-7.69 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.5, 24.7, 25.05, 25.11, 26.9, 29.8, 31.9, 34.7, 42.0, 43.3, 47.2, 52.2, 65.2, 78.0, 100.4, 105.6, 115.4, 116.9, 119.7, 127.3, 127.8, 129.8, 131.7, 131.9, 133.0, 133.6, 133.7, 135.6, 136.9, 138.4, 141.8, 157.5, 158.9, 162.9, 171.2; HRMS (ESI+) calculated for C48H58NaO7Si ([M+Na]+): 797.3850, found 797.3836.
Benzylic Alcohol 89
To a flame-dried 50 mL pressure flask was added a solution of triene 88 (735 mg, 0.95 mmol, 1.00 eq.) in 35 mL of freshly distilled benzene (sodium/benzophenone). The flask was tightly sealed with its white Teflon screw cap, lowered into a pre-heated 75 °C oil bath, and heated at that temperature for 72 hours. After this time, TLC (plate eluted four times in 4:1 hexanes/EtOAc, UV/anisaldehyde) showed essentially complete consumption of the starting triene (Rf = 0.64) and formation of the product of Rf = 0.71. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (eight 1500 μm plates, loaded in CH2Cl2, eluted five times in 85:15 hexanes/EtOAc, silica washed with EtOAc) to give tricycle 70 as a white foam (407 mg, 55%). NMR showed that this was a 3:1 mixture of inseparable endo/exo isomers, which was used directly in the next step.
To a a solution of 70 (204 mg, 0.26 mmol, 1.00 eq.) in 8 mL of THF was added solid TBAT (500 mg, 0.93 mmol, 3.52 eq.). After 12 hours at room temperature, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of two products: the deprotected endo isomer 89 of Rf = 0.24 and the deprotected exo isomer of Rf = 0.15. The reaction was diluted with water and EtOAc, the layers were separated, and the organic phase was washed with water before drying over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (3:1 hexanes/EtOAc → 2:1 → 1:1 hexanes/EtOAc) to give the deprotected endo isomer 89 (98.8 mg, 70%) and the deprotected exo isomer (38.1 mg, 27%).
[α]20D = −75.8 (c 1.76, CH2Cl2); IR (neat) ν 3407, 2947, 2915, 2865, 1720, 1638, 1611, 1509, 1374, 1337, 1271, 1237, 1202, 1173, 1149, 1006, 940 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.83 (1H, q, J = 11.7 Hz), 0.92 (3H, d, J = 6.6 Hz), 0.98 (1H, m), 1.06 (1H, qd, J = 11.5 Hz, 2.9 Hz), 1.30 (3H, s), 1.42-1.48 (3H, m), 1.60 (3H, s), 1.77 (1H, m), 1.93 (1H, td, J = 11.3 Hz, 6.9 Hz), 2.02 (1H, m), 2.13-2.15 (2H, m), 3.19 (1H, m), 3.27 (1H, d, J = 17.1 Hz), 3.31 (1H, dd, J = 11.2 Hz, 6.5 Hz), 3.49 (1H, d, J = 17.1 Hz), 3.69 (3H, s), 4.52 (1H, dd, J = 7.0 Hz, 4.0 Hz), 4.56 (2H, d, J = 5.7 Hz), 5.04 (1H, d, J = 11.3 Hz), 5.05 (1H, d, J = 16.1 Hz), 5.44 (1H, dt, J = 9.8 Hz, 3.5 Hz), 5.76 (1H, ddd, J = 17.7 Hz, 9.6 Hz, 8.2 Hz), 5.97 (1H, d, J = 9.8 Hz), 6.74 (2H, d, J = 8.6 Hz), 7.20 (2H, d, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3): δ 22.5, 25.2, 25.9, 30.4, 32.8, 36.1, 38.0, 41.0, 45.4, 45.6, 49.2, 52.2, 56.5, 65.2, 79.5, 99.1, 105.5, 115.1, 117.4, 128.4, 128.8, 129.3, 133.3, 137.3, 157.8, 162.2, 168.5, 171.7; HRMS (ESI+) calculated for C32H41O7 ([M+H]+): 537.2852, found 537.2852.
Benzylic chloride 90
A 0.8 M solution of methanesulfonyl chloride was prepared by dissolving 312 μL of the neat compound in 5 mL of CH2Cl2. Similarly, a 0.8 M solution of diisopropylethylamine was prepared by dissolving 696 μL of the neat amine in 5 mL of CH2Cl2. To a solution of benzylic alcohol 89 (33.9 mg, 63.2 μmol, 1.00 eq.) in 1.25 mL of CH2Cl2 was added a portion of the amine solution (400 μL, 0.32 mmol, 5.00 eq.) followed by a portion of the methanesulfonyl chloride solution (400 μL, 0.32 mmol, 5.00 eq.), and the reaction was stirred at room temperature for ten hours. After this time, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material and formation of the product of Rf = 0.69. The reaction was quenched with pH 7 phosphate buffer and diluted with CH2Cl2. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2 before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on neutral alumina (3:1 hexanes/EtOAc → 2:1) to afford benzylic chloride 90 as a white foam (30.1 mg, 86%).
[α]20D = +124.5 (c 0.49, CH2Cl2); IR (neat) ν 2919, 2868, 1721, 1634, 1610, 1585, 1511, 1436, 1388, 1375, 1339, 1245, 1202, 1174, 1144, 1005, 932 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.82 (1H, q, J = 11.7 Hz), 0.92 (3H, d, J = 6.6 Hz), 0.97 (1H, m), 1.06 (1H, m), 1.23 (3H, s), 1.39-1.47 (3H, m), 1.60 (3H, s), 1.77 (1H, m), 1.95 (1H, m), 2.01 (1H, m), 2.12 (1H, m), 2.15 (1H, m), 3.2 (1H, m), 3.32 (1H, d, J = 17.4 Hz), 3.34 (1H, m), 3.52 (1H, d, J = 17.1 Hz), 3.70 (3H, s), 4.50 (1H, m), 4.52 (2H, s), 5.05 (2H, m), 5.45 (1H, m), 5.77 (1H, m), 5.97 (1H, d, J = 9.9 Hz), 6.73 (2H, d, J = 8.2 Hz), 7.22 (2H, d, J = 8.2 Hz); 13C NMR (125 MHz, CDCl3): δ 22.5, 25.3, 25.6, 30.4, 30.5, 32.7, 36.0, 38.0, 41.2, 45.4, 45.8, 46.4, 49.2, 52.2, 56.5, 79.6, 99.0, 105.5, 115.2, 117.4, 128.4, 129.3, 129.8, 130.1, 137.2, 158.2, 162.1, 168.5, 171.1; HRMS (ESI+) calculated for C32H40ClO6 ([M+H]+): 555.2513, found 555.2512.
Benzylic stannane 91
A 0.25 M solution of trimethylstannyllithium was prepared as follows: A solution of hexamethylditin (354 mg, 1.08 mmol, 1.10 eq.) in 3.31 mL of THF was cooled to −78 °C, and a 1.6 M solution of methyllithium in ether (613 μL, 0.98 mmol, 1.00 eq.) was added dropwise. After addition was complete, the flask was transferred to an ice bath and stirred at 0 °C for 1.5 hours; the resulting colorless solution of trimethylstannyllithium was then used directly in the next step.
A solution of benzylic chloride 90 (38.1 mg, 68.6 μmol, 1.00 eq.) in 2.5 mL of anhydrous THF was cooled to −40 °C, and an aliquot of the 0.25 M trimethylstannyllithium solution (300 μL, 75 μmol, 1.09 eq.) was added dropwise. The reaction mixture immediately turned bright yellow, and this color faded over the course of 5 minutes. After this time, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed approximately 50% conversion of the starting material (Rf = 0.50) to the desired stannane of Rf = 0.63. Therefore, an additional portion of the stannyllithium reagent was added (150 μL, 37.5 μmol, 0.55 eq.); the bright yellow color reappeared, and the reaction was stirred at −40 °C for another 10 minutes. At this point, TLC showed almost complete conversion of the benzyl chloride to the stannane. The reaction was quenched with pH 7 phosphate buffer and diluted with EtOAc. After warming to room temperature, the layers were separated, and the aqueous phase was extracted with one additional portion of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on neutral alumina (4:1 hexanes/EtOAc) to give stannane 91 as a white foam (27.6 mg, 59%).
[α]20D = +115.3 (c 0.98, CH2Cl2); IR (neat) ν 2947, 2914, 2868, 1724, 1635, 1504, 1435, 1387, 1375, 1338, 1268, 1240, 1205, 1152, 1007, 933, 765 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.00 (9H, t, 2JH-Sn = 52 Hz), 0.81 (1H, q, J = 11.6 Hz), 0.91 (3H, d, J = 6.5 Hz), 0.94 (1H, m), 1.04 (1H, m), 1.23 (3H, s), 1.39-1.43 (3H, m), 1.60 (3H, s), 1.75 (1H, m), 1.91 (1H, m), 1.98 (1H, m), 2.11 (2H, m), 2.19 (2H, s), 3.19 (1H, m), 3.33 (1H, d, J = 17.0 Hz), 3.34 (1H, m), 3.52 (1H, d, J = 17.1 Hz), 3.68 (3H, s), 4.39 (1H, dd, J = 7.2 Hz, 3.6 Hz), 5.03 (1H, d, J = 14.6 Hz), 5.04 (1H, d, J = 11.9 Hz), 5.45 (1H, dt, J = 9.6 Hz, 3.5 Hz), 5.78 (1H, m), 5.96 (1H, d, J = 9.8 Hz), 6.58 (2H, d, J = 8.6 Hz), 6.80 (2H, d, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3): δ −10.1, 18.9, 22.5, 25.3, 25.7, 30.5, 30.6, 32.8, 36.1, 38.0, 41.2, 45.4, 45.5, 45.7, 49.3, 52.2, 56.6, 79.5, 98.7, 105.4, 115.3, 117.2, 127.6, 128.5, 129.3, 135.2, 137.4, 154.3, 162.3, 168.7, 171.7; HRMS (ESI+) calculated for C35H49O6Sn ([M+H]+): 685.2551, found 685.2551.
Carboxylic acid 92
To a solution of benzylic stannane 91 (27.6 mg, 40.4 μmol, 1.00 eq.) in 3 mL of THF and 1.5 mL of water was added solid lithium hydroxide monohydrate (100 mg, 2.38 mmol, 59 eq.). After stirring at room temperature for 36 hours, TLC (100% EtOAc, UV/anisaldehyde) showed complete conversion of the starting material (Rf = 0.90) to the product of Rf = 0.52. The reaction was quenched with 1 M aqueous HCl solution and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with two additional portions of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (1000 μm plate, loaded in CH2Cl2, eluted in 1:3 hexanes/EtOAc, silica washed with EtOAc) to give carboxylic acid 92 as a white foam (21.2 mg, 79%).
[α]20D = +79.2 (c 2.50, CH2Cl2); IR (neat) ν 2918, 2852, 1724, 1633, 1504, 1456, 1388, 1375, 1269, 1239, 1206, 1155, 991, 935, 806, 765 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.00 (9H, t, 2JH-Sn = 52 Hz), 0.82 (1H, q, J = 11.8 Hz), 0.92 (3H, d, J = 6.5 Hz), 0.96 (1H, m), 1.05 (1H, m), 1.24 (3H, s), 1.40-1.42 (3H, m), 1.60 (3H, s), 1.75 (1H, m), 1.93 (1H, m), 2.00 (1H, m), 2.12-2.14 (2H, m), 2.20 (2H, s), 3.25 (1H, m), 3.37 (1H, d, J = 16.6 Hz), 3.41 (1H, m), 3.52 (1H, d, J = 16.5 Hz), 4.39 (1H, dd, J = 7.3 Hz, 3.6 Hz), 5.02 (1H, d, J = 8.8 Hz), 5.03 (1H, d, J = 18.3 Hz), 5.45 (1H, dt, J = 9.6 Hz, 3.3 Hz), 5.73 (1H, dt, J = 17.7 Hz, 9.2 Hz), 5.96 (1H, d, J = 9.9 Hz), 6.58 (2H, d, J = 8.1 Hz), 6.80 (2H, d, J = 7.9 Hz); 13C NMR (125 MHz, CDCl3): δ −10.0, 18.9, 22.5, 25.2, 25.8, 30.6, 31.1, 32.8, 36.0, 38.0, 41.3, 45.5, 45.6, 49.3, 56.5, 79.2, 97.8, 105.9, 115.2, 117.4, 127.6, 128.4, 129.3, 135.2, 137.1, 154.2, 161.9, 163.6, 170.1; HRMS (ESI+) calculated for C34H47O6Sn ([M+H]+): 671.2395, found 671.2399.
Acid chloride 71
A 0.1 M solution of chloroenamine reagent 93 was prepared by dissolving 53 μL of the neat compound in 4.0 mL of anhydrous, degassed chloroform. To a solution of carboxylic acid 92 (4.0 mg, 5.98 μmol, 1.00 eq.) in 500 μL of chloroform was added a portion of the chloroenamine solution (120 μL, 12 μmol, 2.00 eq.), and the reaction mixture was stirred at room temperature for 15 minutes. After this time, a small aliquot of the reaction mixture was quenched with methanol; TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion to the corresponding methyl ester 91 of Rf = 0.63, indicating quantitative formation of acid chloride 71. Formation of acid chloride 71 could also be monitored by NMR. The protons of the methylene group at the alpha position of the acid chloride were deshielded by ~0.5 ppm in the 1H-NMR spectrum relative to those in carboxylic acid 92; similarly, the corresponding methylene carbon was deshielded by 11 ppm in the 13C-NMR spectrum. Furthermore, a strong HMBC signal was observed between the aforementioned methylene protons and a carbon signal at 172 ppm, corresponding to the acid chloride carbonyl group. The crude solution of acid chloride 71 in chloroform was used directly in the subsequent palladium-catalyzed coupling reactions.
Tricycle 96
To a 20 mL pressure tube was added a solution of triene 88 (161 mg, 0.21 mmol, 1.00 eq.) in 5 mL of m-xylene and 1.00 mL of water. The tube was tightly sealed with its white Teflon screw cap and lowered into a pre-heated 160 °C oil bath. After three hours, the reaction mixture was cooled to room temperature, and TLC (4:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material (Rf = 0.26) and formation of the product of Rf = 0.44. The aqueous phase was removed with a pipette, and the organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (two 1500 μm plates, loaded in CH2Cl2, eluted in 95:5 hexanes/EtOAc, silica washed with EtOAc) to give tricycle 96 as a colorless oil (92.5 mg, 72%).
[α]20D = +100.0 (c 0.83, CH2Cl2); IR (neat) ν 2949, 2927, 2856, 1740, 1714, 1610, 1587, 1509, 1428, 1371, 1241, 1169, 1112, 1085, 1007, 924, 824, 741, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.82 (1H, q, J = 11.7 Hz), 0.90 (3H, d, J = 6.7 Hz), 0.92 (1H, m), 1.04 (1H, m), 1.07 (9H, s), 1.37-1.39 (3H, m), 1.71 (1H, m), 1.96-2.06 (4H, m), 2.32 (1H, m), 2.40 (1H, dt, J = 14.6 Hz, 7.2 Hz), 2.62 (1H, dt, J = 18.4 Hz, 7.0 Hz), 2.80 (1H, dt, J = 17.7 Hz, 7.3 Hz), 3.35 (2H, m), 3.53 (3H, s), 4.62 (1H, dd, J = 7.1 Hz, 2.9 Hz), 4.67 (2H, s), 5.01 (1H, d, J = 10.9 Hz), 5.02 (1H, d, J = 15.9 Hz), 5.47 (1H, d, J = 9.9 Hz), 5.60 (1H, dt, J = 17.1 Hz, 8.4 Hz), 5.97 (1H, d, J = 9.9 Hz), 6.77 (2H, d, J = 7.9 Hz), 7.16 (2H, d, J = 8.0 Hz), 7.37 (4H, t, J = 7.4 Hz), 7.42 (2H, t, J = 7.4 Hz), 7.68 (4H, d, J = 7.2 Hz); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.6, 26.9, 27.5, 30.5, 32.7, 36.1, 37.4, 38.1, 43.5, 44.6, 45.4, 49.6, 51.1, 51.7, 56.7, 65.3, 79.1, 115.8, 116.5, 127.3, 127.7, 128.8, 129.3, 129.7, 133.3, 133.6, 135.7, 137.5, 157.2, 173.4, 209.1; HRMS (ESI+) calculated for C44H54NaO5Si ([M+Na]+): 713.3638, found 713.3645.
Diol 97
A solution of tricycle 96 (31.6 mg, 45.7 μmol, 1.00 eq.) in 2 mL of anhydrous toluene was cooled to −78 °C, and a 1.5 M solution of diisobutylaluminum hydride in toluene (260 μL, 0.39 mmol, 8.53 eq.) was added dropwise. After 10 minutes at −78 °C, the flask was transferred to an ice bath and stirred for an additional 30 minutes. After this time, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete consumption of the starting material (Rf = 0.74) and formation of the product of Rf = 0.46. The reaction was quenched at 0 °C with saturated aqueous Rochelle’s salt and diluted with EtOAc. The resulting gelatinous suspension was filtered through Celite, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (500 μm plate, loaded in CH2Cl2, eluted in 3:2 hexanes/EtOAc, silica washed with EtOAc) to give diol 97 as a colorless oil (25.2 mg, 83%). NMR showed that 97 was formed as a 1:1 mixture of diastereomers.
IR (neat) ν 3400, 3071, 3014, 2926, 2857, 1610, 1587, 1509, 1428, 1375, 1298, 1240, 1169, 1112, 1082, 1007, 923, 824, 739, 702 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.83 (1H, m), 0.91 (4H, m), 1.01 (1H, m), 1.08 (9H, s), 1.39-1.40 (3H, m), 1.59-1.97 (8H, m), 2.19 (1H, m), 2.32 (0.5H, m), 2.40 (0.5H, m), 3.07 (0.5H, m), 3.20 (0.5H, m), 3.54-3.64 (2H, m), 3.69 (0.5H, t, J = 8.9 Hz), 3.86 (0.5H, m), 4.70-4.71 (2.5H, m), 4.78 (0.5H, dd, J = 5.8 Hz, 2.7 Hz), 5.07 (0.5H, d, J = 16.9 Hz), 5.11-5.14 (1.5H, m), 5.41 (0.5 H, dt, J = 10.1 Hz, 3.4 Hz), 5.47 (0.5H, m), 5.80 (0.5H, dt, J = 17.8 Hz, 9.4 Hz), 5.90 (1H, m), 5.97 (0.5H, dt, J = 17.9 Hz, 9.5 Hz), 6.88 (1H, d, J = 7.9 Hz), 6.92 (1H, d, J = 8.0 Hz), 7.22 (2H, d, J = 8.0 Hz), 7.36-7.39 (4H, m), 7.41-7.44 (2H, m), 7.67-7.70 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.4, 22.5, 26.9, 29.3, 30.3, 30.8, 31.0, 32.2, 32.4, 32.9, 33.0, 36.2, 37.9, 38.0, 43.5, 44.3, 44.6, 45.1, 45.9, 46.1, 48.1, 50.4, 50.7, 55.6, 56.8, 62.9, 63.1, 65.16, 65.21, 73.1, 73.2, 80.4, 81.4, 116.3, 116.6, 116.9, 117.4, 127.39, 127.44, 127.5, 127.8, 128.0, 129.73, 129.74, 130.1, 130.2, 133.56, 133.58, 133.59, 133.60, 133.9, 134.8, 135.6, 135.7, 138.2, 140.0, 154.6, 156.2; HRMS (ESI+) calculated for C43H56NaO4Si ([M+Na]+): 687.3846, found 687.3840.
Secondary alcohol 98
A 0.1 M solution of TBDPSCl was prepared by dissolving 128 μL of the neat compound in 5 mL of CH2Cl2. Similarly, a 0.1 M solution of imidazole was prepared by dissolving 34 mg of the solid compound in 5 mL of CH2Cl2. To a flask containing neat mixed diol 97 (25.2 mg, 37.9 μmol, 100 eq.) was added a portion of the imidazole solution (758 μL, 75.8 μmol, 2.00 eq.) followed by a portion of the TBDPSCl solution (758 μL, 75.8 μmol, 2.00 eq.). The reaction mixture immediately turned cloudy, and a precipitate formed. After 25 minutes at room temperature, TLC (2:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion of the starting material to the product of Rf = 0.70. The reaction was diluted with CH2Cl2 and quenched with pH 7 phosphate buffer. The layers were separated, and the aqueous phase was extracted with one additional portion of CH2Cl2 before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (1000 μm plate, loaded in CH2Cl2, eluted in 85:15 hexanes/EtOAc, silica washed with EtOAc) to give secondary alcohol 98 as a colorless oil (24.8 mg, 72%). NMR showed that 98 was still a 1:1 mixture of inseparable diastereomers.
IR (neat) ν 3464, 3070, 3014, 2929, 2857, 1611, 1588, 1509, 1472, 1428, 1240, 1111, 1088, 999, 823, 702 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.84 (1H, m), 0.90 (3H, m), 0.93 (1H, m), 0.99 (1H, m), 1.05 (9H, s), 1.09 (9H, s), 1.36-1.40 (3H, m), 1.53-2.02 (8H, m), 2.20 (1H, m), 2.32 (1H, m), 3.06 (0.5H, m), 3.18 (0.5H, m), 3.61-3.68 (2.5H, m), 3.85 (0.5H, m), 4.66-4.68 (2.5H, m), 4.77 (0.5H, m), 5.03 (0.5H, d, J = 17.0 Hz), 5.06-5.10 (1.5H, m), 5.40 (0.5H, ddd, J = 9.6 Hz, 4.5 Hz, 2.8 Hz), 5.47 (0.5H, ddd, J = 9.7 Hz, 4.7 Hz, 2.7 Hz), 5.80 (0.5 H, ddd, J = 17.0 Hz, 10.1 Hz, 8.5 Hz), 5.90 (1H, d, J = 9.4 Hz), 5.99 (0.5H, dt, J = 16.9 Hz, 9.6 Hz), 6.83 (1H, d, J = 8.6 Hz), 6.90 (1H, d, J = 8.6 Hz), 7.19 (1H, d, J = 8.6 Hz), 7.21 (1H, d, J = 8.7 Hz), 7.34-7.43 (12H, m), 7.64-7.70 (8H, m); 13C NMR (125 MHz, CDCl3): δ 19.3, 19.4, 22.5, 22.6, 26.91, 26.94, 28.7, 29.9, 30.8, 31.0, 31.7, 32.0, 33.0, 33.1, 36.2, 36.3, 37.9, 38.1, 43.4, 43.6, 44.0, 44.3, 45.1, 45.7, 46.1, 48.0, 50.6, 50.8, 55.8, 57.0, 64.1, 64.3, 65.19, 65.24, 72.8, 73.0, 80.0, 81.1, 116.0, 116.2, 116.8, 117.1, 127.35, 127.43, 127.6, 127.7, 127.8, 127.9, 129.5, 129.6, 129.71, 129.73, 130.3, 130.5, 133.4, 133.59, 133.60, 133.62, 133.63, 134.00, 134.01, 134.10, 134.11, 134.4, 135.6, 135.7, 138.3, 140.4, 154.9, 156.5; HRMS (ESI+) calculated for C59H75O4Si2 ([M+H]+): 903.5204, found 903.5208.
Ketone 99
To a solution of mixed alcohol 98 (12.4 mg, 13.7 μmol, 1.00 eq.) in 1 mL of CH2Cl2 was added solid Dess–Martin periodinane (20 mg, 47.2 μmol, 3.44 eq.). After 30 minutes, TLC (4:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion of the starting material (Rf = 0.51) to the product of Rf = 0.56. The reaction was diluted with CH2Cl2 and quenched with saturated aqueous Na2S2O3 solution and saturated aqueous NaHCO3 solution. The layers were separated, and the organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (500 μm plate, loaded in CH2Cl2, eluted in 85:15 hexanes/EtOAc, silica washed with acetone) to give ketone 99 as a colorless oil (12 mg, 97%).
[α]20D = +89.6 (c 1.20, CH2Cl2); IR (neat) ν 3070, 2928, 2856, 1712, 1611, 1588, 1509, 1472, 1375, 1240, 1111, 1089, 1007, 923, 823, 702, 614, 505 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.82 (1H, q, J = 11.7 Hz), 0.91 (3H, d, J = 6.6 Hz), 0.94 (1H, m), 1.02 (9H, s), 1.06 (9H, s), 1.07 (1H, m), 1.37-1.41 (3H, m), 1.63 (2H, m), 1.72 (1H, m), 2.00-2.05 (4H, m), 2.41 (1H, dt, J = 17.1 Hz, 7.4 Hz), 2.50 (1H, dt, J = 17.2 Hz, 7.7 Hz), 3.29 (1H, m), 3.35 (1H, dd, J = 10.9 Hz, 6.7 Hz), 3.55 (2H, t, J = 6.1 Hz), 4.64-4.67 (3H, m), 4.94-4.98 (2H, m), 5.46 (1H, dt, J = 9.7 Hz, 3.3 Hz), 5.57 (1H, ddd, J = 16.9 Hz, 10.0 Hz, 8.7 Hz), 5.97 (1H, d, J = 9.7 Hz), 6.75 (2H, d, J = 8.5 Hz), 7.12 (2H, d, J = 8.2 Hz), 7.33-7.42 (12H, m), 7.60-7.62 (4H, m), 7.67-7.68 (4H, m); 13C NMR (125 MHz, CDCl3): δ 19.3, 19.4, 22.6, 26.3, 26.9, 30.6, 32.8, 36.1, 38.1, 39.2, 43.9, 44.5, 45.4, 49.7, 51.1, 56.7, 63.2, 65.2, 79.1, 115.8, 116.2, 127.2, 127.67, 127.72, 128.8, 129.5, 129.6, 129.7, 133.1, 133.6, 133.90, 133.92, 135.6, 135.7, 137.6, 157.2, 211.1; HRMS (ESI+) calculated for C59H72KO4Si2 ([M+K]+): 939.4604, found 939.4600.
Diol 100
A 1.0 M solution of buffered TBAF was prepared by adding 200 μL of glacial acetic acid to 1.5 mL of a 1.0 M solution of TBAF in THF. To a solution of ketone 99 (12 mg, 13.3 μmol, 1.00 eq.) in 1.5 mL of THF was added a portion of the buffered TBAF solution (600 μL, 0.6 mmol, 45 eq.). After stirring at room temperature for ten hours, TLC (1:1 hexanes/EtOAc, UV/anisaldehyde) showed complete conversion of the starting material (Rf = 0.86) to the product of Rf = 0.10. The reaction was quenched with water and diluted with EtOAc. The layers were separated, and the aqueous phase was extracted with one additional portion of EtOAc before the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by preparative TLC (500 μm plate, loaded in CH2Cl2, eluted in 4:1 EtOAc/hexanes, silica washed with acetone) to give diol 100 as a white amorphous solid (5.5 mg, 97%).
[α]20D = +176.2 (c 0.55, CH2Cl2); IR (neat) ν 3349, 2946, 2902, 2866, 2843, 1703, 1612, 1513, 1402, 1249, 1174, 1032, 1006, 918, 814, 719 cm−1; 1H NMR (500 MHz, CDCl3): δ 0.82 (1H, q, J = 11.6 Hz), 0.91 (3H, d, J = 6.7 Hz), 0.94 (1H, m), 1.05 (1H, m), 1.38-1.40 (3H, m), 1.56 (2H, m), 1.74 (1H, m), 1.96-2.05 (4H, m), 2.36 (1H, dt, J = 17.8 Hz, 6.7 Hz), 2.47 (1H, dt, J = 18.0 Hz, 6.8 Hz), 3.27-3.41 (4H, m), 4.57 (2H, s), 4.71 (1H, dd, J = 6.7 Hz, 3.5 Hz), 4.98 (1H, d, J = 9.9 Hz), 5.00 (1H, d, J = 17.1 Hz), 5.45 (1H, dt, J = 9.8 Hz, 3.2 Hz), 5.55 (1H, dt, J = 17.6 Hz, 9.1 Hz), 5.96 (1H, d, J = 9.9 Hz), 6.81 (2H, d, J = 8.1 Hz), 7.22 (2H, d, J = 8.0 Hz); 22.5, 26.0, 30.3, 32.7, 36.1, 38.1, 39.4, 43.8, 44.5, 45.5, 49.5, 51.1, 56.6, 62.2, 65.2, 79.0, 115.9, 116.2, 128.7, 128.9, 129.4, 133.1, 137.5, 157.9, 211.8; HRMS (ESI+) calculated for C27H37O4 ([M+H]+): 425.2692, found 425.2690.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institute of General Medical Sciences (GM065483) to E.J.S., the Natural Sciences and Engineering Research Council of Canada (postdoctoral fellowship for C. A. C.), and Princeton University. We thank Dr. István Pelczer for his assistance with NMR spectroscopy, Dr. John Eng for his help with mass spectrometry, and Lotus Separations for compound purification and analysis. Finally, we also thank Rebecca Lambert for her helpful suggestions in preparing and proofreading this manuscript.
Footnotes
Supporting Information. 1H-NMR and 13C-NMR spectra for all new compounds and full spectral assignments for cyclophanes 10, 24, 26, 30, 43, 44, 45, 46, 49, 50, and 67. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.Isaka M, Rugseree N, Maithip P, Kongsaeree P, Prabpai S, Thebtaranonth Y. Tetrahedron. 2005;61:5577. [Google Scholar]
- 2.Hasegawa A, Koizumi F, Takahashi Y, Ando K, Ogawa T, Hara M, Yoshida M. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu. 2001;43:467. [Google Scholar]
- 3.He HY, Yang HY, Bigelis R, Solum EH, Greenstein M, Carter GT. Tetrahedron Lett. 2002;43:1633. [Google Scholar]
- 4.Shiono Y, Shimanuki K, Hiramatsu F, Koseki T, Tetsuya M, Fujisawa N, Kimura K. Bioorg Med Chem Lett. 2008;18:6050. doi: 10.1016/j.bmcl.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 5.Oikawa H. J Org Chem. 2003;68:3552. doi: 10.1021/jo0267596. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review covering previous synthetic attempts towards the hirsutellones and related natural products see: Nay B, Li X, Ear A. Nat Prod Rep. 2013;30:765. doi: 10.1039/c3np70016j.
- 7.For our previous synthesis of the decahydrofluorene core of hirsutellone B see: Sorensen E, Tilley S, Reber K. Org Lett. 2009;11:701. doi: 10.1021/ol802768p.
- 8.Huang M, Huang C, Liu B. Tetrahedron Lett. 2009;50:2797. [Google Scholar]
- 9.Huang M, Song L, Liu B. Org Lett. 2010;12:2504. doi: 10.1021/ol100692x. [DOI] [PubMed] [Google Scholar]
- 10.Roush W, Halvorsen G. Tetrahedron Lett. 2011;52:2072. doi: 10.1016/j.tetlet.2010.10.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riache N, Ndoye I, Li X, Nay B. Synlett. 2011;18:2685. [Google Scholar]
- 12.Nicolaou K, Sarlah D, Wu T, Zhan W. Angew Chem Int Ed Engl. 2009;48:6870. doi: 10.1002/anie.200903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicolaou K, Sun Y, Sarlah D, Zhan W, Wu T. Org Lett. 2011;13:5708. doi: 10.1021/ol202239u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uchiro H, Kato R, Arai Y, Hasegawa M, Kobayakawa Y. Org Lett. 2011;13:6268. doi: 10.1021/ol202748e. [DOI] [PubMed] [Google Scholar]
- 15.For a review of acylketene generation and reactivity see: Sorensen E, Reber K, Tilley S. Chem Soc Rev. 2009;38:3022. doi: 10.1039/b912599j.
- 16.Boeckman R, Shao P, Wrobleski S, Boehmler D, Heintzelman G, Barbosa A. J Am Chem Soc. 2006;128:10572. doi: 10.1021/ja0581346. [DOI] [PubMed] [Google Scholar]
- 17.Tsuji J. In: Handbook of Organopalladium Chem for Org Syn. Negishi E, editor. Vol. 2. John Wiley & Sons; New York: 2002. pp. 1669–1687. [Google Scholar]
- 18.Näf F, Decorzant R, Thommen W, Willhalm B, Ohloff G., 4 Helv Chim Acta. 1975;58:1016. doi: 10.1002/hlca.19730560425. [DOI] [PubMed] [Google Scholar]
- 19.Bodurow C, Carr M, Moore L. Org Prep Proced Int. 1990;22:109. [Google Scholar]
- 20.Maryanoff B, Reitz A. Chem Rev. 1989;89:863. [Google Scholar]
- 21.Olsen R, Feng X. J Org Chem. 1992;57:5811. [Google Scholar]
- 22.Yoshinari T, Ohmori K, Schrems M, Pfaltz A, Suzuki K. Angew Chem Int Ed Engl. 2010;49:881. doi: 10.1002/anie.200906362. [DOI] [PubMed] [Google Scholar]
- 23.Marshall G, Pietta P, Cavallo P. J Org Chem. 1971;36:3966. doi: 10.1021/jo00824a025. [DOI] [PubMed] [Google Scholar]
- 24.Tanigawa Y, Nishimura K, Kawasaki A, Murahashi S. Tetrahedron Lett. 1982;23:5549. [Google Scholar]
- 25.Trost B, VanVranken D. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 26.Dess D, Martin J. J Org Chem. 1983;48:4155. [Google Scholar]
- 27.Swern D, Mancuso A, Brownfain D. J Org Chem. 1979;44:4148. [Google Scholar]
- 28.Griffith W, Ley S, Whitcombe G, White A. J Chem Soc, Chem Commun. 1987;21:1625. [Google Scholar]
- 29.Hoye T, Danielson M, May A, Zhao H. J Org Chem. 2010;75:7052. doi: 10.1021/jo101598y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Although we did not perform any molecular modeling calculations on macrocycle 25, Nicolaou and Sarlah have obtained a crystal structure for a macrolactam similar to 27. Indeed, the conformation of this compound (at least in the solid state) clearly shows that the reactive centers are too far apart for transannular bond formation to occur readily. Notably, Nicolaou and Sarlah also synthesized an analog of aldehyde 27 lacking the DMB protecting group as part of their earlier studies towards hirsutellone B and were similarly unable to achieve the desired aldol condensation under a variety of acidic and basic conditions. For details see: Sarlah D. PhD dissertation. The Scripps Research Institute; La Jolla, CA: 2011. Adventurous Total Synthesis: Part II. Total Synthesis of Hirsutellones A, B, and C; pp. 241–245.
- 31.Reddy C, Rao N, Srikanth B. Eur J Org Chem. 2010;2:345. [Google Scholar]
- 32.DeMico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. J Org Chem. 1997;62:6974. [Google Scholar]
- 33.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 34.Stille J. Angew Chem Int Ed Engl. 1986;98:504. [Google Scholar]
- 35.Gomez A, Lopez J, Fraserreid B. Synthesis. 1993;10:943. [Google Scholar]
- 36.Birney D, Xu X, Ham S, Huang X. J Org Chem. 1997;62:7114. doi: 10.1021/jo971083d. [DOI] [PubMed] [Google Scholar]
- 37.Adams E, Hiegemann M, Duddeck H, Welzel P. Tetrahedron. 1990;46:5975. [Google Scholar]
- 38.Pollack R. Tetrahedron. 1989;45:4913. [Google Scholar]
- 39.Taylor E, McKillop A. Aldrichimica Acta. 1970;3:4. [Google Scholar]
- 40.Carroll M. J Chem Soc. 1940:704. [Google Scholar]
- 41.Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199. [Google Scholar]
- 42.Burger E, Tunge J. Org Lett. 2004;6:2603. doi: 10.1021/ol049097a. [DOI] [PubMed] [Google Scholar]
- 43.Mander L, Crabtree S, Chu W. Synlett. 1990;3:169. [Google Scholar]
- 44.Guthikonda R, Cama L, Quesada M, Woods M, Salzmann T, Christensen B. J Med Chem. 1987;30:871. doi: 10.1021/jm00388a022. [DOI] [PubMed] [Google Scholar]
- 45.Bruder M, Haseler P, Muscarella M, Lewis W, Moody C. J Org Chem. 2010;75:353. doi: 10.1021/jo902117e. [DOI] [PubMed] [Google Scholar]
- 46.Snider B, Patricia J, Kates S. J Org Chem. 1988;53:2137. [Google Scholar]
- 47.Snider B. Tetrahedron. 2009;65:10738. doi: 10.1016/j.tet.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milstein D, Stille J. J Am Chem Soc. 1978;100:3636. [Google Scholar]
- 49.Djerassi C. Chem Rev. 1948;43:271. doi: 10.1021/cr60135a004. [DOI] [PubMed] [Google Scholar]
- 50.VanRheenen V, Kelly R, Cha D. Tetrahedron Lett. 1976;17:1973. [Google Scholar]
- 51.Yeom C, Kim H, Lee S, Kim B. Synlett. 2007;1:146. [Google Scholar]
- 52.Appel R. Angew Chem Int Ed Engl. 1975;14:801. [Google Scholar]
- 53.Alnajjar M, Kuivila H. J Am Chem Soc. 1985;107:416. [Google Scholar]
- 54.Ghosez L, Devos A, Remion J, Frisquehesbain A, Colens A. J Chem Soc, Chem Commun. 1979;24:1180. [Google Scholar]
- 55.Love B, Jones E. J Org Chem. 1999;64:3755. doi: 10.1021/jo982433e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.