

Abstract
The increasing incidence of inducible chromosomal AmpC β-lactamases within the clinic is a growing concern because these enzymes deactivate a broad range of even the most recently developed β-lactam antibiotics. As a result, new strategies are needed to block the action of this antibiotic resistance enzyme. Presented here is a strategy to combat the action of inducible AmpC by inhibiting the β-glucosaminidase NagZ, which is an enzyme involved in regulating the induction of AmpC expression. A divergent route facilitating the rapid synthesis of a series of N-acyl analogues of 2-acetamido-2-deoxynojirimycin is reported here. Among these compounds are potent NagZ inhibitors that are selective against functionally related human enzymes. These compounds reduce minimum inhibitory concentration values for β-lactams against a clinically relevant Gram-negative bacterium bearing inducible chromosomal AmpC β-lactamase, Pseudomonas aeruginosa. The structure of a NagZ–inhibitor complex provides insight into the molecular basis for inhibition by these compounds.
Keywords: carbohydrates, enzymes, inhibitors, selectivity, synthesis
Introduction
Resistance to even the most recent generation of β-lactams is steadily developing as various resistance mechanisms disseminate through the bacterial domain. Resistance mechanisms to β-lactams are varied, but the most important involve the β-lactamases—enzymes that deactivate β-lactam antibiotics by cleaving the cyclic amide moiety. One specific class of these enzymes, inducible chromosomal AmpC β-lactamases,1–3 is increasingly problematic in many Gram-negative bacteria, because these enzymes deactivate a broad range of even the most recent β-lactam antibiotics,4–6 and are resistant to clinically available β-lactamase inhibitors.7, 8 As a result, new strategies for blocking the action of this class of enzyme are of considerable interest.9
AmpC β-lactamase expression depends on the activity of a number of proteins that are engaged in peptidoglycan metabolism. The peptidoglycan is an essential component of the bacterial cell and is a highly cross-linked hetero-polymer that forms an exoskeleton around the organism, defining its shape and protecting it from osmotic lysis.10 During normal cell division, a considerable amount of the peptidoglycan is degraded and recycled.10,11 The resulting GlcNAc-1,6-anhydroMurNAc-peptide degradation products have their non-reducing GlcNAc residue removed by a cytosolic β-glucosaminidase known as NagZ.12,13 The resulting products are N-acetyl-d-glucosamine and a series of 1,6-anhydroMurNAc-tri-, tetra- and pentapeptides (Scheme 1A). These 1,6-anhydroMurNAc catabolic fragments activate the transcription of inducible ampC by binding to the transcriptional regulator AmpR.14 To prevent the continuous expression of ampC, another molecule, UDP-MurNAc-pentapeptide, which is a biosynthetic building block of the cell wall derived from these catabolic products, is involved in repressing ampC transcription by binding to AmpR.14,15 The relative concentrations of these molecules enable bacteria to sense β-lactams and so regulate AmpC expression.2
Scheme 1.
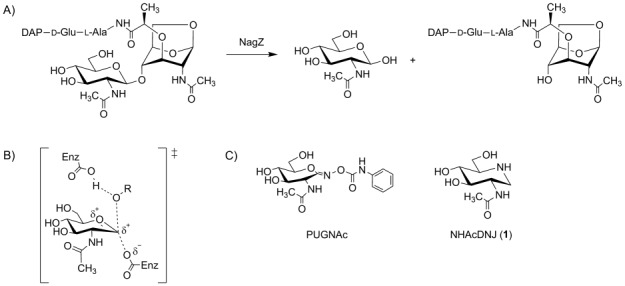
A) NagZ catalyzed hydrolysis of peptidoglycan cell-wall fragments releases the series of 1,6-anhydroMurNAc peptide inducer molecules that activate transcription of ampC expression (tripeptide shown). B) The putative transition state of the NagZ catalyzed hydrolysis of N-acetylglucosaminides (denoted by ≠). C) Structures of known inhibitors of NagZ enzymes.
One strategy to combat inducible AmpC would be to inhibit NagZ using small-molecule inhibitors. NagZ, a member of family GH3 (for an overview of the CAZy classification system of glycoside hydrolases see http://www.cazy.org16) uses a two-step, double-displacement mechanism involving the formation and breakdown of a covalent glycosyl-enzyme intermediate via oxocarbenium-ion-like transition states (Scheme 1B).17–20 By inhibiting NagZ, the formation of the inducer molecules comprising 1,6-anhydroMurNAc peptides would be impeded thus leading to reduced AmpC production and increased sensitivity to β-lactams. The approach of targeting NagZ has received recent validation using both chemical,21,22 and genetic studies,23–25 while structural studies of NagZ are now enabling the design of improved inhibitors.26
The main focus of NagZ inhibitor design has centred around the known β-glucosaminidase inhibitor, O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino N-phenylcarbamate (PUGNAc, Scheme 1C).27 Despite its potency for NagZ,21 a downside to using this molecule in a complex biological context is that it lacks selectivity for NagZ over important human enzymes. PUGNAc has been demonstrated to inhibit family GH84 human O-GlcNAcase (OGA),28,29 family GH20 human β-hexosaminidases30 and family GH89 hexosaminidases related to NAGLU.31 An area gaining increasing attention in carbohydrate enzymology is the need for inhibitors with improved selectivity between functionally related enzymes.32 Therefore, in an effort to overcome problems associated with concomitant inhibition of these enzymes, a series of PUGNAc derivatives were prepared through modification of the pendant N-acyl chain21,33 and were found to be selective for NagZ as well as useful at reducing AmpC β-lactamase expression.21,23
Another molecule that suffers from the same selectivity problem, yet has been used as an inhibitor of β-hexosaminidases, is 2-acetamido-2-deoxynojirimycin (NHAcDNJ, 1, Scheme 1C). This compound is known to potently inhibit hexosaminidases from families GH2034,35 and GH8931 and has recently been found to inhibit NagZ from E. coli.22 Based on these observations we envisaged that making modifications to the pendant N-acyl chain of 1 might yield new potent and selective inhibitors for NagZ enzymes. Such compounds might be valuable tools, not only by inhibiting NagZ and thus rendering increased susceptibility to β-lactams of Gram-negative bacteria harbouring inducible ampC, but also by aiding improved understanding of the binding of selective inhibitors to this family of enzymes and the role played by NagZ in peptidoglycan recycling.
Results and Discussion
Multiple syntheses of 1 previously described in the literature use N-acetyl-d-glucosamine as a starting material.36–40 To prepare the series of target compounds we envisaged that a divergent synthesis would facilitate their rapid preparation. Accordingly, N-acetyl-d-glucosamine was not a viable starting material since we aimed to generate a panel of compounds with various N-acyl groups. We felt, due to the nature of the chemical transformations necessary, that an azido group at C-2 would be stable throughout the synthesis enabling us to prepare a common synthetic intermediate (2, Scheme 2), which would be amenable to rapid diversification to generate the desired panel of N-acyl compounds. In addition, the 5,6-alkene could be used in the preparation of the desired iminosugars, through their intermediate ulososides. This general approach has shown value in the preparation of 2-acetamido-1,2-dideoxynojirimycin-lysine hybrids41 and other iminosugars.42
Scheme 2.
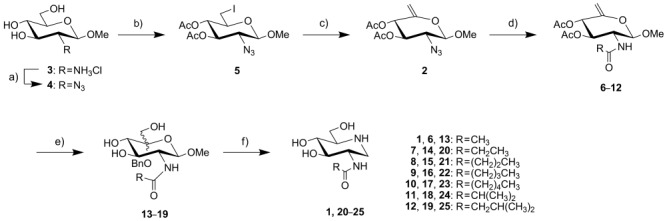
Reagents: a) ImSO2N3⋅HCl, K2CO3, CuSO4, MeOH; b) i: TsCl, pyridine, CH2Cl2; ii: NaI, DMF; iii: Ac2O, C5H5N; c) DBU, THF; d) i: PBu3, THF, H2O; ii: (RCO)2O; e) i: 3-chloroperbenzoic acid, CH2Cl2, BnOH; ii: NaOMe, MeOH; f) NH4OAc, Pd(OH)2/C, H2, MeOH/H2O (15:1).
Starting from the readily accessible hydrochloride 3, available in three steps from d-glucosamine hydrochloride,43 the azido group was introduced to protect the amine moiety using an azido-transfer reagent44 to give the triol 445 (Scheme 2). With triol 4 in hand, a one-pot activation at O-6, using a tosylate group followed by displacement of the tosylate with sodium iodide and in situ acetylation gave the iodide 5 in excellent yield. Elimination of hydroiodic acid across C-5/6 was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in THF to give the desired intermediate alkene 2. Treatment of the alkene 2 with tributylphosphine in THF/H2O, followed by acylation with the appropriate acyl anhydride gave the series of amides 6–12. Oxidation of the alkenes 6–12 with 3-chloroperbenzoic acid provided the presumed intermediate ulososides which were deprotected to give the triols 13–19. Finally, debenzylation by hydrogenolysis and in situ reductive amination of 13–19 with ammonium acetate in the presence of hydrogen gratifyingly gave the desired iminosugars 1 and 20–25 in excellent overall yield, exclusively as the d-gluco-configured materials.
It has been previously established that 1 is a potent competitive inhibitor of lysosomal family GH20 β-hexosaminidases, the lysosomal family GH89 α-hexosaminidase NAGLU, and E. coli NagZ. The Ki value for the human lysosomal family GH20 β-hexosaminidases is 540 nM35 (determined against β-hexosaminidase B) and for human NAGLU it is 450 nM.46 As previously discussed, developing selective inhibitors of NagZ is important to block NagZ function in bacteria but also to ensure that there is no concomitant inhibition of these human enzymes.
With the synthesised panel of inhibitors in hand, we evaluated them against representative NagZ enzymes found in Vibrio cholerae (VcNagZ) and Salmonella typhimurium (StNagZ) and found them to be potent competitive inhibitors of these enzymes (Table 1). What was also of interest was that increasing the N-acyl chain length leads to a greater increase in Ki value for both β-hexosaminidase B and NAGLU as compared to NagZ enzymes, consistent with those observed for the N-acyl PUGNAc analogues and presumably a consequence of the more spacious active site found in NagZ enzymes.21 Furthermore these compounds were also poor inhibitors of OGA, with the parent compound showing a Ki value of 23 μM, consistent with an independent report for this enzyme.35 We find that the selectivity ratio for the NagZ enzymes improves as the chain length increases; this is illustrated best for compound 21, which shows over 50-fold selectivity for VcNagZ (>20-fold for StNagZ) over the human enzymes, whilst retaining potency for the NagZ enzymes. To gain a more detailed understanding of the molecular basis for the inhibition of NagZ enzymes by 21, which we deemed to be the most potentially promising compound in this series in terms of potency as well as selectivity, we determined the three-dimensional structure of StNagZ in complex with 21 at 1.45 Å resolution (FigureFigure 1).
Table 1.
Inhibition constants and selectivity ratio (S.R.) of inhibitors for O-GlcNAcase, β-hexosaminidase B, NAGLU, VcNagZ and StNagZ.
| Compound | O-GlcNAcase Ki | β-Hexosaminidase B | NAGLU Ki | VcNagZ Ki | StNagZ Ki | S.R. | S.R. |
|---|---|---|---|---|---|---|---|
| [μM] | Ki [μM] | [μM] | [μM] | [μM] | (Ki OGA/Ki VcNagZ) | (Ki OGA/Ki StNagZ) | |
| 1 | 23 | 0.5435 | 0.4546 | 8.5 | 25.2 | 2.8 | 0.94 |
| 20 | 130 | 61 | 96 | 4.2 | 45.4 | 31 | 2.9 |
| 21 | >500 | 1460 | >1000 | 9.4 | 23.2 | >53 | >22 |
| 22 | >1000 | >5000 | >1000 | 110 | 47.3 | >9 | >21 |
| 23 | >1000 | >5000 | >1000 | 1135 | 102 | >0.9 | >10 |
| 24 | >1000 | 670 | 800 | 15.8 | 24.6 | 63 | >40 |
| 25 | >1000 | >5000 | >1000 | 555 | 294 | >1.8 | >3.4 |
Figure 1.
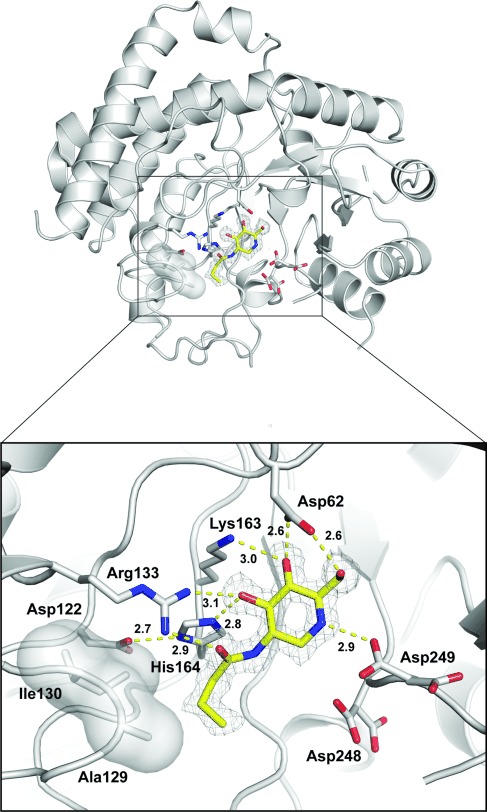
Crystal structure of StNagZ bound to 21. Active-site residues and 21 are drawn as sticks with oxygen and nitrogen atoms shown in red and blue, respectively. Carbon atoms of the enzyme are shown in grey, while the carbon atoms of the inhibitor are yellow. The hydrophobic surface formed by side chains of Ala129 and Ile130, against which the butyl chain sits, is shown in grey. The catalytic nucleophile, Asp248, is shown in stick format. Occupancy values for the dual conformations of Asp248 are 0.48/0.52, and 0.53/0.47 for Asp249 (flipped in or out towards the inhibitor, respectively). Hydrogen bonds are shown as yellow dashed lines. Electron density around 21 is a maximum-likelihood-weighted omit map (Fobs−Fcalcd) contoured to 2.5σ.
With the enzyme–inhibitor complex we find that the pseudo-glycoside ring structure of 21 adopts a relaxed 4C1 chair conformation that closely resembles that observed for the reaction product GlcNAc. Accordingly, similar hydrogen-bonding interactions are observed between 21 and residues in the active site (for a detailed description of the structure and mechanism of NagZ, see ref. 20). However, we also note that the NH group within the ring of 21, which replaces the endocyclic oxygen of GlcNAc, forms a hydrogen bond with the side-chain of Asp249, which likely confers the increased binding affinity relative to GlcNAc. The butyl moiety of the N-acyl group of 21 appears to form hydrophobic interactions with a small surface created by the side chains of Ala129 and Ile130 adopting a similar conformation as that seen in the VcNagZ structures bound to PUGNAc-derived inhibitors.21,26 Finally, the structure shows the butyl chain is exposed to solvent on the outer side, supporting the idea that the relatively open active-site arrangement of NagZ accounts for the much greater selectivity demonstrated toward the bacterial hydrolases by the NHAcDNJ-derived inhibitors with more bulky modified N-acyl chains.
We next set out to obtain insight into whether these compounds could increase the susceptibility of bacteria harbouring inducible AmpC β-lactamase to β-lactams. We therefore evaluated them against Pseudomonas aeruginosa PA01, a Gramnegative bacterium that harbours a chromosomally inducible AmpC β-lactamase.47 This opportunistic pathogen is problematic for patients suffering from cystic fibrosis, severe burns and pulmonary disease.48–50 Importantly, P. aeruginosa PA01 contains a functional NagZ and strains lacking the nagZ gene are known to have increased susceptibility to β-lactams, supporting the validity of this strain in such β-lactam susceptibility assays.23,24
A series of β-lactam antibiotics, cefoxitin, ceftazidime, ampicillin, the monobactam aztreonam and the carbapenem imipenem, were chosen as they are commonly used in clinical antibiotic susceptibility experiments. Using minimal inhibitory concentration (MIC) assays we found that cultures treated with the selective inhibitor 21 are more susceptible to these β-lactam antibiotics when compared to control cultures that were not treated with 21 (Table 2).
Table 2.
Susceptibility of P. aeruginosa PA01 against various β-lactam antibiotics.
| Antibiotic | MIC [μgmL−1][a] | Clearing radius [mm][b] | ||
|---|---|---|---|---|
| −21 | +21 | −21 | +21 | |
| Ceftazidime | 2 | 0.5 | 12.2 | 14.3 |
| Aztreonam | 1 | 0.5 | 13.8 | 16.0 |
| Imipenem | 8 | 4 | 11.9 | 13.3 |
| Ampicillin | 512 | 128 | ||
| Cefoxitin | 2048 | 512 | ||
MIC determined by standard serial dilution.
Susceptibility determined in an agar diffusion assay using 6 mm filter disks loaded with 30 μg of antibiotic with or without 21. The zone of clearance was measured after incubation overnight.
To use a separate assessment of antibiotic susceptibility we took the more potent β-lactams, ceftazidime, aztreonam and imipenem and assessed them in agar diffusion assays (Table 2). In accord with the MIC data we found that agar-diffusion assays gave similar results, revealing enhanced susceptibility to β-lactams in the presence of 21. As a control we evaluated 21 as an inhibitor of bacterial growth but observed no differences in growth rates even at a concentration of 1 mM (data not shown), indicating that the inhibitor, on its own, is not antibacterial or bacteriostatic. Of note is that the concentration of 21 required to give rise to the increased susceptibility observed is fivefold less than the concentration of the PUGNAc analogue O-(2-deoxy-2-N-2-ethylbutyryl-d-glucopyranosylidene)amino N-phenylcarbamate (EtBuPUG) used previously to induce a similar effect.23 Together, these results suggest that 21 might gain more ready access to the cytosol, either through a transporter protein or passive diffusion as compared to EtBuPUG.
Conclusions
The incidence of inducible chromosomal AmpC β-lactamases is increasing and since these enzymes deactivate a broad range of β-lactam antibiotics they are a pressing health issue. We have devised a divergent route that enables the rapid synthesis of a series of potent and selective NHAcDNJ-based inhibitors bearing different N-acyl groups that target NagZ, an important enzyme in the regulation of AmpC activity. One of these compounds reduces MIC values for β-lactams against a clinically relevant Gram-negative bacterium bearing inducible chromosomal AmpC β-lactamase, P. aeruginosa. Using a structure of a NagZ–inhibitor complex we provide insight into the molecular basis for the selectivity and potency of this inhibitor. It is anticipated that further structure-guided refinement will lead to candidates with increased potency for NagZ and, using the strategy outlined here, systematic elaboration of other β-N-acetylglucosaminidase inhibitor scaffolds might also yield selective and potent inhibitors for NagZ.
Experimental Section
General methods for chemical synthesis: 1H and 13C NMR spectra were recorded on a Bruker ARX500 (500 MHz for 1H and 125 MHz for 13C) or a Bruker AV600 (600 MHz for 1H and 150 MHz for 13C; chemical shifts quoted relative to CHCl3 for CDCl3 and CH3OH for D2O where appropriate) spectrometer. Mass spectra were recorded with a VG-Autospec spectrometer using the fast atom bombardment technique, with 3-nitrobenzyl alcohol as a matrix. Elemental analyses of all synthesised compounds used in enzyme assays were performed at the Australian National University Microanalytical Facility. Flash chromatography was performed on silica gel (BDH) with the specified solvents. Thin-layer chromatography (TLC) was effected on Merck silica gel 60 F254 aluminium-backed plates that were stained by heating (>200°C) with 5% sulfuric acid in EtOH. Percentage yields for chemical reactions as described are quoted only for those compounds that were purified by recrystallization or by column chromatography, and purity was assessed using TLC or 1H NMR spectroscopy.
Methyl 3,4-di-O-acetyl-2-azido-2-deoxy-6-iodo-β-d-glucopyranoside (5): p-Toluenesulfonyl chloride (3.9 g, 20 mmol) was added to a solution of azide 445 (4.0 g, 18 mmol) in pyridine (20 mL) and CH2Cl2 (80 mL) and the solution was stirred (RT, 6 h). The reaction was quenched by the addition of water and the resultant mixture stirred (1 h). The organic layer was then collected and was washed with water (1×50 mL), aq. HCl (1M, 1×50 mL), saturated aq. NaHCO3 solution (1×50 mL), brine (1×20 mL), dried (MgSO4), filtered and concentrated. The residue was then dissolved in DMF (60 mL) and to it was added NaI (8.0 g, 53 mmol) and the resultant mixture stirred (100°C, 4 h). The mixture was then concentrated and the resultant residue diluted with EtOAc (150 mL) and was washed with water (2×20 mL), brine (1×20 mL), dried (MgSO4), filtered and concentrated. The residue was then dissolved in a mixture of CH2Cl2 (30 mL) and pyridine (10 mL) and Ac2O (10 mL) was added. The resultant solution was left at RT overnight. The mixture was quenched with MeOH and concentrated. The resultant residue was then diluted with CH2Cl2 (100 mL) and washed with water (2×20 mL), aq. HCl (1M, 1×50 mL), saturated aq. NaHCO3 solution (1×50 mL), brine (1×20 mL), dried (MgSO4), filtered and concentrated. Flash chromatography of the resultant residue (EtOAc/hexane 3:7) yielded 5 as a colourless oil (4.8 g, 64%, over three steps). Rf=0.7 (EtOAc/hexane 2:3); 1H NMR (600 MHz, CDCl3): δ=4.98 (dd, J=9.3, 9.3 Hz, 1H), 4.80 (dd, J=9.5, 9.5 Hz, 1H), 4.31 (d, J=8.0 Hz, 1H), 3.63 (s, 3H), 3.49–3.44 (m, 2H), 3.29 (dd, J=2.7, 11.0 Hz, 1H), 3.14 (dd, J=8.4, 11.0 Hz, 1H), 2.07 (s, 3H), 2.04 ppm (s, 3H); 13C NMR (150 MHz, CDCl3): δ=169.9, 169.6, 102.5, 73.2, 72.3, 72.2, 63.8, 57.4, 20.64, 20.63, 2.8 ppm; νmax=2114 cm−1 (N3); HRMS: m/z calcd: 414.0162 [M+H]+, found: 414.0156; elemental analysis calcd (%) for C11H16IN3O6: C 31.98, H 3.90, N 10.17; found: C 31.93, H 3.92, N 10.15.
Methyl 3,4-di-O-acetyl-2-azido-2,6-dideoxy-β-d-xylo-hex-5-enoside (2): DBU (4.9 mL, 33 mmol) was added to iodide 5 (4.5 g, 11 mmol) in THF (40 mL) and the mixture refluxed (2 h). The mixture was then concentrated and the resultant residue diluted with EtOAc (150 mL) and was washed with water (2×40 mL), ice-cold aq. HCl (1M, 1×50 mL), saturated aq. NaHCO3 solution (1×50 mL), brine (1×50 mL), dried (MgSO4), filtered and concentrated. Flash chromatography of the resultant residue (EtOAc/hexane 3:7) yielded 2 as a colourless oil (2.5 g, 80%). Rf=0.4 (EtOAc/hexane 1:4); 1H NMR (500 MHz, CDCl3): δ=5.41 (ddd, J=1.6, 1.6, 10.0 Hz, 1H), 4.93 (dd, J=9.2, 9.2 Hz, 1H), 4.82 (dd, J=1.6, 1.6 Hz, 1H), 4.59 (dd, J=1.6, 1.6 Hz, 1H), 4.37 (d, J=7.2 Hz, 1H), 3.59 (s, 3H), 2.08 (s, 3H), 2.07 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ=169.6, 169.3, 103.2, 97.5, 71.4, 69.2, 63.4, 57.3, 20.6 ppm; νmax=2108 cm−1 (N3); HRMS: m/z calcd: 286.1039 [M+H]+, found: 286.1028; elemental analysis calcd (%) for C11H15N3O6: C 46.32, H 5.30, N 14.73; found: C 46.28, H 5.33, N 14.80.
General procedure for formation of methyl 3,4-di-O-acetyl-2-acylamido-2,6-dideoxy-β-d-xylo-hex-5-enosides (6–12): Tributyl phosphine (0.2 mL, 0.8 mmol) was added to azide 2 (200 mg, 0.7 mmol) in a solution of THF (5 mL) and H2O (0.5 mL) at 0°C and the solution stirred. This was followed by the addition of the appropriate acyl anhydride (3 equiv) and the mixture stirred (5 h). Concentration followed by flash chromatography of the residue (EtOAc/hexane 7:3) gave the desired compounds 6–12 in yields ranging from 52% to 73%.
Methyl 2-acetamido-3,4-di-O-acetyl-2,6-dideoxy-β-d-xylo-hex-5-enoside (6): Yield: 60%. 1H and 13C NMR spectra were consistent with those found in the literature.41
Methyl 3,4-di-O-acetyl-2,6-dideoxy-2-propamido-β-d-xylo-hex-5-enoside (7): Yield: 73%; Rf=0.3 (EtOAc/hexane 7:3); 1H NMR (600 MHz, CDCl3): δ=5.81 (d, J=8.8 Hz, 1H), 5.61 (ddd, J=1.3, 1.3, 7.5 Hz, 1H), 4.94 (dd, J=6.0, 7.5 Hz, 1H), 4.78 (dd, J=1.3, 1.3 Hz, 1H), 4.66 (d, J=4.0 Hz, 1H), 4.57 (dd, J=1.3, 1.3 Hz, 1H), 4.24 (ddd, J=4.0, 6.0, 8.8 Hz, 1H), 3.48 (s, 3H), 2.24–2.19 (m, 2H), 2.11 (s, 3H), 2.06 (s, 3H), 1.14 ppm (t, J=7.5 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ=173.3, 170.4, 168.9, 150.5, 102.3, 97.3, 71.4, 68.6, 56.2, 52.1, 29.6, 20.8, 20.7, 9.5 ppm; HRMS: m/z calcd: 316.1396 [M+H]+, found: 316.1371; elemental analysis calcd (%) for C14H21NO7: C 53.33, H 6.71, N 4.44; found: C 53.30, H 6.66, N 4.51.
Methyl 3,4-di-O-acetyl-2-butamido-2,6-dideoxy-β-d-xylo-hex-5-enoside (8): Yield: 70%; Rf=0.4 (EtOAc/hexane 7:3); 1H NMR (600 MHz, CDCl3): δ=5.76 (d, J=9.3 Hz, 1H), 5.61 (d, J=7.5 Hz, 1H), 4.95 (dd, J=6.3, 6.4 Hz, 1H), 4.79 (s, 1H), 4.66 (d, J=4.1 Hz, 1H), 4.57 (s, 1H), 4.24 (ddd, J=4.1, 7.5, 9.3 Hz, 1H), 3.48 (s, 3H), 2.16 (t, J=8.0 Hz, 2H), 2.12 (s, 3H), 2.06 (s, 3H), 1.68–1.63 (m, 2H), 0.94 ppm (t, J=7.3 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ=172.5, 170.4, 169.0, 150.6, 102.4, 97.2, 71.4, 68.6, 56.2, 52.2, 38.6, 20.84, 20.8, 18.9, 13.6 ppm; HRMS: m/z calcd: 330.1553 [M+H]+, found: 330.1564; elemental analysis calcd (%) for C15H23NO7: C 54.70, H 7.04, N 4.25; found: C 54.65, H 7.07, N 4.33.
Methyl 3,4-di-O-acetyl-2,6-dideoxy-2-valeramido-β-d-xylo-hex-5-enoside (9): Yield: 67%; Rf=0.5 (EtOAc/hexane 7:3); 1H NMR (600 MHz, CDCl3): δ=5.79 (d, J=8.6 Hz, 1H), 5.60 (d, J=7.5 Hz, 1H), 4.95 (dd, J=6.4, 7.3 Hz, 1H), 4.78 (s, 1H), 4.65 (d, J=4.1 Hz, 1H), 4.57 (s, 1H), 4.23 (ddd, J=4.1, 6.4, 8.6 Hz, 1H), 3.48 (s, 3H), 2.19–2.16 (m, 2H), 2.12 (s, 3H), 2.05 (s, 3H), 1.61–1.56 (m, 2H), 1.36–1.29 (m, 2H), 0.90 ppm (t, J=6.8 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ=172.7, 170.4, 169.0, 150.6, 102.4, 97.2, 71.4, 68.6, 56.2, 52.2, 36.7, 27.5, 22.2, 20.82, 20.8, 13.7 ppm; HRMS: m/z calcd: 344.1709 [M+H]+, found: 344.1722; elemental analysis calcd (%) for C16H25NO7: C 55.97, H 7.34, N 4.08; found: C 55.89, H 7.39, N 4.09.
Methyl 3,4-di-O-acetyl-2,6-dideoxy-2-hexamido-β-d-xylo-hex-5-enoside (10): Yield: 52%; Rf=0.7 (EtOAc/hexane 7:3); 1H NMR (500 MHz, CDCl3): δ=5.76 (d, J=8.7 Hz, 1H), 5.60 (dt, J=1.1, 1.1, 7.6 Hz, 1H), 4.95 (dd, J=6.3, 7.4 Hz, 1H), 4.78 (d, J=1.1 Hz, 1H), 4.65 (d, J=4.1 Hz, 1H), 4.57 (d, J=1.1 Hz, 1H), 4.24 (ddd, J=4.1, 6.3, 8.7 Hz, 1H), 3.49 (s, 3H), 2.19–2.16 (m, 2H), 2.12 (s, 3H), 2.06 (s, 3H), 1.65–1.59 (m, 4H), 1.35–1.25 (m, 4H), 0.89 ppm (t, J=6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ=172.7, 170.4, 169.0, 150.6, 102.4, 97.2, 71.4, 68.6, 56.2, 52.2, 36.6, 31.3, 25.2, 22.3, 20.84, 20.8, 13.9 ppm; HRMS: m/z calcd: 358.1866 [M+H]+, found: 358.1857; elemental analysis calcd (%) for C17H27NO7: C 57.13, H 7.61, N 3.92; found: C 57.01, H 7.53, N 3.99.
Methyl 3,4-di-O-acetyl-2,6-dideoxy-2-isobutamido-β-d-xylo-hex-5-enoside (11): Yield: 54%; Rf=0.45 (EtOAc/hexane 7:3); 1H NMR (500 MHz, CDCl3): δ=5.85 (d, J=8.4 Hz, 1H), 5.60 (d, J=7.5 Hz, 1H), 4.95 (dd, J=6.3, 6.3 Hz, 1H), 4.79 (s, 1H), 4.65 (d, J=4.0 Hz, 1H), 4.57 (s, 1H), 4.22 (ddd, J=4.0, 6.3, 8.4 Hz, 1H), 3.48 (s, 3H), 2.38–2.33 (m, 1H), 2.12 (s, 3H), 2.05 (s, 3H), 1.15 (d, J=6.9 Hz, 3H), 1.14 ppm (d, J=6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ=176.5, 170.4, 168.9, 150.5, 102.4, 97.5, 71.3, 68.6, 56.2, 51.9, 35.5, 20.83, 20.8, 19.4, 19.3 ppm; HRMS: m/z calcd: 330.1553 [M+H]+, found: 330.1570; elemental analysis calcd (%) for C15H23NO7: C 54.70, H 7.04, N 4.25; found: C 54.63, H 7.09, N 4.28.
Methyl 3,4-di-O-acetyl-2,6-dideoxy-2-isovalermido-β-d-xylo-hex-5-enoside (12): Yield: 68%; Rf=0.5 (EtOAc/hexane 7:3); 1H NMR (600 MHz, CDCl3): δ=5.75 (d, J=8.9 Hz, 1H), 5.60 (dt, J=1.3, 1.3, 7.6 Hz, 1H), 4.96 (dd, J=6.5, 7.5 Hz, 1H), 4.79 (d, J=1.3 Hz, 1H), 4.64 (d, J=4.2 Hz, 1H), 4.57 (d, J=1.3 Hz, 1H), 4.24 (ddd, J=4.2, 6.5, 8.9 Hz, 1H), 3.48 (s, 3H), 2.12 (s, 3H), 2.10–2.04 (m, 3H), 2.06 (s, 3H), 0.96–0.93 ppm (m, 6H); 13C NMR (150 MHz, CDCl3): δ=172.1, 170.4, 169.0, 150.6, 102.5, 97.1, 71.4, 68.7, 56.2, 52.2, 46.0, 26.1, 22.35, 22.3, 20.82, 20.8 ppm; HRMS: m/z calcd: 344.1709 [M+H]+, found: 344.1729; elemental analysis calcd (%) for C16H25NO7: C 55.97, H 7.34, N 4.08; found: C 56.02, H 7.41, N 4.04.
General procedure for formation of methyl (5R/S)-2-acylamino-5-C-benzyloxy-2-deoxy-β-d-xylo-hexopyranosides (13–19): To a 1% solution of 6–12 (0.5 mmol) in a mixture of CH2Cl2/benzyl alcohol (1:1 v/v, 12 mL), 3-chloroperbenzoic acid (70%, 0.6 mmol) was added, and the mixture was stirred (2 h). The mixture was then diluted with CH2Cl2 (20 mL) and washed with saturated aq. NaHCO3 solution (1×50 mL), dried (MgSO4), filtered and concentrated. The resulting residue was chromatographed (EtOAc) to yield a residue that was dissolved in MeOH and treated with sodium methoxide (10 mg) and the solution stirred (RT, 30 min). The mixture was quenched with resin (Amberlite IR-120, H+), filtered and concentrated to give 13–19 as a mixture of diastereomers at C-5 from which the major (5S) epimer was purified by flash chromatography (MeOH/CHCl3 1:9) in yields ranging from 53% to 68% over two steps.
Methyl (5R/S)-2-acetamido-5-C-benzyloxy-2-deoxy-β-d-xylo-hexopyranoside (13): Yield: 68%. 1H and 13C NMR spectra of the (5S) epimer were consistent with those found in the literature.41
Methyl (5R/S)-5-C-benzyloxy-2-deoxy-2-propamido-β-d-xylo-hexopyranoside (14): Yield: 64%. (5S) epimer—Rf=0.2 (EtOAc); 1H NMR (500 MHz, CD3OD): δ=7.45–7.42 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.23 (m, 1H), 4.76 (ABq, J=10.9 Hz, 1H), 4.71–4.68 (m, 2H), 4.02 (dd, J=7.0, 9.7 Hz, 1H), 3.89–3.83 (m, 3H), 3.71 (dd, J=7.9, 9.6 Hz, 1H), 3.44 (s, 3H), 2.26–2.21 (m, 2H), 1.12 ppm (t, J=7.6 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=177.4, 140.0, 129.2, 129.0, 128.3, 102.9, 101.9, 76.0, 73.6, 64.6, 62.3, 56.8, 56.3, 30.4, 10.3 ppm; HRMS: m/z calcd: 356.1709 [M+H]+, found: 356.1711; elemental analysis calcd (%) for C17H25NO7: C 57.45, H 7.09, N 3.94; found: C 57.28, H 7.01, N 3.99.
Methyl (5R/S)-5-C-benzyloxy-2-butamido-2-deoxy-β-d-xylo-hexopyranoside (15): Yield: 53%. (5S) epimer—Rf=0.25 (EtOAc); 1H NMR (500 MHz, CD3OD): δ=7.45–7.41 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.22 (m, 1H), 4.75 (ABq, J=9.0 Hz, 1H), 4.71–4.68 (m, 2H), 4.03 (dd, J=7.4, 9.6 Hz, 1H), 3.90–3.83 (m, 3H), 3.71 (dd, J=8.1, 9.3 Hz, 1H), 3.44 (s, 3H), 2.20 (t, J=6.4 Hz, 2H), 1.69–1.62 (m, 2H), 0.96 ppm (t, J=7.4 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=176.5, 140.0, 129.1, 129.0, 128.3, 102.8, 101.9, 76.1, 73.5, 64.6, 62.3, 56.8, 56.3, 39.3, 20.3, 13.9 ppm; HRMS: m/z calcd: 370.1866 [M+H]+, found: 370.1889; elemental analysis calcd (%) for C18H27NO7: C 58.52, H 7.37, N 3.79; found: C 58.49, H 7.51, N 3.71.
Methyl (5R/S)-5-C-benzyloxy-2-deoxy-2-valeramido-β-d-xylo-hexopyranoside (16): Yield: 68%. (5S) epimer—Rf=0.3 (EtOAc); 1H NMR (600 MHz, CD3OD): δ=7.45–7.43 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.22 (m, 1H), 4.76 (ABq, J=10.9 Hz, 1H), 4.71–4.68 (m, 2H), 4.02 (dd, J=7.3, 9.6 Hz, 1H), 3.89–3.82 (m, 3H), 3.71 (dd, J=8.1, 9.3 Hz, 1H), 3.44 (s, 3H), 2.24–2.20 (m, 2H), 1.62–1.58 (m, 2H), 1.40–1.35 (m, 2H), 0.94 ppm (t, J=7.3 Hz, 3H); 13C NMR (150 MHz, CD3OD): δ=176.7, 140.0, 129.2, 129.0, 128.3, 102.9, 101.9, 76.1, 73.6, 64.6, 62.3, 56.8, 56.3, 37.2, 29.1, 23.2, 14.1 ppm; HRMS: m/z calcd: 384.2022 [M+H]+, found: 384.2010; elemental analysis calcd (%) for C19H29NO7: C 59.52, H 7.62, N 3.65; found: C 59.64, H 7.59, N 3.61.
Methyl (5R/S)-5-C-benzyloxy-2-deoxy-2-hexamido-β-d-xylo-hexopyranoside (17): Yield: 61%. (5S) epimer—Rf=0.4 (EtOAc); 1H NMR (500 MHz, CD3OD): δ=7.45–7.41 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.21 (m, 1H), 4.76–4.70 (m, 3H), 4.03 (dd, J=7.2, 9.5 Hz, 1H), 3.90–3.84 (m, 3H), 3.73 (dd, J=8.1, 9.6 Hz, 1H), 3.44 (s, 3H), 2.24–2.20 (m, 2H), 1.66–1.59 (m, 2H), 1.40–1.30 (m, 4H), 0.91 ppm (t, J=6.8 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=176.7, 140.0, 129.1, 129.0, 128.3, 102.8, 101.8, 76.0, 73.4, 64.5, 62.3, 56.8, 56.3, 37.3, 32.3, 26.6, 23.4, 14.3 ppm; HRMS: m/z calcd: 398.2179 [M+H]+, found: 398.2171; elemental analysis calcd (%) for C20H31NO7: C 60.44, H 7.86, N 3.52; found: C 60.51, H 7.93, N 3.51.
Methyl (5R/S)-5-C-benzyloxy-2-deoxy-2-isobutamido-β-d-xylo-hexopyranoside (18): Yield: 55%. (5S) epimer—Rf=0.3 (EtOAc); 1H NMR (500 MHz, CD3OD): δ=7.45–7.42 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.21 (m, 1H), 4.76 (ABq, J=11.0 Hz, 1H), 4.71–4.68 (m, 2H), 4.00 (dd, J=6.9, 9.3 Hz, 1H), 3.90–3.82 (m, 3H), 3.73 (dd, J=7.8, 9.3 Hz, 1H), 3.44 (s, 3H), 2.48–2.42 (m, 1H), 1.14 (d, J=6.7 Hz, 3H), 1.13 ppm (d, J=6.7 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=180.5, 140.0, 129.2, 129.0, 128.3, 102.9, 102.0, 76.0, 73.5, 64.6, 62.2, 56.8, 56.2, 36.5, 20.0, 19.8 ppm; HRMS: m/z calcd: 370.1866 [M+H]+, found: 370.1872; elemental analysis calcd (%) for C18H27NO7: C 58.52, H 7.37, N 3.79; found: C 58.50, H 7.31, N 3.75.
Methyl (5R/S)-5-C-benzyloxy-2-deoxy-2-isovaleramido-β-d-xylo-hexopyranoside (19): Yield: 64%. (5S) epimer—Rf=0.4 (EtOAc); 1H NMR (600 MHz, CD3OD): δ=7.45–7.42 (m, 2H), 7.33–7.29 (m, 2H), 7.25–7.21 (m, 1H), 4.75 (ABq, J=11.0 Hz, 1H), 4.72–4.69 (m, 2H), 4.04 (dd, J=7.4, 9.7 Hz, 1H), 3.90–3.83 (m, 3H), 3.72 (dd, J=8.1, 9.5 Hz, 1H), 3.44 (s, 3H), 2.12–2.05 (m, 3H), 0.98–0.94 ppm (m, 6H); 13C NMR (150 MHz, CD3OD): δ=176.0, 140.0, 129.1, 129.0, 128.3, 102.8, 101.8, 76.1, 73.5, 64.6, 62.3, 56.7, 56.2, 46.7, 27.5, 22.8, 22.6 ppm; HRMS: m/z calcd: 384.2022 [M+H]+, found: 384.2024, elemental analysis calcd (%) for C19H29NO7: C 59.52, H 7.62, N 3.65; found: C 59.60, H 7.65, N 3.70.
General procedure for formation of 2-acylamido-1,5-imino-1,2,5-trideoxy-d-glucitols (1, 20–25): To a solution of 13–19 in MeOH/H2O (15:1 v/v, 0.03M), NH4HCO2 (1 equiv) and 20% Pd(OH)2/C (0.1 equiv) were added, and the heterogeneous reaction mixture was stirred under hydrogen at RT and ambient pressure (1 atm, 48 h). After filtration and evaporation, the resulting residue was chromatographed (CHCl3/MeOH/conc. NH3 12:8:1) to yield the desired compounds 1, 20–25 in yields ranging from 48 to 63%.
2-Acetamido-1,5-imino-1,2,5-trideoxy-d-glucitol (1): Yield: 58%. 1H and 13C NMR spectra were consistent with those found in the literature.39
1,5-Imino-2-propamido-1,2,5-trideoxy-d-glucitol (20): Yield: 52%; Rf=0.25 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (600 MHz, CD3OD): δ=3.86–3.80 (m, 2H), 3.71 (dd, J=5.9, 11.4 Hz, 1H), 3.38–3.34 (m, 2H), 3.21 (dd, J=4.7, 12.5 Hz, 1H), 2.71–2.66 (m, 1H), 2.56 (dd, J=11.9, 11.9 Hz, 1H), 2.26–2.20 (m, 2H), 1.12 ppm (t, J=7.6 Hz, 3H); 13C NMR (150 MHz, CD3OD): δ=177.5, 76.7, 72.6, 62.5, 61.4, 52.3, 47.8, 30.1, 10.3 ppm; HRMS: m/z calcd: 219.1345 [M+H]+, found: 219.1344; elemental analysis calcd (%) for C9H18N2O4: C 49.53, H 8.31, N 12.84; found: C 49.51, H 8.26, N 12.89.
2-Butamido-1,5-imino-1,2,5-trideoxy-d-glucitol (21): Yield: 63%; Rf=0.4 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (500 MHz, CD3OD): δ=3.90–3.85 (m, 2H), 3.75 (dd, J=5.3, 11.5 Hz, 1H), 3.48 (dd, J=9.7, 9.7 Hz, 1H), 3.43 (dd, J=9.4, 9.4 Hz, 1H), 3.21 (dd, J=4.8, 12.7 Hz, 1H), 2.81–2.76 (m, 1H), 2.64 (dd, J=11.9, 11.9 Hz, 1H), 2.28–2.20 (m, 2H), 1.65–1.59 (m, 2H), 0.91 ppm (t, J=7.5 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=178.4, 75.9, 71.7, 61.2, 60.9, 51.6, 47.0, 38.7, 19.9, 13.6 ppm; HRMS: m/z calcd: 233.1501 [M+H]+, found: 233.1516; elemental analysis calcd (%) for C10H20N2O4: C 51.71, H 8.68, N 12.06; found: C 51.77, H 8.59, N 12.13.
1,5-Imino-1,2,5-trideoxy-2-valeramido-d-glucitol (22): Yield: 61%; Rf=0.5 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (500 MHz, CD3OD): δ=3.82 (dd, J=2.9, 11.1 Hz, 1H), 3.77 (ddd, J=4.7, 10.6, 11.0 Hz, 1H), 3.65 (dd, J=6.0, 11.1 Hz, 1H), 3.30–3.25 (m, 2H), 3.14 (dd, J=4.8, 12.4 Hz, 1H), 2.56–2.51 (m, 1H), 2.44 (dd, J=12.1, 12.1 Hz, 1H), 2.25–2.19 (m, 2H), 1.63–1.56 (m, 2H), 1.40–1.34 (m, 2H), 0.93 ppm (t, J=7.3 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=176.7, 77.3, 73.5, 62.7, 62.3, 53.2, 48.7, 36.9, 29.1, 23.3, 14.1 ppm; HRMS: m/z calcd: 247.1658 [M+H]+, found: 247.1651; elemental analysis calcd (%) for C11H22N2O4: C 53.64, H 9.00, N 11.37; found: C 53.57, H 9.03, N 11.29.
2-Hexamido-1,5-imino-1,2,5-trideoxy-d-glucitol (23): Yield: 48%; Rf=0.55 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (600 MHz, CD3OD): δ=3.81 (dd, J=3.1, 11.2 Hz, 1H), 3.74 (ddd, J=4.7, 11.0, 11.0 Hz, 1H), 3.64 (dd, J=4.1, 11.1 Hz, 1H), 3.28–3.23 (m, 2H), 3.11 (dd, J=4.7, 12.3 Hz, 1H), 2.50–2.47 (m, 1H), 2.40 (dd, J=11.9, 11.9 Hz, 1H), 2.25–2.19 (m, 2H), 1.64–1.59 (m, 2H), 1.37–1.29 (m, 4H), 0.90 ppm (t, J=6.8 Hz, 3H); 13C NMR (150 MHz, CD3OD): δ=176.7, 77.5, 73.8, 62.7, 62.6, 53.5, 48.9, 37.2, 32.5, 26.7, 23.4, 14.3 ppm; HRMS: m/z calcd: 261.1814 [M+H]+, found: 261.1807; elemental analysis calcd (%) for C12H24N2O4: C 55.36, H 9.29, N 10.76; found: C 55.31, H 9.25, N 10.68.
1,5-Imino-2-isobutamido-1,2,5-trideoxy-d-glucitol (24): Yield: 54%; Rf=0.4 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (500 MHz, CD3OD): δ=3.81 (dd, J=3.0, 11.2 Hz, 1H), 3.71 (ddd, J=4.8, 10.9, 10.9 Hz, 1H), 3.64 (dd, J=5.0, 11.2 Hz, 1H), 3.34–3.30 (m, 1H), 3.24 (dd, J=9.6, 9.6 Hz, 1H), 3.07 (dd, J=4.9, 12.4 Hz, 1H), 2.50–2.44 (m, 2H), 2.39 (dd, J=12.3, 12.3 Hz, 1H), 1.10 ppm (t, J=7.8 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ=180.9, 77.5, 73.8, 62.8, 62.5, 53.6, 48.8, 36.4, 20.1, 19.8 ppm; HRMS: m/z calcd: 233.1501 [M+H]+, found: 233.1511; elemental analysis calcd (%) for C10H20N2O4: C 51.71, H 8.68, N 12.06; found: C 51.83, H 8.51, N 12.16.
1,5-Imino-2-isovaleramido-1,2,5-trideoxy-d-glucitol (25): Yield: 63%; Rf=0.5 (CHCl3/MeOH/conc. NH3 12:8:1); 1H NMR (600 MHz, CD3OD): δ=3.81–3.74 (m, 2H), 3.64 (dd, J=6.1, 10.5 Hz, 1H), 3.27–3.20 (m, 2H), 3.10 (dd, J=4.4, 12.3 Hz, 1H), 2.47–2.43 (m, 1H), 2.39 (dd, J=11.7, 11.7 Hz, 1H), 2.11–2.03 (m, 3H), 0.94 ppm (m, 6H); 13C NMR (150 MHz, CD3OD): δ=175.9, 77.7, 74.1, 62.9, 62.8, 53.8, 48.8, 46.5, 27.4, 22.8, 22.6 ppm; HRMS: m/z calcd: 247.1658 [M+H]+, found: 247.1661; elemental analysis calcd (%) for C11H22N2O4: C 53.64, H 9.00, N 11.37; found: C 53.59, H 8.98, N 11.31.
Kinetic analysis of inhibitors: Assays against O-GlcNAcase, VcNagZ and StNagZ were performed in NaPi buffer (50 mM, NaCl (100 mM), pH 6.5) and for β-hexosaminidase B citrate buffer (50 mM, NaCl (100 mM), pH 4.25) using 4-methylumbelliferyl N-acetyl-β-d-glucosaminide as substrate. For NAGLU, assays were performed in acetate buffer (100 mM, pH 4.3), containing bovine serum albumin (0.5 mgmL−1) using 4-methylumbelliferyl N-acetyl-α-d-glucosaminide as substrate. Release of 4-methylumbelliferone was monitored continuously using a fluorimeter plate reader for OGA, VcNagZ and StNagZ, and a 30 min stopped-assay procedure was used for β-hexosaminidase B and NAGLU (quenched with fourfold excess of quenching buffer, glycine (200 mM), pH 10.75). For OGA, the inhibitors were preincubated with the enzyme for 10 min before the addition of the substrate. Readings were taken at excitation and emission wavelengths of 368 nm and 450 nm respectively. Assays contained substrate at the previously determined Km value of the substrate for the enzyme, and the enzyme typically at a concentration of 100–200 nM. Inhibitors were added at a range of concentrations encompassing their Ki values. The rates at each inhibitor concentration were plotted and the best fit line through the points ascertained. The −1/Ki was taken as the point where the line of best fit intersected with 1/Vmax.
NagZ crystallization, structure determination and refinement: StNagZ plasmid construction, expression and purification have been previously described.20 StNagZ crystals were grown at RT using the hanging drop vapor-diffusion method by mixing equal volumes of reservoir buffer (25% PEG1000, MES (0.1M), pH 6.5) and protein solution (6 mgmL−1) in crystallization buffer (NaCl (150 mM), BisTris (20 mM), pH 6.5). A single StNagZ crystal was soaked overnight in a drop containing reservoir buffer and 21 at a concentration of 30 mM to obtain the protein–inhibitor complex. The 25% PEG1000 present in the buffer was sufficient for cryo-protection, and crystals were harvested by flash-cooling in liquid nitrogen. X-ray diffraction data were collected at beamline 08ID-1 at the Canadian Light Source (Saskatoon, Canada). Diffraction data were integrated using XDS51 and scaled and merged using SCALA52 (see Table S1). The structure was solved by molecular replacement using the program PHASER53 and the crystal structure of native unliganded StNagZ (PDB ID: 4GVG). Subsequent rounds of refinement were performed using phenix.refine and COOT.54,55 A ligand restraint file was generated for 21 using PHENIX eLBOW and the inhibitor was initially fit into electron density using PHENIX Ligandfit.54 Solvent molecules were added using phenix.refine and final refinement was performed using COOT and phenix.refine. Stereochemical quality of the final model was assessed by using MolProbity.56 The final refinement statistics are presented in Table S1.
Determination of the Minimal Inhibitory Concentration of β-lactams: Cultures were prepared by inoculating Mueller–Hinton broth (5 mL) with a small amount of a glycerol stock of P. aeruginosa PA01 and then were grown at 37°C to an OD600 value of ≈0.5. 96-well plates containing a range of concentrations of β-lactams varying by factors of 2 were prepared. Each well contained 80 μL of the antibiotic in Mueller–Hinton broth and the volume was made up to 100 μL by addition of either 20 μL of 21 (1 mM in H2O) or 20 μL H2O. These broths were inoculated with the culture (100 μL) and allowed to incubate at 37°C for 18 h. The optical density at 595 nm was measured for all cultures and the MIC determined from the concentration of antibiotic at which no growth was observed. All MIC determinations were performed in triplicate.
Agar diffusion tests: A culture of P. aeruginosa PA01 was prepared as described above. The cells were harvested by centrifugation (13000 rpm, 3 min). The cells were then resuspended in Mueller–Hinton broth (2 mL) and 500 μL of this suspension was used to inoculate the appropriate mixtures of inhibitor and Mueller–Hinton broth. Culture A contained Mueller–Hinton broth (500 μL) and 21 (500 μM in Mueller–Hinton broth, 500 μL). Culture B contained Mueller–Hinton broth (1000 μL). These mixtures were then cultured for 60 min at 37°C. Mueller–Hinton broth agar plates (1.5% agar) were streaked with the bacterial culture. Antibiotic discs (6 mm diameter) previously loaded with 21 (500 μM, 10 μL) or H2O alone, were placed on the agar plates. After incubation overnight at 37°C, the diameter of the inhibition zone was measured. All determinations were performed in triplicate.
Acknowledgments
K.A.S. thanks the Australian Research Council for support and the Centre for Microscopy, Characterization and Analysis, The University of Western Australia, which are supported by University, State and Federal Government funding. D.J.V. and B.L.M. thank the Canadian Institutes of Health Research (MOP 97818) and Cystic Fibrosis Canada for funding. J.P.B. was supported by a postdoctoral fellowship from the Manitoba Health Research Council (MHRC). B.L.M. holds a Manitoba Research Chair in Structural Biology. D.J.V. is a Canada Research Chair in Chemical Glycobiology and a EWR Steacie Memorial Fellow. G.E.W. was supported by a fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC) and T.M.G. was supported by a Wellcome Trust Sir Henry Wellcome Postdoctoral fellowship and a research trainee award from MSFHR. We also thank Veronica Larmour for technical assistance and Shaun Labiuk and the staff of the Canadian Light Source (CLS) beamline 08ID-1 for assistance with data collection. The CLS is supported by the NSERC, National Research Council, the Canadian Institutes of Health Research (CIHR), and the University of Saskatchewan.
References
- [1].Sanders CC. Annu. Rev. Microbiol. 1987;41:573. doi: 10.1146/annurev.mi.41.100187.003041. [DOI] [PubMed] [Google Scholar]
- [2].Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. EMBO J. 1994;13:4684. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jacoby G. Clin. Microbiol. Rev. 2009;22:161. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jacoby GA, Munoz-Price LS. N. Engl. J. Med. 2005;352:380. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- [5].Reisbig MD, Hanson ND. J. Antimicrob. Chemother. 2002;49:557. doi: 10.1093/jac/49.3.557. [DOI] [PubMed] [Google Scholar]
- [6].Bradford PA, Urban C, Mariano N, Projan SJ, Rahal JJ, Bush K. Antimicrob. Agents Chemother. 1997;41:563. doi: 10.1128/aac.41.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tondi D, Morandi F, Bonnet R, Costi MP, Shoichet BK. J. Am. Chem. Soc. 2005;127:4632. doi: 10.1021/ja042984o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eidam O, Romagnoli C, Dalmasso G, Barelier S, Caselli E, Bonnet R, Shoichet BK, Prati F. Proc. Natl. Acad. Sci. USA. 2012;109:17448. doi: 10.1073/pnas.1208337109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fisher JF, Meroueh SO, Mobashery S. Chem. Rev. 2005;105:395. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- [10].Vollmer W, Holtje JV. Curr. Opin. Microbiol. 2001;4:625. doi: 10.1016/s1369-5274(01)00261-2. [DOI] [PubMed] [Google Scholar]
- [11].Park JT. J. Bacteriol. 1993;175:7. doi: 10.1128/jb.175.1.7-11.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cheng Q, Li H, Merdek K, Park JT. J. Bacteriol. 2000;182:4836. doi: 10.1128/jb.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Votsch W, Templin MF. J. Biol. Chem. 2000;275:39032. doi: 10.1074/jbc.M004797200. [DOI] [PubMed] [Google Scholar]
- [14].Jacobs C, Frere J-M, Normark S. Cell. 1997;88:823. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- [15].Uehara T, Park JT. J. Bacteriol. 2002;184:4233. doi: 10.1128/JB.184.15.4233-4239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Henrissat B, Davies G. Curr. Opin. Struct. Biol. 1997;7:637. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- [17].Vocadlo DJ, Mayer C, He S, Withers SG. Biochemistry. 2000;39:117. doi: 10.1021/bi991958d. [DOI] [PubMed] [Google Scholar]
- [18].Vocadlo DJ, Withers SG. Biochemistry. 2005;44:12809. doi: 10.1021/bi051121k. [DOI] [PubMed] [Google Scholar]
- [19].Stubbs KA, Scaffidi A, Debowski AW, Mark BL, Stick RV, Vocadlo DJ. J. Am. Chem. Soc. 2008;130:327. doi: 10.1021/ja0763605. [DOI] [PubMed] [Google Scholar]
- [20].Bacik J-P, Whitworth GE, Stubbs KA, Vocadlo DJ, Mark BL. Chem. Biol. 2012;19:1471. doi: 10.1016/j.chembiol.2012.09.016. [DOI] [PubMed] [Google Scholar]
- [21].Stubbs KA, Balcewich M, Mark BL, Vocadlo DJ. J. Biol. Chem. 2007;282:21382. doi: 10.1074/jbc.M700084200. [DOI] [PubMed] [Google Scholar]
- [22].Yamaguchi T, Blázquez B, Hesek D, Lee M, Llarrull LI, Boggess B, Oliver AG, Fisher JF, Mobashery S. ACS Med. Chem. Lett. 2012;3:238. doi: 10.1021/ml2002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Asgarali A, Stubbs KA, Oliver A, Vocadlo DJ, Mark BL. Antimicrob. Agents Chemother. 2009;53:2274. doi: 10.1128/AAC.01617-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zamorano L, Reeve TM, Deng L, Juan C, Moya B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. Antimicrob. Agents Chemother. 2010;54:3557. doi: 10.1128/AAC.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huang Y-W, Hu R-M, Lin C-W, Chung T-C, Yang T-C. Antimicrob. Agents Chemother. 2012;56:1936. doi: 10.1128/AAC.05645-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Balcewich M, Stubbs KA, He Y, James T, Davies GJ, Vocadlo DJ, Mark BL. Protein Sci. 2009;18:1541. doi: 10.1002/pro.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beer D, Maloisel JL, Rast DM, Vasella A. Helv. Chim. Acta. 1990;73:1918. [Google Scholar]
- [28].Dong DL, Hart GW. J. Biol. Chem. 1994;269:19321. [PubMed] [Google Scholar]
- [29].Haltiwanger RS, Grove K, Philipsberg GA. J. Biol. Chem. 1998;273:3611. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- [30].Miller DJ, Gong X, Shur BD. Development. 1993;118:1279. doi: 10.1242/dev.118.4.1279. [DOI] [PubMed] [Google Scholar]
- [31].Ficko-Blean E, Stubbs KA, Nemirovsky O, Vocadlo DJ, Boraston AB. Proc. Natl. Acad. Sci. USA. 2008;105:6560. doi: 10.1073/pnas.0711491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gloster TM, Vocadlo DJ. Nat. Chem. Biol. 2012;8:683. doi: 10.1038/nchembio.1029. [DOI] [PubMed] [Google Scholar]
- [33].Stubbs KA, Zhang N, Vocadlo DJ. Org. Biomol. Chem. 2006;4:839. doi: 10.1039/b516273d. [DOI] [PubMed] [Google Scholar]
- [34].Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. J. Biol. Chem. 2004;279:13478. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ho CW, Popat SD, Liu TW, Tsai KC, Ho MJ, Chen WH, Yang AS, Lin CH. ACS Chem. Biol. 2010;5:489. doi: 10.1021/cb100011u. [DOI] [PubMed] [Google Scholar]
- [36].Fleet GWJ, Smith PW, Nash RJ, Fellows LE, Parekh RB, Rademacher TW. Chem. Lett. 1986:1051. [Google Scholar]
- [37].Fleet GWJ, Fellows LE, Smith PW. Tetrahedron. 1987;43:979. [Google Scholar]
- [38].Kappes E, Legler G. J. Carbohydr. Chem. 1989;8:371. [Google Scholar]
- [39].Furneaux RH, Gainsford GJ, Lynch GP, Yorke SC. Tetrahedron. 1993;49:9605. [Google Scholar]
- [40].Granier T, Vasella A. Helv. Chim. Acta. 1998;81:865. [Google Scholar]
- [41].Steiner AJ, Schitter G, Stutz AE, Wrondigg TM, Tarling CA, Withers SG, Mahuran DJ, Tropak MJ. Tetrahedron: Asymmetry. 2009;20:832. doi: 10.1016/j.tetasy.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gandy MN, Piggott MJ, Stubbs KA. Aust. J. Chem. 2010;63:1409. [Google Scholar]
- [43].Billing JF, Nilsson UJ. Tetrahedron. 2005;61:863. [Google Scholar]
- [44].Goddard-Borger ED, Stick RV. Org. Lett. 2007;9:3797. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- [45].Nilsson KGI, Pan H, Larsson-Lorek U. J. Carbohydr. Chem. 1997;16:459. [Google Scholar]
- [46].Zhao K-W, Neufeld EF. Protein Expression Purif. 2000;19:202. doi: 10.1006/prep.2000.1230. [DOI] [PubMed] [Google Scholar]
- [47].Sanders CC, Sanders WE., Jr Clin. Infect. Dis. 1992;15:824. doi: 10.1093/clind/15.5.824. [DOI] [PubMed] [Google Scholar]
- [48].Govan JR, Deretic V. Microbiol. Rev. 1996;60:539. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lyczak JB, Cannon CL, Pier GB. Clin. Microbiol. Rev. 2002;15:194. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nagaki M, Shimura S, Tanno Y, Ishibashi T, Sasaki H, Takishima T. Chest. 1992;102:1464. doi: 10.1378/chest.102.5.1464. [DOI] [PubMed] [Google Scholar]
- [51].Kabsch W. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Evans P. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- [53].McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Crystallogr. D Biol. Crystallogr. 2005;61:458. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- [54].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Emsley P, Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [56].Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.