

Abstract
Mitochondrial dysfunction has been implicated in the aetiology of many complex diseases, as well as the ageing process. Much of the research on mitochondrial dysfunction has focused on how mitochondrial damage may potentiate pathological phenotypes. The purpose of this review is to draw attention to the less well-studied mechanisms by which the cell adapts to mitochondrial perturbations. This involves communication of stress to the cell and successful induction of quality control responses, which include mitophagy, unfolded protein response, upregulation of antioxidant and DNA repair enzymes, morphological changes, and if all else fails apoptosis. The mitochondrion is an inherently stressful environment and we speculate that dysregulation of stress signaling or an inability to switch on these adaptations during times of mitochondrial stress may underpin mitochondrial dysfunction and hence amount to pathological states over time.
1. Introduction
Approximately 1.45 billion years ago, gram negative bacteria were engulfed by primitive eukaryotic cells giving rise to the mitochondrion [1–3]. However, the complex relationship between this organelle and its host is not fully understood, and the critical role that mitochondria play in various disease states has only been appreciated in recent years. Nuclear encoded proteins coordinate with mitochondrially encoded proteins for the biogenesis and maintenance of the complete mitoproteome. In return, mitochondria produce 90% of the cells ATP. Despite this elegant symbiosis, the inherent differences between mitochondria and the rest of the cell can lead to complications that may ultimately have pathological consequences. For instance, mtDNA release can stimulate an inflammatory response in the host cell [4]. Mitochondria possess a harsh protein folding environment, due to the high levels of reactive oxygen species (ROS), and the fact that more than 99% of mitochondrial proteins need to be transported from the cytosol into the mitochondria and correctly folded. In addition to proteotoxic stress, mitochondria are highly susceptible to DNA mutations from ROS and a high DNA replication error rate, which is confounded by less sophisticated DNA repair mechanisms [5].
Being the site of programmed cell death and energy metabolism, the cells survival is ultimately dependent on precise coordination between mitochondria and the rest of the cell. Consequently there are a number of mitochondrial stress signals that are communicated to the rest of the cell that stimulate cellular adaptions, which support this organelle-host symbiotic relationship. This is an emerging area in mitochondrial biology that has not been well studied to date. We propose that inability for the cell to perceive and respond to mitochondrial stress may be a platform for mitochondrial dysfunction (Figure 1). Mitochondrial dysfunction is likely to be at least partly involved in the aetiology of complex diseases of ageing, including Parkinson's disease (PD) [6], Alzheimer's disease (AD) [7], pancreatic β-cell failure [8], and insulin resistance (IR) [9], as well as the ageing process itself [10]. As such, it is critical to better understand the molecular mechanisms coordinating mitochondrial stress signaling with cellular adaptations.
Figure 1.
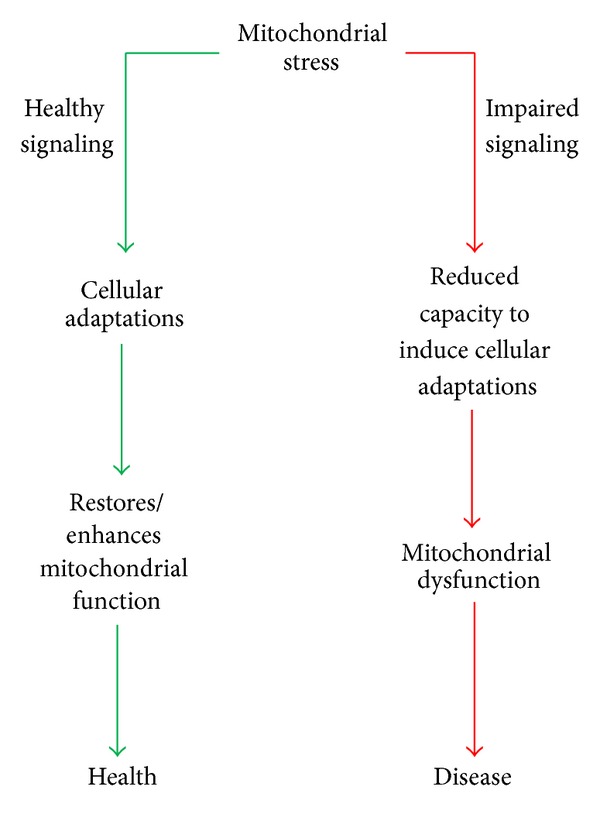
Hypothesis of responses to mitochondrial stress in health and disease. In times of mitochondrial stress, signals are sent to the cell which promote cellular adaptations that restore or possibly enhance mitochondrial function to maintain health (green arrows). In situations where mitochondrial stress signals are not relayed to other parts of the cell or the cell does not respond stress signals (red arrows), there is a failure for cellular adaptations which may consequently result in mitochondrial dysfunction and the development of disease.
Much of the previous literature linking mitochondrial dysfunction with disease has focused on the linear view that mitochondrial stress causes damage, which subsequently causes disease. However, mitochondria respond to various stresses by inducing a complex array of cellular responses and adaptations to reduce the impact of subsequent stressors. The purpose of this review is to summarise the literature surrounding cellular adaptations and quality control processes to cope with mitochondrial stress, which may be potential pharmacological targets.
2. Mitochondrial Stress Signals
It is clear that mitochondria are particularly vulnerable to endogenous stress and are major sensors of environmental stress, such as diet and toxic substances. Mitochondria signal stress by membrane depolarization, alterations in adenine nucleotide levels, ROS production, Ca2+ fluxes, permeability transition pore opening, and perhaps secretion of proteins/peptides (Figure 2). Here we will discuss how different stress signals promote the development of cellular adaptations, through retrograde signalling from the mitochondria to the nucleus, posttranslational modifications or activation of proteins and other mechanisms.
Figure 2.
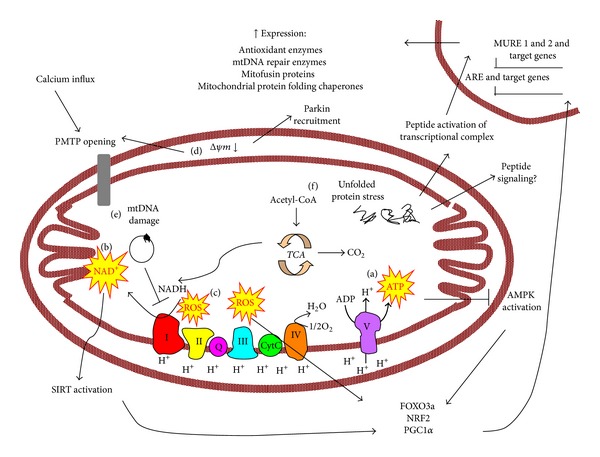
Schematic summary of mitochondrial stress signaling and cellular adaptations. ATP (a), NAD+ (b), and ROS (c) are outputs of the electron transport chain and oxidative phosphorylation that may function as stress signals. NAD+ can activate sirtuins (SIRT) and increased AMP/ATP ration can activate AMPK, which activate transcription of antioxidant defences, mitochondrial DNA repair enzymes, and other target genes important in mitochondrial biogenesis and metabolism through transcription factors FOXO3a, NRF2, and the transcriptional coactivator PGC1α. ROS can also directly activate these transcriptional regulators. Mitochondrial DNA (mtDNA) damage can potentiate oxphos dysfunction (e) and hence lead to the above responses. Loss of innermitochondrial membrane potential (d) can lead to PMTP opening or parkin recruitment and hence mitophagy. Through peptide export, unfolded protein stress (f) can activate a transcriptional complex which acts on the MURE1, MURE2, and CHOP elements to induce the transcription of mitochondrial protein folding chaperones.
2.1. Impaired Oxidative Phosphorylation
Oxidative phosphorylation (oxphos) is the process whereby electrons gained during substrate oxidation are transported along the electron transport chain (ETC), coupled with the pumping of protons across the innermitochondrial membrane and the generation of a proton gradient, which is used as the driving force for ATP production at complex V (ATP synthase) in conjunction with oxygen consumption at complex IV. Defective oxphos reduces bioenergetic capacity and is a sign of cellular stress. Perturbations in oxphos can lead to reduced ATP production, changes in redox status, innermitochondrial membrane depolarization, and excessive reactive oxygen species (ROS) generation.
2.1.1. Alterations in Energy Intermediates
ATP Levels. Oxphos is a multistep process that effectively results in ATP production from fuel substrates. ATP is a major output of oxphos function (Figure 2(a)) and hence the level of ATP and the ratio of ATP with other adenine nucleotides are a signal of mitochondrial function as well as cellular energy charge. During conditions of compromised mitochondrial function there are shifts in the ratio of adenine nucleotides and the best characterised cellular adaptation of an increased AMP/ATP ratio, resulting from defective oxphos, is the activation of the enzyme AMP-activated protein kinase (AMPK). This is a key energy-sensing kinase that reprograms cellular metabolism through phosphorylation of numerous target proteins.
Through alterations in the activity of downstream targets, AMPK activation rapidly stimulates a shift to catabolic metabolism and over the longer term increases mitochondrial biogenesis and oxidative capacity. Stimulation of AMPK has been shown to increase the expression of PGC1α and metabolic enzymes in skeletal muscle [11, 12] with increased fatty acid oxidation [12, 13] and glycogen synthesis [13]. Consistent with an important role for AMPK in energy transduction, oxidative capacity is reduced in myocardial tissue when AMPK activity is lost [14].
In addition to regulating mitochondrial substrate oxidation, AMPK has other effects on mitochondrial parameters. Metformin treatment activates AMPK activity in conjunction with inducing PGC1α and mitofusin protein 2 (Mfn2) protein expression in myocardial tissue [15]. A gain of function AMPK mutation in skeletal muscle has also been reported to increase the expression of mitochondrial fusion/fission proteins, Mfn2, optic atrophy 1 (OPA1), and dynamin-related protein 1 (Drp1) [16] which implicates AMPK in the regulation of mitochondrial dynamics as well as content. Furthermore, AMPK functionally prevents high-glucose induced mitochondrial fission in endothelial cells [17], highlighting that AMPK activity promotes mitochondrial quality control processes, as well as stimulating mitochondrial metabolism. In line with this, AMPK may function in the regulation of mitophagy, the autophagic clearance of mitochondria, through phosphorylation of an autophagy gene [18]. These latter two functions suggest AMPK influences quality control processes as well as regulation of mitochondrial oxidative metabolism.
NAD + Levels. In addition to ATP production, mitochondrial oxphos involves electrons being stripped out of fuel substrates and carried on NAD+, making NAD+ a signal of mitochondrial oxphos function (Figure 2(b)). The NAD+/NADH ratio, which signals redox status of the cell, can in turn be altered with changes in substrate flux and oxphos function. The redox ratio has been known for a long time to be an important feedback regulator of substrate metabolism, but in recent years it has become evident that NAD+ directly influences the activity of a number of enzymes that may affect quality control processes.
NAD+ activates Poly(ADP-ribose) polymerase 1 (PARP1) which is important for maintenance and repair of genomic DNA [19] and also stimulates transcription of NRF1 [20]. These dual actions of PARP1 activation suggest that low-grade bioenergetic stress and subsequent changes in the redox ratio may enhance the coordination of nuclear genomic integrity with regulation of mitochondrial function.
Another important group of enzymes regulated by changes in the NAD+/NADH ratio are the sirtuin (SIRT) family of NAD+-dependent deacetylases. Through alterations in posttranslational modifications (e.g., acetylation) on lysine residues, SIRT proteins influence a number of metabolic processes and cellular functions.
SIRT1 is known to regulate mitochondrial function in an anterograde, or nucleus to mitochondria fashion, signaling in response to redox status [21]. SIRT1 is activated by an increase in NAD+ and via deacetylation of the master regulator of mitochondrial biogenesis, PGC1α, and has been reported to increase mitochondrial bioenergetic capacity [22, 23]. In addition to regulating mitochondrial quantity, SIRT1 has been reported to regulate mitochondrial quality in response to redox status, by stimulating mitophagy [24]. SIRT3 is another important member of the sirtuin family likely to regulate mitochondrial function in response to redox status. SIRT3 is localised in the mitochondrion and has been shown to influence mitochondrial function more directly by deacetylating a range of enzymes involved in metabolic pathways in mitochondria [25, 26]. Alterations in SIRT3 activity can alter the response to stress pathways involving the MPTP and SIRT3 may also function in ameliorating oxidative stress in response to mitochondrial redox status, as it deacetylates and activates the antioxidant enzyme MnSOD [27], as well as FOXO3a [28], a transcription factor which promotes cellular antioxidant defences, which is described in more detail below.
2.1.2. Mitochondrial Membrane Depolarization
In addition to changes in ATP and nutrient intermediates, alterations in oxphos can induce a stress response via changes in membrane potential. More specifically, the innermitochondrial membrane becomes polarized during oxphos and loss of the membrane potential or depolarization can act as a signal (Figure 2(d)), promoting mitophagy and mitochondrial permeability transition pore (MPTP) opening. These processes and their potential role in pathological states are discussed below.
Mitophagy. Mitochondrial depolarization can induce lysozomal degradation of mitochondria in a process called mitophagy. A major pathway mediating mitophagy has been identified which involves the PD associated proteins PTEN-induced putative kinase 1 (PINK1) and parkin. Parkin is an E3 ubiquitin ligase which translocates to mitochondria upon membrane depolarization in a PINK1-dependent process [29]. PINK1 actually binds to depolarized mitochondria, which recruits parkin to ubiquitinylated mitochondrial proteins [30]. Mitophagy is a quality control pathway which enables defective mitochondria to be cleared without impeding cell survival.
Defective mitophagy coincides with cell models of neurological diseases including AD [31] and PD [32], as well as pancreatic β-cell dysfunction [33], cardiomyopathy [34], and hepatic IR [35]. These findings strongly implicate reduced capacity to clear defective mitochondria with complex disease. However, the lack of mammalian in vivo models in this area means that it is unclear if these cellular effects are physiologically relevant or whether it is cause or consequence of disease. Another feature of these studies that makes it difficult to discern the relevance of reduced autophagic clearance of mitochondria in the aetiology of chronic disease is that defective mitophagy is usually defined as reduced parkin translocation in response to the uncoupling agents CCCP/FCCP under nonphysiological circumstances [36, 37]. Little research exists on levels of basal mitophagy in disease models. One study used colocalisation of mitochondria and lysozomes under normal state to signify basal mitophagy and found a reduction in basal mitophagy in fibroblasts derived from human patients with a mutation in PD associated protein DJ-1 [38], suggesting that impaired mitophagy may be a feature of human Parkinsonism. Further investigation of basal mitophagy in vivo in mammalian disease models would greatly facilitate insight into the role of defective mitophagy in disease development.
In all, mitophagy is a quality control process that enables adaptation to oxphos stress and may be protective against complex disease development. However, it is not currently understood how this process is switched on during physiologically normal mitochondrial membrane depolarization.
Permeability Transition Changes. Membrane depolarization can also initiate cell death by opening of the MPTP. This pore is a protein complex of VDAC, cyclophilin D (CypD), and adenine nucleotide translocase that can open and close and allows small substances to cross the mitochondrial membrane in response to membrane depolarization [39]. Opening of the MPTP causes mitochondrial swelling which can lead to cell death. Opening can be caused by ROS, mitochondrial calcium influxes from the ER, as well as membrane depolarization, but other factors may also influence the susceptibility of the MPTP to open in response to these stimuli [40]. Increased susceptibility of opening of the MPTP is linked with pathological states [41] and conversely, delayed response or inhibition of MPTP is associated with cell survival and reduced neurodegeneration [42].
Mitochondrial perturbations can lead to MPTP resistance as a cellular adaption. MPTP opening can be inhibited by NAD+, a signal of low energy status by SIRT3 deacetylation of CypD [43]. Inhibition of MPTP opening by CypD deficiency may further increase membrane depolarization by induction of uncoupling proteins, which have been proposed to offset high-fat diet induced obesity by increasing basal metabolic rate [44].
The phospholipid composition of the mitochondrial membrane also affects PMTP opening and now there is emerging evidence indicating that fatty acids and ceramides play a role in regulating the activity of MPTP which links lipids with mitochondrial dysfunction. Omega 3 fatty acid incorporation into the mitochondrial membrane delays MPTP opening [45]. Ceramides, a type of sphingolipids that are associated with IR, may disrupt healthy mitochondrial signaling by sensitizing the MPTP [46–48]. This suggests that a possible mechanism by which ceramides elicit IR is through potentiation of mitochondrial stress.
Membrane depolarization may result in either mitophagy or apoptosis depending on the extent of damage, which improves cellular or whole body functioning by cleansing damaged organelles or cells, respectively.
2.1.3. Mitochondrial Reactive Oxygen Species Production
Mitochondria are a major site of ROS production (Figure 2(c)), as byproducts of normal oxphos function which can be further increased when there are oxphos defects [49]. ROS can readily diffuse out of the mitochondrial membrane and elicit cellular responses, making them active signaling molecules through activation of redox sensitive proteins, including transcription factors and transcriptional coactivators, making them key players in eliciting retrograde signaling.
Oxidative damage has been linked with a number of complex diseases, including neurological disorders [50, 51], cardiovascular diseases [52], and insulin resistance [53]. Furthermore, human polymorphisms in the antioxidant enzymes MnSOD and GPX are associated with increased risk of multiple cancers [54–57], cardiovascular disease [58], and diabetes [59].
Much of the literature surrounding ROS induced damage has incubated cultured cells in supraphysiological concentrations of exogenous hydrogen peroxide or has used high doses of pharmacological inhibitors of complex I and III for short periods of time [60–62]. These are major chemical insults that induce extensive cell damage and apoptosis and do not model endogenous, physiologically normal levels of mitochondrial ROS production during chronic mitochondrial stress. For instance, when oxphos dysfunction was modelled by DNA gamma polymerase mutation, resulting in complex I and III defects and an accompanying increase in ROS production, there was no decrease in cell survival because the cells were rescued by induction of selenoproteins GSH and GPX [63]. This was mediated by the zinc finger protein ZNF143 which is required for transcription of a protein required for the incorporation of selenocysteine into selenoproteins [63]. Another disease-related process which involves ROS is that of preconditioning, where a mild insult induces adaptations that protect against subsequent stresses. For example, low-grade ROS plays a role in inducing PKC epsilon translocation, which is involved in neuroprotective ischemic preconditioning, whereas larger ROS insults activate PKC delta, which causes apoptosis and neurodegeneration [64]. In line with these findings in neurons, mitochondrial ROS also have preconditioning roles in cardiomyocytes [65].
Extending on the idea that oxidative stress is involved in preconditioning phenomena, there is evidence that mild mitochondrial ROS stressors may actually improve lifespan, through a process termed “mitochondrial hormesis” (mitohormesis). This theory describes the link between low-grade mitochondrial stressors and enhanced cellular function [66]. Mitohormesis has been proposed to increase life span in C. elegans in response to low-grade arsenic exposure [67] and low-glucose availability [68]. Low-grade mitochondrial stressors have also been reported to protect neuronal cells against a secondary large stress by maintaining mitochondrial membrane potential [69], and low-dose complex I inhibition improves mitochondrial capacity and antioxidant defences in neuronal cells [70], implicating mitohormesis in neuroprotection. Although mitohormesis has been demonstrated to increase lifespan in C. elegans and improve functioning in mammalian cell culture, in vivo mammalian studies are lacking.
There are other examples where ROS induces cellular adaptations to clear subsequent ROS insults or improve mitochondrial functioning, which highlights an important negative feedback role for these molecules. One major mechanism that switches on antioxidant defences in the cell is the activation of antioxidant response element (ARE). ARE is a cis-acting enhancer sequence that controls expression of a variety of antioxidant enzymes. A ROS-mediated signaling cascade that results in activation of this enhancer has been identified, which provides evidence for cellular adaptation in response to ROS signals, where nuclear respiratory factor 2 (NRF2) transcription factor activates the ARE enhancer to switch on an antioxidant gene program [71]. Oxidation of lipids, especially membrane lipids, is one of the mechanisms proposed in the free radical theory of ageing, but products of lipid oxidation may actually function in cell signaling to cause adaptations. For instance, the oxidised lipid metabolite 4-hydroxynonenal promotes NRF2 activation of the ARE inducing expression of antioxidant genes [72]. In addition to ARE, ROS activates the transcription factor FOXO3a [73], which induces expression of MnSOD [74] catalase [75] and PrxIII [76] which have antioxidant functions in the mitochondria. Overall, these responses are protective against oxidative damage suggesting that low-grade ROS constitutes an important stress signal that can lead to adaptations that ameliorate a major insult later and protect against disease processes.
ROS not only induce antioxidant responses in a negative feedback type mechanism, but also can stimulate cellular adaptations that improve mitochondrial capacity in general. ROS increases mitochondrial biogenesis and mitochondrial DNA content in fibroblasts [77, 78]. Hydrogen peroxide treatment increases peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) and PGC1β promoter activity and expression, and in addition to stimulating mitochondrial biogenesis, these transcriptional coactivators also stimulate the expression of multiple antioxidant defences [79, 80]. In vivo overexpression of pgc1β in rat skeletal muscle resulted in increased expression of antioxidant enzymes and an overall reduction in oxidative damage [81], highlighting its role in inducing antioxidant defenses. Nuclear respiratory factor 1 (NRF1) is a transcription factor associated with mitochondrial biogenesis and was found to be activated and subsequently activate mitochondrial transcription factor A (TFAM) in a redox-dependent pathway [82], which is again consistent with the notion that nontoxic levels of ROS can improve mitochondrial function. Additionally, the autophagy gene family Atg14 are regulated by ROS [83] and hence there may even be a role of mitochondrial ROS in stimulating mitophagy. Collectively these findings suggest that ROS are important signalling molecules that not only stimulate the expression of antioxidant systems to protect against future oxidative insults, but also can modify other aspects of mitochondrial function.
Overall, there has been a large focus in the literature on the effects of exogenous ROS in vitro. Increased understanding of the mechanisms that cells use to respond to mitochondrial oxidative stress in vivo is less thoroughly investigated to date and may be relevant in understanding the pathogenesis of complex disease, particularly as a reduced capacity to mount and antioxidant response is linked with a number of disease states [55, 56, 58, 59], as well as the ageing process [84].
2.2. Mitochondrial Morphology Transitions Are Important Responses to Mitochondrial Stress Signals
Mitochondria are dynamic organelles that constantly undergo morphological changes in the opposing processes of fusion and fission. In fusion, multiple mitochondria form one elongated mitochondrion, whereas, in fission, a mitochondrion generates small, fragmented mitochondria. Mitochondrial fission and division occurs when Drp-1 is recruited from the cytosol to divide the mitochondrion [85]. Fusion of mitochondria involves mitofusin proteins 1 and 2 (Mfn1 and Mfn2) anchoring outer mitochondrial membranes and OPA1 anchoring the innermitochondrial membranes [85].
Changes in mitochondrial morphology are known to occur in response to mitochondrial stressors and may be a cellular adaptation to regulate cell survival. The pro-survival protein Pim-1 and apoptotic protein PUMA have opposing roles in Drp-1 localisation to the mitochondria [86], suggesting that alterations in mitochondrial dynamics may be an integral part of the apoptotic program. In general, mitochondrial fusion occurs in response to mitochondrial stress and elongated mitochondria are thought to be pro-survival by increasing resistance to apoptosis and mitophagy. Mitochondrial elongation can be induced by ROS [87] as well as increase resistance to ROS [88], suggesting another feedback mechanism by which signals may allow the cell to adapt to mitochondrial oxidative stress. Mitochondrial fission, on the other hand, occurs in response to major stressors and primes mitochondria for mitophagy and apoptosis. Major stressors which promote mitochondrial fission and hence fragmentation include cyclosporine A [89] as well as excessive ceramides in yeast [90] and cardiomyocytes [91]. Additionally, short, high-dose incubations of hydrogen peroxide [92] and palmitate [93] induce mitochondrial fragmentation in C2C12 myotubes. Mitochondrial fission can increase mitochondrial uncoupling to ameliorate ROS production [94] as well as stimulate mitophagy [95, 96] and apoptosis [97–100] in order to let the cell adapt and respond to these major stressors.
Dysregulation of mitochondrial quality control processes has been implicated in pathological states and ageing. Down-regulation of Drp1 and mitochondrial elongation can be caused by a pharmacological DNA damage inducer [88] and cellular senescence [101], which mechanistically links ageing with reduced mitochondrial bioenergetics. Overexpression of AD associated protein, Tau, prevents Drp1 localisation to mitochondria [102] demonstrating that localisation as well as expression of mitochondrial morphology regulators is important in cellular quality control. Metabolic health may also be influenced by alterations in mitochondrial morphology with skeletal muscle expression of Mfn-2 being increased with exercise [103] and downregulated in obese mice [104] suggesting a link between energy metabolism and mitochondrial morphology transitions.
Overall the evidence in the literature indicates that the inherent plasticity in mitochondrial morphology provides the capacity for changes in this organelle in response to the prevailing environmental conditions, which provides a platform for rapidly changing bioenergetics and apoptotic state of the mitochondrion in response to specific stress signals.
2.3. Mitochondrial Genomic Stress Signals DNA Repair
Mitochondrial DNA (mtDNA) is double stranded and organised into a circular structure which encodes 37 genes, of which 13 are subunits of the ETC, 2 are rRNAs, and 22 are tRNAs [105]. mtDNA is susceptible to DNA damage and mutations from replication error, ROS, and basic DNA repair machinery [5, 106]. As mtDNA encodes for critical subunits of the ETC, mtDNA mutations often result in defective oxphos and increased ROS production [107] and hence act as a mitochondrial stress signal (Figure 2(e)).
mtDNA repair enzymes play an important role in responding to different stresses with increased activity of these enzymes that are protecting against oxidative stress induced apoptosis [108], palmitate induced IR and ROS production [109], and cardiac fibrosis [110]. Conversely, deficits in mitochondrial DNA repair have been shown to potentiate neurodegeneration [111] and age-related macular degeneration [112], which emphasises the potential importance of dysregulated cellular adaptation to mtDNA damage in the pathogenesis of disease.
Interestingly, many of the cellular adaptations to oxphos stress also lead to increased expression of mtDNA repair enzyme 8-oxoguanine glycosylase (OGG1), the major mitochondrial DNA repair enzyme. For example, the NRF2, part of the ROS induced antioxidant adaptation, can bind to the OGG1 promoter region to induce OGG1 expression [113], reinforcing the notion that, in response to mitochondrial stress, multiple cellular repair and adaptive responses are induced. The antioxidant enzyme MnSOD can also interact with DNA polymerase gamma to promote repair of mtDNA lesions [114], further linking antioxidant defences with mtDNA repair. There may also be a role for mitochondrial dynamics and degradation in mtDNA damage adaptation. Knockdown of mitochondrial fission, fusion, and mitophagy genes results in reduced capacity to clear mtDNA lesions [115] which reveals a role for mitophagy and morphology transitions in mtDNA cleansing. Mitochondrial OGG1 activity is also increased by exercise [116] which parallels upregulation of other mitochondrial quality control processes with physical activity.
Thus, while mtDNA does not have the same level of protection and repair as nDNA, there are a number of feedback mechanisms in place to respond to mitochondrial genotoxic stress.
2.4. Mitochondrial Proteotoxic Stress Signaling
It is well known that aberrant protein folding and subsequent formation of toxic oligomeric intermediates, such as amyloids, are implicated in chronic diseases of ageing. Given that mitochondrial dysfunction is also widely implicated in complex diseases of ageing and mitochondria are vulnerable to proteotoxic stress, it is possible that mitochondrial specific protein misfolding is a component of chronic disease. Consistent with this idea, accumulation of β-amyloid specifically in the mitochondrial fraction has been associated with severe mitochondrial dysfunction and cell death [117].
2.4.1. Mitochondrial Unfolded Protein Response
The cell has a quality control pathway to adapt to mitochondrial proteotoxic stress called the mitochondrial unfolded protein response (mtUPR) (Figure 2(f)). mtUPR was first identified as a mitochondrial quality control process over 10 years ago in mammalian cells induced by overexpression of a mutant version of OTC which does not fold properly [118]. This resulted in increased expression of mitochondrial import proteins, folding chaperones and heat shock proteins, and the ATP-dependent mitochondrial protease ClpP [118]. Upregulation of these genes during mtUPR induction was found to occur via activation of the CHOP, MURE1, and MURE2 elements that induces the transcription of a number of proteins including mitochondrial heat shock proteins and other mitochondrial quality control proteins [119], but the mechanisms that lead to the activation of the CHOP, MURE1, and MURE2 remained elusive.
The topic was approached in a simpler model organism, C. elegans, by Haynes et al. which facilitated some further characterisation of steps involved in this pathway [120]. mtUPR activation was demonstrated to be dependent on the activity of ATP-dependent mitochondrial protease ClpP [120], suggesting a role for mitochondrial peptides in the process. Further, transcriptional activation of mitochondrial folding chaperones is dependent on nuclear translocation of the ubiquitin-like protein 5 (ubl-5) [121], a bZip transcription factor, and homeodomain containing transcription factor, DVE [122]. Nuclear localisation of this transcription complex is dependent on activity of the mitochondrial peptide transporter, HAF-1, and hence a model was put forth where mitochondrial peptide efflux during proteotoxic stress activates the transcription of mitochondrial heat shock proteins [122].
Although HAF-1 is homologous to mammalian ATP binding cassette proteins, mitochondrially located ATP binding cassette proteins have roles in heme transport, whereas ATP binding cassette proteins with peptide efflux roles are located elsewhere in the cell [123], so thus far mitochondrial peptide export has not been confirmed in mammalian systems. Additionally, upregulation of nuclear encoded mitochondrial protein folding chaperones does not necessarily imply that they actually enter the mitochondrion and get folded themselves. Interestingly, during mtUPR activation, the import efficiency of activating transcription factor associated with stress-1 (ATFS-1) is reduced which allows more to enter the nucleus and activate transcription of mitochondrial quality control proteins [124]. Despite an emerging pathway, the molecular mechanisms by which peptides actually activate assembly of this transcription complex are unclear.
mtUPR is clearly a control process providing cellular adaptation to mitochondrial proteotoxic stress and there is some evidence suggesting a role for the activation of this pathway in chronic disease and ageing. Increased NAD+ concentrations have been implicated in longevity, notably through the activation of SIRT proteins or related homologues. Pharmacological increases in NAD+, increased hsp-6 reporter activity, and lifespan in C. elegans independently of the C. elegans SIRT1 homologue [125] reveal mtUPR induction as an additional potential antiageing pathway of NAD+ activity. Aged wild type mice display higher Hsp60 protein expression in midbrain than young mice, but this age-dependent effect is diminished in DJ-1 knockout mice [126] providing further evidence that mtUPR induction may be an important adaptation for healthy ageing, as well as implicating mtUPR in PD. Furthermore, overexpression of the mitochondrial chaperone TRAP1 ameliorates α-synuclein toxicity [127], which reinforces the idea that upregulation of mitochondrial protein quality control activity may prevent disease, especially PD. Another peculiar piece of evidence that suggests that mtUPR induction is an important adaptation to mitochondrial stress is that the activation of mtUPR appears to increase resistance to the cytotoxic effects of statins, which specifically perturb mitochondrial homeostasis [128]. Interestingly, Hsp60 is overexpressed in cancer cells and prevents cyclophilin D mediated permeability transition and hence promotes cancer cell survival and tumour growth [129]. These findings not only suggest that suppression of mitochondrial apoptosis could be a mechanism by which Hsp60 is pro-survival but also highlight how dysregulation may lead to disease.
2.4.2. Cell-to-Cell Signals
Transcriptomic analysis of ATFS-1-dependent genes demonstrates signaling genes and transcriptional regulators in addition to mitochondrial quality control genes [124], suggesting that mtUPR activation may result in diverse cell signaling pathways. Recent work has also shown that mitochondrial dysfunction in neurons of C. elegans induces mtUPR induction in the gut, which provides evidence for cellular adaptations to mitochondrial stress in a noncell autonomous manner [130]. Indeed the implications of this study were that there may be secreted signals that are induced in response to mitochondrial stress and signal to distal tissues. These signaling molecules, termed mitokines, may be proteins involved in retrograde signaling, mitochondrially derived peptides, or perhaps even nucleic acid-based molecules. An example of this phenomena is the peptide humanin, which was protective against AD [131] and believed to be encoded by mtDNA, as it is 99% similar to mitochondrial 16S rRNA and is not present in mtDNA-depleted cells [132]. It has been posited that there may be more mitochondrially derived or induced peptides which could play diverse cellular signalling roles [133]. An example of a mitochondrially induced stress signal comes from a study that showed impaired oxphos function in muscle-caused secretion of fibroblast growth factor 21, hence acting as a mitochondrial stress-responsive hormone [134]. Overall these studies suggest that mitochondrial stress in one tissue may cause adaptive response in other tissues through the secretion of peptides or proteins, and it has also been shown that the mitochondrial genome encodes long noncoding RNA molecules [135]; however, whether these can move out of the mitochondria to function in cell signaling is unknown.
3. Conclusions
Mitochondria should not simply be thought of as isolated organelles that generate energy. They have a complex relationship with the rest of the cell requiring back and forth coordination of two genomes and constant communication about the bioenergetic status of the cell. Dysregulation of mitochondrial communication processes and reduced cellular capacity to activate mitochondrial quality control processes in times of stress may therefore be important part of the molecular basis for the mitochondrial component of complex disease.
Acknowledgments
Work in the laboratories of the authors is supported by funding from the National Health and Medical Research Council of Australia (NHMRC), the Australian Research Council (ARC), and the Diabetes Australia Research Trust. Jayne Alexandra Barbour is supported by an Australian Postgraduate Award scholarship and Nigel Turner is supported by an ARC Future Fellowship.
Conflict of Interests
The authors declare that there are no conflict of interests regarding the publication of this paper.
References
- 1.Gray MW. Origin and evolution of organelle genomes. Current Opinion in Genetics and Development. 1993;3(6):884–890. doi: 10.1016/0959-437x(93)90009-e. [DOI] [PubMed] [Google Scholar]
- 2.Emelyanov VV. Rickettsiaceae, rickettsia-like endosymbionts, and the origin of mitochondria. Bioscience Reports. 2001;21(1):1–17. doi: 10.1023/a:1010409415723. [DOI] [PubMed] [Google Scholar]
- 3.Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. The archaebacterial origin of eukaryotes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(51):20356–20361. doi: 10.1073/pnas.0810647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0049767.e49767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286(1):127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 6.Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiology of Disease. 2013;51:35–42. doi: 10.1016/j.nbd.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu XM, Huang HC, Jiang ZF. Mitochondrial dysfunction and cellular metabolic deficiency in Alzheimer's disease. Neuroscience Bulletin. 2012;28(5):631–640. doi: 10.1007/s12264-012-1270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Experimental Diabetes Research. 2012;2012:11 pages. doi: 10.1155/2012/703538.703538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J-A, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circulation Research. 2008;102(4):401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bratic A, Larsson NG. The role of mitochondria in aging. The Journal of Clinical Investigation. 2013;123(3):951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fillmore N, Jacobs DL, Mills DB, Winder WW, Hancock CR. Chronic AMP-activated protein kinase activation and a high-fat diet have an additive effect on mitochondria in rat skeletal muscle. Journal of Applied Physiology. 2010;109(2):511–520. doi: 10.1152/japplphysiol.00126.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee WJ, Kim M, Park H-S, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochemical and Biophysical Research Communications. 2006;340(1):291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Vitzel KF, Bikopoulos G, Hung S, et al. Chronic treatment with the AMP-kinase activator AICAR increases glycogen storage and fatty acid oxidation in skeletal muscles but does not reduce hyperglucagonemia and hyperglycemia in insulin deficient rats. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0062190.e62190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stride N, Larsen S, Treebak JT, et al. 5′-AMP activated protein kinase is involved in the regulation of myocardial β-oxidative capacity in mice. Frontiers in Physiology. 2012;3, article 33 doi: 10.3389/fphys.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittington HJ, Hall AR, McLaughlin CP, Hausenloy DJ, Yellon DM, Mocanu MM. Chronic metformin associated cardioprotection against infarction: not just a glucose lowering phenomenon. Cardiovascular Drugs and Therapy. 2013;27(1):5–16. doi: 10.1007/s10557-012-6425-x. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Roves PM, Osler ME, Holmström MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase γ3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. The Journal of Biological Chemistry. 2008;283(51):35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]
- 17.Bhatt MP, Lim YC, Kim YM, Ha KS. C-peptide activates AMPKα and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013;62(11):3851–3862. doi: 10.2337/db13-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Molecular Aspects of Medicine. 2013;34(6):1124–1137. doi: 10.1016/j.mam.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hossain MB, Ji P, Anish R, Jacobson RH, Takada S. Poly(ADP-ribose) polymerase 1 interacts with nuclear respiratory factor 1 (NRF-1) and plays a role in NRF-1 transcriptional regulation. The Journal of Biological Chemistry. 2009;284(13):8621–8632. doi: 10.1074/jbc.M807198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cantó C, Houtkooper RH, Pirinen E, et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metabolism. 2012;15(6):838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α . The Journal of Biological Chemistry. 2005;280(16):16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 23.Cantó C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang S, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. The Journal of Biological Chemistry. 2012;287(23):19304–19314. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hebert AS, Dittenhafer-Reed KE, Yu W, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Molecular Cell. 2013;49(1):186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giralt A, Villarroya F. SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochemical Journal. 2012;444(1):1–10. doi: 10.1042/BJ20120030. [DOI] [PubMed] [Google Scholar]
- 27.Tao R, Coleman MC, Pennington JD, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular Cell. 2010;40(6):893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobs KM, Pennington JD, Bisht KS, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. International Journal of Biological Sciences. 2008;4(5):291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Developmental Cell. 2012;22(2):320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khandelwal PJ, Herman AM, Hoe H-S, Rebeck GW, Moussa CE-H. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Aβ in AD models. Human Molecular Genetics. 2011;20(11):2091–2102. doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batlevi Y, la Spada AR. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiology of Disease. 2011;43(1):46–51. doi: 10.1016/j.nbd.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell T, Johnson MS, Ouyang X, et al. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita+/Ins2-derived β-cells. American Journal of Physiology—Endocrinology and Metabolism. 2013;305:E585–E599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottlieb RA, Mentzer RM, Jr., Linton P-J. Impaired mitophagy at the heart of injury. Autophagy. 2011;7(12):1573–1574. doi: 10.4161/auto.7.12.18175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H-Y, Han J, Cao SY, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia. Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. The Journal of Biological Chemistry. 2009;284(45):31484–31492. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nature Communications. 2013;4, article 1410 doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. The Journal of Biological Chemistry. 2012;287(48):40652–40660. doi: 10.1074/jbc.M112.419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krebiehl G, Ruckerbauer S, Burbulla LF, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0009367.e9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Frontiers in Physiology. 2013;4, article 95 doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circulation Research. 2012;111(9):1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 41.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circulation Research. 2003;93(4):292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 42.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiology of Aging. 2011;32(3):398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hafner AV, Dai J, Gomes AP, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2(12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D deficiency prevents diet-induced obesity in mice. FEBS Letters. 2011;585(4):677–682. doi: 10.1016/j.febslet.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 45.O’Shea KM, Khairallah RJ, Sparagna GC, et al. Dietary ω-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. Journal of Molecular and Cellular Cardiology. 2009;47(6):819–827. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6(3):118–125. doi: 10.1016/j.mito.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. The Journal of Biological Chemistry. 2002;277(30):26796–26803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.di Paola M, Zaccagnino P, Montedoro G, Cocco T, Lorusso M. Ceramide induces release of pro-apoptotic proteins from mitochondria by either a Ca2+-dependent or a Ca2+-independent mechanism. Journal of Bioenergetics and Biomembranes. 2004;36(2):165–170. doi: 10.1023/b:jobb.0000023619.97392.0c. [DOI] [PubMed] [Google Scholar]
- 49.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seet RCS, Lee C-YJ, Lim ECH, et al. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radical Biology and Medicine. 2010;48(4):560–566. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 51.Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer’s disease. Biochimica et Biophysica Acta. 2000;1502(1):139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 52.Popolo A, Autore G, Pinto A, Marzocco S. Oxidative stress in patients with cardiovascular disease and chronic renal failure. Free Radical Research. 2013;47(5):346–356. doi: 10.3109/10715762.2013.779373. [DOI] [PubMed] [Google Scholar]
- 53.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. The Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang S, Wang F, Shi X, et al. Association between manganese superoxide dismutase (MnSOD) Val-9Ala polymorphism and cancer risk—a meta-analysis. European Journal of Cancer. 2009;45(16):2874–2881. doi: 10.1016/j.ejca.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 55.Hong Z, Tian C, Zhang X. GPX1 gene Pro200Leu polymorphism, erythrocyte GPX activity, and cancer risk. Molecular Biology Reports. 2013;40(2):1801–1812. doi: 10.1007/s11033-012-2234-3. [DOI] [PubMed] [Google Scholar]
- 56.Chen J, Cao Q, Qin C, et al. GPx-1 polymorphism (rs1050450) contributes to tumor susceptibility: evidence from meta-analysis. Journal of Cancer Research and Clinical Oncology. 2011;137(10):1553–1561. doi: 10.1007/s00432-011-1033-x. [DOI] [PubMed] [Google Scholar]
- 57.Jin F, Xiong W-J, Jing J-C, Feng Z, Qu L-S, Shen X-Z. Evaluation of the association studies of single nucleotide polymorphisms and hepatocellular carcinoma: a systematic review. Journal of Cancer Research and Clinical Oncology. 2011;137(7):1095–1104. doi: 10.1007/s00432-010-0970-0. [DOI] [PubMed] [Google Scholar]
- 58.Tian C, Liu T, Fang S, Du X, Jia C. Association of C47T polymorphism in SOD2 gene with coronary artery disease: a case-control study and a meta-analysis. Molecular Biology Reports. 2012;39(5):5269–5276. doi: 10.1007/s11033-011-1324-y. [DOI] [PubMed] [Google Scholar]
- 59.Tian C, Fang S, Du X, Jia C. Association of the C47T polymorphism in SOD2 with diabetes mellitus and diabetic microvascular complications: a meta-analysis. Diabetologia. 2011;54(4):803–811. doi: 10.1007/s00125-010-2004-5. [DOI] [PubMed] [Google Scholar]
- 60.Tyurina YY, Winnica DE, Kapralova VI, Kapralov AA, Tyurin VA, Kagan VE. LC/MS characterization of rotenone induced cardiolipin oxidation in human lymphocytes: implications for mitochondrial dysfunction associated with Parkinson's disease. Molecular Nutrition & Food Research. 2013;57(8):1410–1422. doi: 10.1002/mnfr.201200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leuner K, Schütt T, Kurz C, et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxidants and Redox Signaling. 2012;16(12):1421–1433. doi: 10.1089/ars.2011.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yen H-C, Chen F-Y, Chen S-W, Huang Y-H, Chen Y-R, Chen C-W. Effect of mitochondrial dysfunction and oxidative stress on endogenous levels of coenzyme Q10 in human cells. Journal of Biochemical and Molecular Toxicology. 2011;25(5):280–289. doi: 10.1002/jbt.20387. [DOI] [PubMed] [Google Scholar]
- 63.Lu W, Chen Z, Zhang H, Wang Y, Luo Y, Huang P. ZNF143 transcription factor mediates cell survival through upregulation of the GPX1 activity in the mitochondrial respiratory dysfunction. Cell Death & Disease. 2012;3, article e422 doi: 10.1038/cddis.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez-Pinzon MA, Dave KR, Raval AP. Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxidants and Redox Signaling. 2005;7(9-10):1150–1157. doi: 10.1089/ars.2005.7.1150. [DOI] [PubMed] [Google Scholar]
- 65.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. The Journal of Biological Chemistry. 1998;273(29):18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 66.Tapia PC. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Medical Hypotheses. 2006;66(4):832–843. doi: 10.1016/j.mehy.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 67.Schmeisser S, Schmeisser K, Weimer S, et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell. 2013;12(3):508–517. doi: 10.1111/acel.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metabolism. 2007;6(4):280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 69.Bironaite D, Westberg JA, Andersson LC, Venalis A. A variety of mild stresses upregulate stanniocalcin-1 (STC-1) and induce mitohormesis in neural crest-derived cells. Journal of the Neurological Sciences. 2013;329(1-2):38–44. doi: 10.1016/j.jns.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 70.Yuyun X, Jinjun Q, Minfang X, et al. Effects of low concentrations of rotenone upon mitohormesis in SH-SY5Y cells. Dose Response. 2013;11(2):270–280. doi: 10.2203/dose-response.12-005.Gao. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of Biological Chemistry. 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang Y, Li W, Kong AN. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell & Bioscience. 2012;2(1, article 40) doi: 10.1186/2045-3701-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonello S, Zähringer C, BelAiba RS, et al. Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(4):755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 74.Kops GJPL, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 75.Tan W-Q, Wang K, Lv D-Y, Li P-F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. The Journal of Biological Chemistry. 2008;283(44):29730–29739. doi: 10.1074/jbc.M805514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jeong HJ, Jeong HW, Song SS, et al. Upregulation of peroxiredeoxin III in the hippocampus of acute immobilization stress model rats and the Foxo3a-dependent expression in PC12 cells. Cellular and Molecular Neurobiology. 2011;31(7):1041–1046. doi: 10.1007/s10571-011-9703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee H-C, Yin P-H, Lu C-Y, Chi C-W, Wei Y-H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochemical Journal. 2000;348, part 2:425–432. [PMC free article] [PubMed] [Google Scholar]
- 78.Lee H-C, Yin P-H, Chi C-W, Wei Y-H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. Journal of Biomedical Science. 2002;9(6, part 1):517–526. doi: 10.1007/BF02254978. [DOI] [PubMed] [Google Scholar]
- 79.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 80.Valle I, Álvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovascular Research. 2005;66(3):562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 81.Wright LE, Brandon AE, Hoy AJ, et al. Amelioration of lipid-induced insulin resistance in rat skeletal muscle by overexpression of Pgc-1β involves reductions in long-chain acyl-CoA levels and oxidative stress. Diabetologia. 2011;54(6):1417–1426. doi: 10.1007/s00125-011-2068-x. [DOI] [PubMed] [Google Scholar]
- 82.Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. The Journal of Biological Chemistry. 2006;281(1):324–333. doi: 10.1074/jbc.M508805200. [DOI] [PubMed] [Google Scholar]
- 83.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO Journal. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ungvari Z, Bailey-Downs L, Gautam T, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-kB activation in the nonhuman primate macaca mulatta. Journals of Gerontology A. 2011;66(8):866–875. doi: 10.1093/gerona/glr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Din S, Mason M, Völkers V, et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(15):5969–5974. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chou CH, Lin C, Yang M, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0049112.e49112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang DB, Garden GA, Kinoshita C, et al. Declines in Drp1 and parkin expression underlie DNA damage-induced changes in mitochondrial length and neuronal death. Journal of Neuroscience. 2013;33(4):1357–1365. doi: 10.1523/JNEUROSCI.3365-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicology Letters. 2013;218(1):30–38. doi: 10.1016/j.toxlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 90.Aerts AM, Zabrocki P, François IEJA, et al. Ydc1p ceramidase triggers organelle fragmentation, apoptosis and accelerated ageing in yeast. Cellular and Molecular Life Sciences. 2008;65(12):1933–1942. doi: 10.1007/s00018-008-8129-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parra V, Eisner V, Chiong M, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovascular Research. 2008;77(2):387–397. doi: 10.1093/cvr/cvm029. [DOI] [PubMed] [Google Scholar]
- 92.Fan X, Hussien R, Brooks GA. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radical Biology and Medicine. 2010;49(11):1646–1654. doi: 10.1016/j.freeradbiomed.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jheng H-F, Tsai P-J, Guo S-M, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Molecular and Cellular Biology. 2012;32(2):309–319. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galloway CA, Lee H, Nejjar S, et al. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. 2012;61(8):2093–2104. doi: 10.2337/db11-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frank M, Duvezin-Caubet S, Koob S, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochimica et Biophysica Acta. 2012;1823(12):2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 96.Dagda RK, Cherra SJ, III, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. The Journal of Biological Chemistry. 2009;284(20):13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guo X, Chen K-H, Guo Y, Liao H, Tang J, Xiao R-P. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circulation Research. 2007;101(11):1113–1122. doi: 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- 98.Kariya S, Sawada K, Kobayashi T, et al. Combination treatment of hydrogen peroxide and X-rays induces apoptosis in human prostate cancer PC-3 cells. International Journal of Radiation Oncology, Biology, Physics. 2009;75(2):449–454. doi: 10.1016/j.ijrobp.2009.04.092. [DOI] [PubMed] [Google Scholar]
- 99.Palermo V, Falcone C, Mazzoni C. Apoptosis and aging in mitochondrial morphology mutants of S. cerevisiae. Folia Microbiologica. 2007;52(5):479–483. doi: 10.1007/BF02932107. [DOI] [PubMed] [Google Scholar]
- 100.Peng L, Men X, Zhang W, et al. Dynamin-related protein 1 is implicated in endoplasmic reticulum stress-induced pancreatic β-cell apoptosis. International Journal of Molecular Medicine. 2011;28(2):161–169. doi: 10.3892/ijmm.2011.684. [DOI] [PubMed] [Google Scholar]
- 101.Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. Journal of Cell Science. 2010;123, part 6:917–926. doi: 10.1242/jcs.059246. [DOI] [PubMed] [Google Scholar]
- 102.DuBoff B, Gotz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75(4):618–632. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cartoni R, Léger B, Hock MB, et al. Mitofusins 1/2 and ERRα expression are increased in human skeletal muscle after physical exercise. Journal of Physiology. 2005;567, part 1:349–358. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism: a novel regulatory mechanism altered in obesity. The Journal of Biological Chemistry. 2003;278(19):17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 105.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics. 2005;6(5):389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environmental and Molecular Mutagenesis. 2010;51(5):440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 107.Indo HP, Davidson M, Yen H-C, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7(1-2):106–118. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 108.Cheng Y, Ren X, Gowda AS, et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death & Disease. 2013;4, article e731 doi: 10.1038/cddis.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yuzefovych LV, Solodushko VA, Wilson GL, Rachek LI. Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology. 2012;153(1):92–100. doi: 10.1210/en.2011-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN. Cardiac overexpression of 8-oxoguanine dna glycosylase 1 protects mitochondrial dna and reduces cardiac fibrosis following transaortic constriction. American Journal of Physiology—Heart and Circulatory Physiology. 2011;301(5):H2073–H2080. doi: 10.1152/ajpheart.00157.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Progress in Neurobiology. 2011;94(2):166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang Y, Zhang L, Zhang L, Bai J, Ge HY, Liu P. Expression changes in DNA repair enzymes and mitochondrial DNA damage in aging rat lens. Molecular Vision. 2010;16:1754–1763. [PMC free article] [PubMed] [Google Scholar]
- 113.Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13(1, article 253) doi: 10.1186/1471-2407-13-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bakthavatchalu V, Dey S, Xu Y, et al. Manganese superoxide dismutase is a mitochondrial fidelity protein that protects Polγ against UV-induced inactivation. Oncogene. 2012;31(17):2129–2139. doi: 10.1038/onc.2011.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bess AS, Crocker TL, Ryde IT, Meyer JN. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Research. 2012;40(16):7916–7931. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Radak Z, Atalay M, Jakus J, Boldogh I, Davies K, Goto S. Exercise improves import of 8-oxoguanine DNA glycosylase into the mitochondrial matrix of skeletal muscle and enhances the relative activity. Free Radical Biology and Medicine. 2009;46(2):238–243. doi: 10.1016/j.freeradbiomed.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cha M-Y, Han S-H, Son SM, et al. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034929.e34929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO Journal. 2002;21(17):4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial Unfolded Protein Response (mtUPR) and cognate promoter elements. PLoS ONE. 2007;2(9, article e874) doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell. 2007;13(4):467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 121.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174(1):229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular Cell. 2010;37(4):529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. Journal of Lipid Research. 2001;42(7):1007–1017. [PubMed] [Google Scholar]
- 124.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154(2):430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Raman AV, Chou VP, Atienza-Duyanen J, di Monte DA, Bellinger FP, Manning-Boğ AB. Evidence of oxidative stress in young and aged DJ-1-deficient mice. FEBS Letters. 2013;587(10):1562–1570. doi: 10.1016/j.febslet.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 127.Butler EK, Voigt A, Lutz AK, et al. The mitochondrial chaperone protein TRAP1 mitigates α-synuclein toxicity. PLoS Genetics. 2012;8(2) doi: 10.1371/journal.pgen.1002488.e1002488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(15):5981–5986. doi: 10.1073/pnas.1218778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ghosh JC, Siegelin MD, Dohi T, Altieri DC. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Research. 2010;70(22):8988–8993. doi: 10.1158/0008-5472.CAN-10-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hashimoto H, Niikura T, Tajima H, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(11):6336–6341. doi: 10.1073/pnas.101133498. Erratum in Proceedings of the National Academy of Sciences of the United States of America, vol. 98, no. 22, p. 12854, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tajima H, Niikura T, Hashimoto Y, et al. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neuroscience Letters. 2002;324(3):227–231. doi: 10.1016/s0304-3940(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 133.Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides? Trends in Endocrinology & Metabolism. 2013;24(5):222–228. doi: 10.1016/j.tem.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim KH, Jeong YT, Oh H, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature Medicine. 2013;19(1):83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 135.Rackham O, Shearwood A-MJ, Mercer TR, Davies SMK, Mattick JS, Filipovska A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA. 2011;17(12):2085–2093. doi: 10.1261/rna.029405.111. [DOI] [PMC free article] [PubMed] [Google Scholar]