

Abstract
Glycoproteins in focus
Metabolic labeling of azido sugars combined with two-photon fluorescence lifetime imaging microscopy enables the visualization of specific glycoforms of endogenous proteins. This method can be utilized to detect glycosylated proteins in both cell culture and intact human tissue slices.
Keywords: bioorthogonal chemistry, fluorescence, glycosylation, metabolic incorporation, prostate cancer
A cell surface protein’s glycosylation state can profoundly influence its biological capabilities and can report on the physiological state of the underlying cell.1–7 Thus, visualization of particular protein glycoforms is an important though challenging goal. Most protein-directed imaging methods (e.g. GFP tags, fluorescent antibodies) are not sensitive to a protein’s glycosylation state. Our lab has previously developed a method for imaging certain glycan structures on native glycoproteins by metabolic labeling with functionalized sugars. But, this glycan-targeted imaging method cannot reveal the identities of proteins to which the labeled glycans are attached.8–10 Imaging of a specific protein glycoform will require integration of both the protein and the glycan’s identities.
Other groups have recently made strides towards this goal. Söderberg and coworkers used proximity ligation to detect a specific glycoform of the tumor maker MUC2.11 More recently, Haga et al. used azido sugar labeling of GFP-tagged proteins to image cell surface glycoproteins by Förster Resonance Energy Transfer (FRET) fluorescence microscopy.12 Due to their reliance on GFP-tagged proteins however, this method cannot image endogenous glycoproteins or proteins that are not amenable to fluorescent protein fusion. Even so, there are some limitations to a traditional FRET-based technique. The distance between the donor and acceptor fluorophores in a FRET experiment is related to the efficiency of energy transfer and typically precludes the use of two large macromolecules, such as IgGs (>10 nm).13 Another compounding factor for imaging specific protein glycoforms is the discrepancy between protein copy number and glycan abundance. The difference in abundance between common types of glycans and a specific protein can be orders of magnitude on the cell surface.14 This large difference in relative number can complicate analyses in imaging applications. For example, in a typical FRET-based experiment, the donor fluorophore is excited and emission from the acceptor fluorophore is monitored. In the case of high acceptor fluorophore concentration, acceptor bleedthrough can occur causing a false positive FRET signal (SI Figure 1).
Herein, we present a new approach to image endogenous protein glycoforms using a combination of azido sugar labeling and 2-photon fluorescence lifetime imaging microscopy (FLIM). We rely on a small (<7 nm) targeting moiety, an Fab fragment, to introduce the donor fluorophore and locate the protein component. We applied our previously developed glycan labeling strategy to introduce the acceptor fluorophore. In this scheme, cells were first incubated with an azido sugar, peracetylated N-azidoacetylmannosamine (Ac4ManNAz), which is processed by the cellular machinery and incorporated into glycoproteins as azido sialic acid (SiaNAz). Subsequent bioorthogonal reaction with a cyclooctyne-fluorophore15,16 conjugate delivers the acceptor fluorophore within minimal distance (Figure 1).
Figure 1.
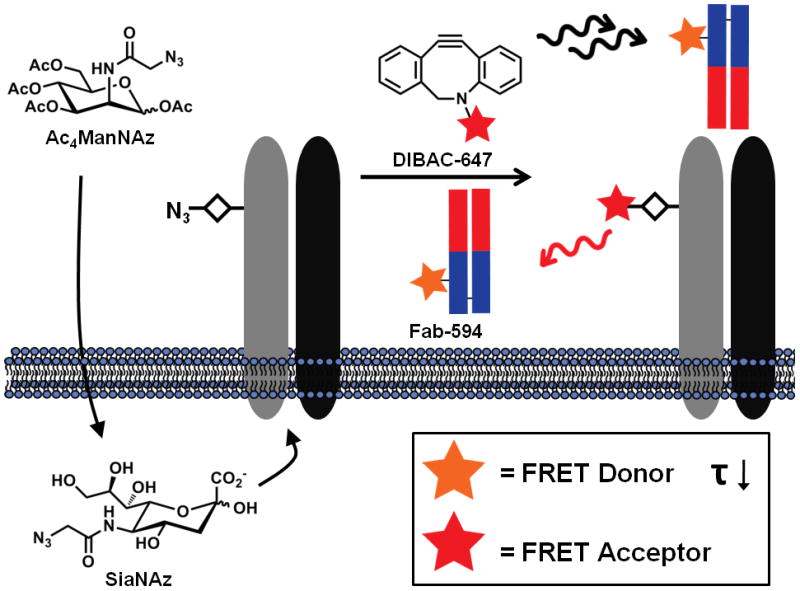
A technique for imaging the glycosylation state of glycoproteins through the use of metabolic labeling and 2-photon fluorescence lifetime imaging microscopy (FLIM). Cells or tissues are fed the azido sugar, Ac4ManNAz, which is metabolized to SiaNAz and SiaNAz is then incorporated in sialylated glycoproteins. The azide functionality is reacted with an appropriate bioorthogonal probe bearing an acceptor fluorophore to report on the localization of sialic acid residues. In a subsequent step, the protein scaffold of the glycoprotein is targeted with the use of a donor fluorophore-labeled Fab fragment. The presence of both the FRET donor and acceptor fluorophores on a specific glycoprotein is monitored by changes in the fluorescence lifetime (τ) of the donor after two-photon excitation on a per pixel basis.
A common method for circumventing acceptor bleedthrough is to focus on the donor fluorophore’s emission in a FRET experiment. Energy transfer between the donor and acceptor fluorophore results in two major changes to the donor’s physical properties. The first is reduction in emission from the donor. Imaging this photon reduction in a population of cells requires normalization by photobleaching of the acceptor to reveal the maximum amount of donor emission, a difficult and tedious task when the field of view contains numerous cells. The other change for the donor fluorophore upon energy transfer is a decrease in fluorescence lifetime.17 This time-dependent property is advantageous since no further experimentation or sample manipulation is necessary. We sought to utilize the decrease in fluorescence lifetime of the donor fluorophore associated with FRET to monitor the sialylation state of a given glycoprotein through 2-photon FLIM.
Overexpression of the integrin αVβ3 subtype is observed in a variety of cancers and is often correlated with invasiveness due to its pro-angiogenic function.18 Integrin αVβ3 possesses four reported and nine potential N-glycosylation sites on the α subunit and two reported and four potential sites on the β subunit. Recently, several reports have suggested that integrin αVβ3’s glycosylation state can dramatically alter its migratory and angiogenic functions. In two separate reports, sialylation of integrin αVβ3 was found to be required for cell proliferation, migration, and regeneration in wound healing assays.19,20 Panjwani and coworkers have shown that αVβ3’s glycosylation state also affects VEGF- and bFGF-mediated angiogenesis through an interaction with galectin-3.21 Motivated by the importance of αVβ3’s glycosylation state, we chose to pursue this integrin as a proof-of-concept glycoprotein for imaging its sialylation status.
We first investigated whether endogenous integrin αVβ3 is sialylated in the U87MG glioblastoma cell line, which is known to express the heterodimer complex at high levels.22 αVβ3 was immunoprecipitated from U87MG lysate, and the presence of sialic acid on both subunits was assessed by lectin blotting. α2,3- and α2,6-linked sialosides can be distinguished by blotting with Maackia amurensis agglutinin (MAA) and Sambucus nigra lectins (SNA), respectively. After immunoprecipitation, we observed MAA binding to both integrin subunits, suggesting they are sialylated with α2,3-linked residues (Figure 2A).
Figure 2.
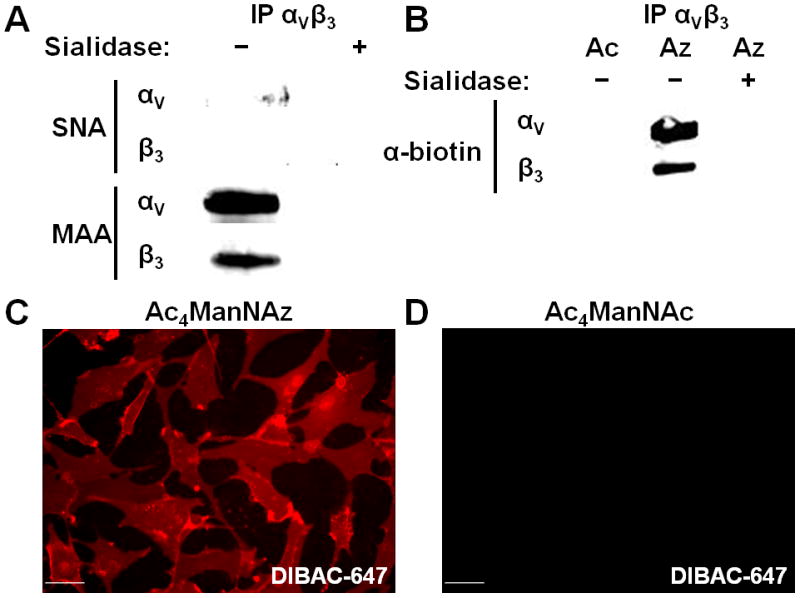
Incorporation of SiaNAz into integrin αVβ3 and U87MG cell surface glycoconjugates. (A) Lectin blot of α and β subunits of immunoprecipitated αVβ3 with α2,6 (SNA) and α2,3 (MAA) sialic acid binding lectins in the presence or absence of sialidase. (B) Reactivity of αVβ3 subunits with phosphine-biotin. Cells were fed either Ac4ManNAz (Az) or Ac4ManNAc (Ac) for 3 days and incubated with phosphine-biotin, a probe for the presence of azides, in the presence or absence of sialidase. Fluorescence microscopy of U87MG cells treated with either Ac4ManNAz (C) or Ac4ManNAc (D) and then DIBAC-647 (C,D). Scale bar: 50 μm.
Our FRET scheme is predicated on SiaNAz incorporation into the N-glycans of integrin αVβ3 (Figure 1). To confirm this, we supplemented cells with either Ac4ManNAz or Ac4ManNAc for 3 days. We then incubated cells with a biotin-conjugated phosphine, a bioorthogonal probe for the azido functionality.9 Cell lysates were subjected to immunoprecipitation with an anti-integrin αVβ3 antibody and SiaNAz was detected by Western blotting. Azide-dependent labeling was observed for both the alpha and beta chains of integrin αVβ3 (Figure 2B), suggesting that SiaNAz is present on both subunits of the heterodimer. We also confirmed that the azide-specific signal was indeed due to SiaNAz residues by digesting the lysates with sialidase and observed reduced signal on both integrin subunits (Figure 2B).
We next turned our attention to the selection of the proper FRET pair for the dual labeling strategy. Neuronal cell lines, such as U87MG, are known to exhibit strong flavin-and riboflavin-associated autofluorescence centered at 540–550 nm.23 To avoid cellular autofluorescence, we focused on donor fluorophores with emission above 600 nm. We ultimately chose the Alexa Fluor 594 (594) and Alexa Fluor 647 (647) fluorophore combination since Alexa Fluor 594’s emission maximum is 617 nm, and the Förster radius for the pair is 8.5 nm (see Supporting Information).
Preparation of the donor Fab fragment conjugate commenced by treating the monoclonal antibody against integrin αVβ3 (LM609 clone) with a peptidase that cleaves the full length IgG between the Fab and Fc segments. The Fab fragment was purified from Fc and unreacted IgG by incubation with protein A resin, and in a subsequent step the lysine residues were reacted with Alexa Fluor 594 N-hydroxy succinimidyl ester to furnish the Fab-594 conjugate (see Supporting Information). The acceptor cyclooctyne was also synthesized with commercially available reagents, dibenzoazacyclooctyne (DIBAC) amine and Alexa Fluor 647 N-hydroxy succinimidyl ester (see Supporting Information). The ease of construction of both the donor, Fab-594, and the acceptor, DIBAC-647, targeting moieties underscores the versatility of this method and its applicability to other glycoprotein targets.
After treatment of U87MG cells with Fab-594, we observed specific cellular labeling of membrane-associated αVβ3 that could be inhibited by the presence of full-length anti-αVβ3 IgG (SI Figure 2). We also sought to determine if DIBAC-647 was a faithful bioorthogonal probe for azides. Towards this end, we fed cells either Ac4ManNAz or Ac4ManNAc for 3 days and then incubated them with DIBAC-647. Cell surface labeling occurred only in cells administered Ac4ManNAz (Figure 2C,D).
After confirming the labeling of αVβ3’s protein backbone with the FRET donor Fab-594 and SiaNAz residues with the FRET acceptor DIBAC-647, we focused on monitoring FRET using two-photon FLIM. The two-photon FLIM FRET technique entails exciting the donor fluorophore using near-IR/IR femtosecond pulses and monitoring the time-dependent fluorescence decay in the nanosecond regime.17 In the case when an acceptor fluorophore is in close proximity to the donor fluorophore, the excited state of the donor is depleted via energy transfer, which ultimately results in the reduced fluorescence lifetime (τ) of the donor molecule. We observed that Fab-594 had an in vitro characteristic τ of 3.09 ns, which was determined by fitting Fab-594’s fluorescence decay to a single exponential (SI Figure 3). In FLIM, the fluorescence emission for each pixel of an image is monitored in a time-dependent manner. The emission data are then fitted to extract the τ values, and these are displayed as a heat-map image.17
FLIM images were obtained by incubating U87MG cells with either Ac4ManNAz or Ac4ManNAc for 3 days and then labeling cells with DIBAC-647 and Fab-594, sequentially. Cells that were treated with Ac4ManNAc had an average τ of 2.91 ns (Figure 3A). The cell surface regions had a relatively constant τ as represented by the deep blue color in the image in 3A. On the other hand, in cells treated with Ac4ManNAz, the average τ decreased to 2.60 ns (Figure 3B), which is consistent with cell surface FRET24 between the Fab-594 and DIBAC-647; the cell surface τ appeared far more heterogeneous than in 3A as evident by green and yellow clusters. Collectively, these data indicate that cell surface integrin αVβ3 is sialylated on U87MG cells. We further analyzed the τ values by constructing a histogram of cell surface τ for both Ac4ManNAc and Ac4ManNAz cell populations (Figure 3C). The decrease in τ values from cells labeled with only the Fab-594 donor versus cells with both the Fab-594 donor and DIBAC-647 acceptor was also accompanied by a switch from a unimodal (red) to a multimodal distribution (blue). These changes may reflect variations in the amount of sialylation on individual integrin αVβ3 molecules. As well, cells lacking Fab-594 but labeled with the DIBAC-647 acceptor displayed minimal signal (Figure 4A).
Figure 3.
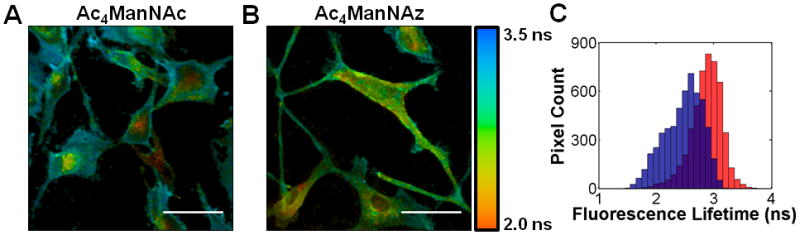
FLIM imaging of sialylated integrin αVβ3. 2-Photon FLIM images (A,B) of U87MG cells treated with either Ac4ManNAc (A) or Ac4ManNAz (B), followed by DIBAC-647 and Fab-594. FLIM images are the heat maps of τ values determined per pixel, see accompanying legend (right). (C) Histogram of cell surface τ values for cells treated with either Ac4ManNAc (red bars) or Ac4ManNAz (blue bars) and then incubated with DIBAC-647 and Fab-594. Scale bar: 50 μm.
Figure 4.
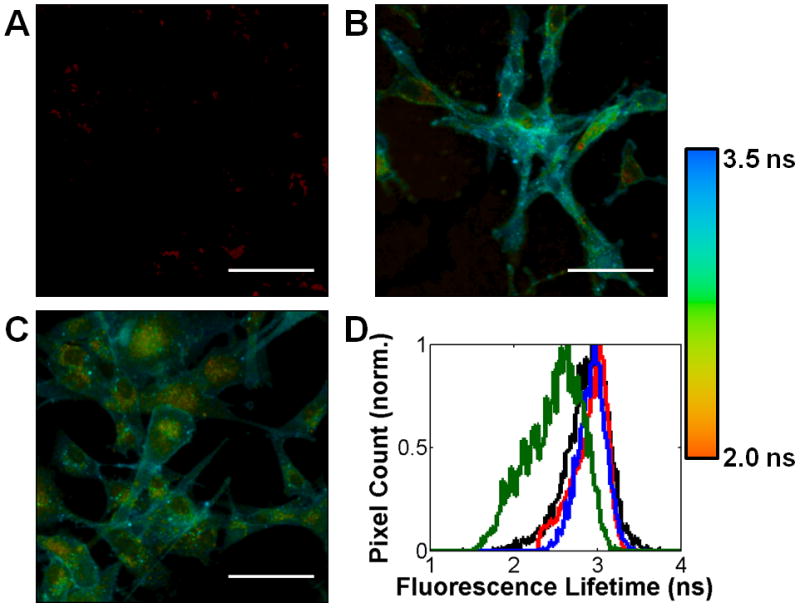
FLIM FRET is dependent on the proximity of the acceptor dye to Fab-594 and the target protein’s glycosylation state. FLIM images of (A) Ac4ManNAz-fed U87MG cells treated with DIBAC-647 alone, (B) U87MG cells modified with DPPE-647 and then incubated with Fab-594 and (C) Ac4ManNAz-fed U87MG cells first treated with sialidase then followed by incubation with DIBAC-647 and Fab-594. FLIM images are the heat maps of τ values determined per pixel, see accompanying legend (right). (D) Histogram of cell surface τ values for cells treated with either Ac4ManNAc (black line), DPPE-647 (red line), Ac4ManNAz and sialidase (blue line), or Ac4ManNAz (green) and then incubated with DIBAC-647 (black, blue, and green) and Fab-594 (black, red, blue, and green). Scale bar: 50 μm.
We further sought to evaluate whether the observed FRET arose from SiaNAz residues on integrin αVβ3 itself or alternatively, from nearby sialic acid-modified glycoproteins or glycolipids. We reasoned that by adding an acceptor fluorophore to cell surfaces non-specifically, while guaranteeing that the acceptor moiety was not bound to our integrin-of-interest, we could differentiate between sialylated integrin-dependent FRET and background FRET. Cells were first incubated with dipalmitoyl phosphatidylethanolamine (DPPE)-647, which incorporates the acceptor into cell membranes uniformly, and then followed with Fab-594 labeling. No decrease in τ values was observed in this case as cell surfaces appeared to maintain a similar hue to cells that only possess the Fab-594 fragment (Figure 4B,D). Thus, we reasoned that sialic acid residues on nearby proteins or lipids do not influence the FRET signal originating from αVβ3 complexes. We also tested whether the observed FRET was a sialylation state-dependent phenomenon. We treated Ac4ManNAz-fed U87MG cells with a sialidase to cleave SiaNAz residues from cell surface glycoconjugates. The cells were further reacted with DIBAC-647 and incubated with Fab-594. Sialidase treatment abolished the decrease in τ (Figure 4C,D), indicating that SiaNAz residues on integrin αVβ3 were critical for the observed FRET.
A unique attribute of this platform is that it can monitor the glycosylation state of endogeneous glycoproteins as well as image deep within a specimen, owing to the two-photon mode of excitation.
We have previously shown that human prostate adenocarinoma tissue slices can metabolically incorporate azido sugars into their glycoproteins.25 Prostate cancers are known to undergo an upregulation in integrin αVβ3,26 which provided us an opportunity to apply our imaging technique to human tissue slices. Grade 3–4 precision-cut adenocarinoma tissue slices derived from an 8-mm core of a radical prostatectomy were cultured for 3 days in either Ac4ManNAz or Ac4ManNAc. We first determined that αVβ3 immunoprecipitated from tissue lysate was recognized by MAA (SI Figure 5A), similar to αVβ3 derived from U87MG lysate, and incorporated SiaNAz residues into their glycans (SI Figure 5B). Using fluorescence microscopy, Fab-594 and DIBAC-647 were found to label prostate tissue slices in a αVβ3-(SI Figure 6) and azide-dependant (SI Figure 7) manner, respectively. We next applied our FLIM FRET strategy to the tissue slices by first staining with DIBAC-647 and then Fab-594. Ac4ManNAc-treated tissue slices displayed the characteristic Fab-594 τ (Figure 5A,D), as evident by the blue color. The Ac4ManNAz-treated tissue slices had a decreased τ (Figure 5B,D) that could be abrogated by treatment with sialidase (Figure 5C,D), again consistent with FRET and the presence of sialylated integrin αVβ3 embedded within the tissue slices. These data show that we can probe protein glycosylation with increasing molecular precision.
Figure 5.
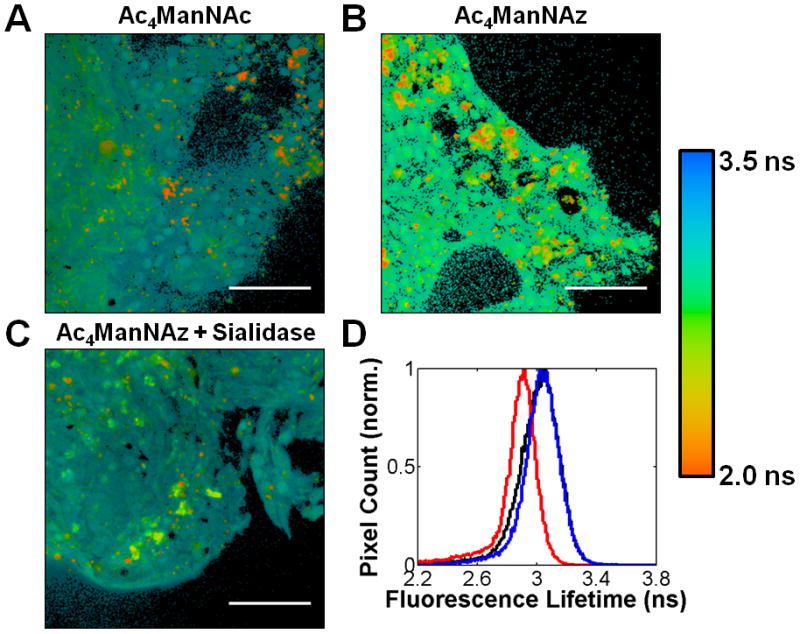
FLIM images and lifetime histogram of grade 3–4 human prostate adenocarinoma tissue slices. Tissue slices were incubated with either Ac4ManNAc (A), Ac4ManNAz (B), or Ac4ManNAz and sialidase (C) then treated with DIBAC-647 and Fab-594. FLIM images are the heat maps of τ values determined per pixel, see accompanying legend (right). (D) Histogram of τ values for tissues treated with either Ac4ManNAc (black line), Ac4ManNAz and sialidase (blue line), or Ac4ManNAz (red) and then incubated with DIBAC-647 and Fab-594. Scale bar: 50 μm.
In conclusion, we have developed a FLIM FRET-based method for visualizing the glycosylation state of specific glycoproteins. The method overcomes intrinsic limits of classic FRET imaging, which can be undermined by bleedthrough. As well, we can now image discrete protein glycoforms in whole tissue slices. The method therefore adds to a growing toolkit for characterizing cell surface glycomes with molecular precision. We plan on utilizing this technique in the future for imaging the cognate cell surface ligands for secreted lectins as well as for diagnostic purposes in monitoring the glycosylation status of specific glycoproteins.
Supplementary Material
Acknowledgments
We thank Jason Hudak and Geoffrey O’Donoghue for helpful discussions and for critical reading of the manuscript and Holly Aaron (UC Berkeley Molecular Imaging Center) for technical assistance. This work was funded by grants from the NIH to C.R.B. (GM066047 and GM058867), to S.L.M. (2T32DK007217-37), and to A.dlZ. (1DP5OD012179-01). B.B. and D.R.S. were supported by NSF predoctoral fellowships. A.dlZ. was supported by the Damon Runyon Cancer Research Foundation (DFS-#06-13) and was a Damon Runyon Fellow (DRG-2094-11).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Brian Belardi, Departments of Chemistry and Molecular and Cell Biology, Howard Hughes Medical Institute, University of California, Berkeley, CA 94720 (USA).
Prof. Adam de la Zerda, Departments of Chemistry and Molecular and Cell Biology, Howard Hughes Medical Institute, University of California, Berkeley, CA 94720 (USA). Department of Structural Biology, Stanford University School of Medicine, Stanford, CA 94305 (USA)
David R. Spiciarich, Departments of Chemistry and Molecular and Cell Biology, Howard Hughes Medical Institute, University of California, Berkeley, CA 94720 (USA)
Sophia L. Maund, Department of Urology, Stanford University School of Medicine, Stanford, CA 94305 (USA)
Prof. Donna M. Peehl, Department of Urology, Stanford University School of Medicine, Stanford, CA 94305 (USA)
Prof. Carolyn R. Bertozzi, Email: crb@berkeley.edu, Departments of Chemistry and Molecular and Cell Biology, Howard Hughes Medical Institute, University of California, Berkeley, CA 94720 (USA)
References
- 1.Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, et al. Proc Natl Acad Sci. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 3.Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 4.Cha S-K, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang C-L. Proc Natl Acad Sci. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 6.Marth JD, Grewal PK. Nat Rev Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 8.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 9.Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 10.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conze T, Carvalho AS, Landegren U, Almeida R, Reis CA, David L, Söderberg O. Glycobiology. 2010;20:199–206. doi: 10.1093/glycob/cwp161. [DOI] [PubMed] [Google Scholar]
- 12.Haga Y, Ishii K, Hibino K, Sako Y, Ito Y, Taniguchi N, Suzuki T. Nat Commun. 2012;3:907. doi: 10.1038/ncomms1906. [DOI] [PubMed] [Google Scholar]
- 13.Galbraith CG, Galbraith JA. J Cell Sci. 2011;124:1607–1611. doi: 10.1242/jcs.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor N.Y: 2008. [PubMed] [Google Scholar]
- 15.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 16.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levitt JA, Matthews DR, Ameer-Beg SM, Suhling K. Curr Opin Biotechnol. 2009;20:28–36. doi: 10.1016/j.copbio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Meyer A, Auernheimer J, Modlinger A, Kessler H. Curr Pharm Des. 2006;12:2723–2747. doi: 10.2174/138161206777947740. [DOI] [PubMed] [Google Scholar]
- 19.Kremser ME, Przybyło M, Hoja-Łukowicz D, Pocheć E, Amoresano A, Carpentieri A, Bubka M, Lityńska A. Biochim Biophys Acta. 2008;1780:1421–1431. doi: 10.1016/j.bbagen.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Chiodelli P, Urbinati C, Mitola S, Tanghetti E, Rusnati M. J Biol Chem. 2012;287:20456–20466. doi: 10.1074/jbc.M111.337139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markowska AI, Liu FT, Panjwani N. J Exp Med. 2010;207:1981–1993. doi: 10.1084/jem.20090121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 23.Benson RC, Meyer RA, Zaruba ME, McKhann GM. J Histochem Cytochem. 1979;27:44–48. doi: 10.1177/27.1.438504. [DOI] [PubMed] [Google Scholar]
- 24.Belardi B, O’Donoghue GP, Smith AW, Groves JT, Bertozzi CR. J Am Chem Soc. 2012;134:9549–9552. doi: 10.1021/ja301694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubbard SC, Boyce M, McVaugh CT, Peehl DM, Bertozzi CR. Bioorg Med Chem Lett. 2011;21:4945–4950. doi: 10.1016/j.bmcl.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goel HL, Li J, Kogan S, Languino LR. Endocr Relat Cancer. 2008;15:657–664. doi: 10.1677/ERC-08-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.