

Statins are considered very effective in reducing cardiovascular morbidity and mortality in high-risk patients. However, although adherence to statins improves morbidity and mortality (1), it remains suboptimal (2). One of the most important causes of nonadherence is the so-called statin intolerance, mainly because of muscle-related symptoms. These symptoms most often consist of myalgia unaccompanied by significant creatine kinase (CK) elevations. Less often, myositis (elevated CK >10 times the upper limit of normal) or rhabdomyolysis (CK level >10,000 IU/L or accompanied by significant elevation in creatinine level) develops.
In randomized controlled trials, the incidence of statin myopathy is ~1.5–5.0% (3). However, this low incidence may be misleading for several reasons. First, in most studies patients with a history of statin intolerance were excluded. Other studies had a single-blinded statin run-in phase, and patients experiencing muscle-related symptoms or CK elevations during this phase were excluded. Patients who tend to be at risk for developing muscle-related symptoms, such as women, elderly patients, and patients with significant comorbidity, who comprise a large proportion of statin-treated patients in real-life settings, are underrepresented in randomized controlled trials. Some studies have defined muscle-related effects by elevated CK levels only, disregarding myalgia. Last but not least, patients enrolled in studies might be motivated and so minimize reporting of mild myalgias, thus leading to underestimation of the magnitude of the problem.
Data concerning real-life incidence of statin-related myopathy are scarce. In the Prediction of Muscular Risk in Observational Conditions (PRIMO) study (4), 7,924 patients receiving high-dosage statin therapy in an outpatient setting in France were asked about muscle-related symptoms. Overall, muscular symptoms were reported by 10.5% of the patients. A weakness of this study is that it lacked a comparison/control group not treated with statins. In a study of adults aged ≥40 years who participated in National Health and Nutrition Examination Survey 1999–2004, the unadjusted prevalence of musculoskeletal pain was significantly higher for statin users (23 vs. 18% among those not using statins, P = 0.02) (5). After confounders were controlled for, among those without arthritis statin use was associated with a significantly higher prevalence of musculoskeletal pain in any region (adjusted prevalence ratios 1.33 [95% CI 1.06–1.67]). This study highlights the difficulty of relating myalgia to statin use in a relatively frequent occurrence of such symptoms in the population.
This review will deal with the possible mechanisms of muscle-related symptoms associated with statins and the ways to manage them. Other side effects of statins, such as hepatotoxicity, hemorrhagic stroke, cognitive decline, peripheral neuropathy, diabetes, insomnia, tendinitis, arthralgia, arthritis, cataract, etc., are beyond the scope of this review and have previously been described (6). Statins can produce two distinct forms of myotoxicity: toxic and immune mediated.
TOXIC MYOPATHY
The precise mechanisms underlying toxic statin myopathy are unknown, but several hypotheses have been suggested. These effects may be aggravated by conditions that increase statin blood level, such as concomitant medications interfering with statin metabolism via inhibition of CYP3A4, CYP2C9, glucuronidation, or other processes (7).
Genetic factors
In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) study, a genome-wide scan yielded a strong association of simvastatin-associated myopathy with the rs4363656 single nucleotide polymorphism located within SLCO1B1 on chromosome 12 (8). This association was replicated in the Heart Protection Study. The odds ratios for the development of simvastatin-induced myopathy were 4.5 and 16.9 for heterozygous and homozygous C allele transitions at this single nucleotide polymorphism. SLCO1B1 encodes the organic anion–transporting polypeptide (OATP)1B1, which has been shown to regulate the hepatic uptake of statins. Since all statins require hepatic transporters for their transmembrane flux, it was postulated that polymorphisms in this gene would affect serum levels of all statins and thus the risk for myopathy. However, in one relatively small study, SLCO1B1 genotypes, which were associated with simvastatin-induced myopathy in SEARCH, were not associated with atorvastatin-induced musculoskeletal effects (9). Thus, it seems that the importance of the SLCO1B1 genotypes depends on the specific statin used. Hydrophilic statins are thought to be actively transported into hepatocytes by expressing OATP, whereas lipophilic statins diffuse nonselectively into extrahepatic tissues such as muscle (10). A recent study demonstrated that expression of human OATP2B1 in human skeletal muscle myoblast cells by adenoviral vectors increased intracellular accumulation and toxicity of statins in an in vitro model (11).
In this regard, it is interesting to note that in the PRIMO study, the most hydrophilic statins (pravastatin and fluvastatin) were least likely to cause myalgia, whereas simvastatin, the most lipophilic one, was most likely to be associated with muscular adverse effects (4). Cerivastatin, the most lipophilic statin, was removed from the market owing to an unacceptably high rate of rhabdomyolysis. Details about the lipophilicity, potency, metabolism, and drug interactions of the various statins are provided in Table 1.
Table 1.
Selected properties of various statins
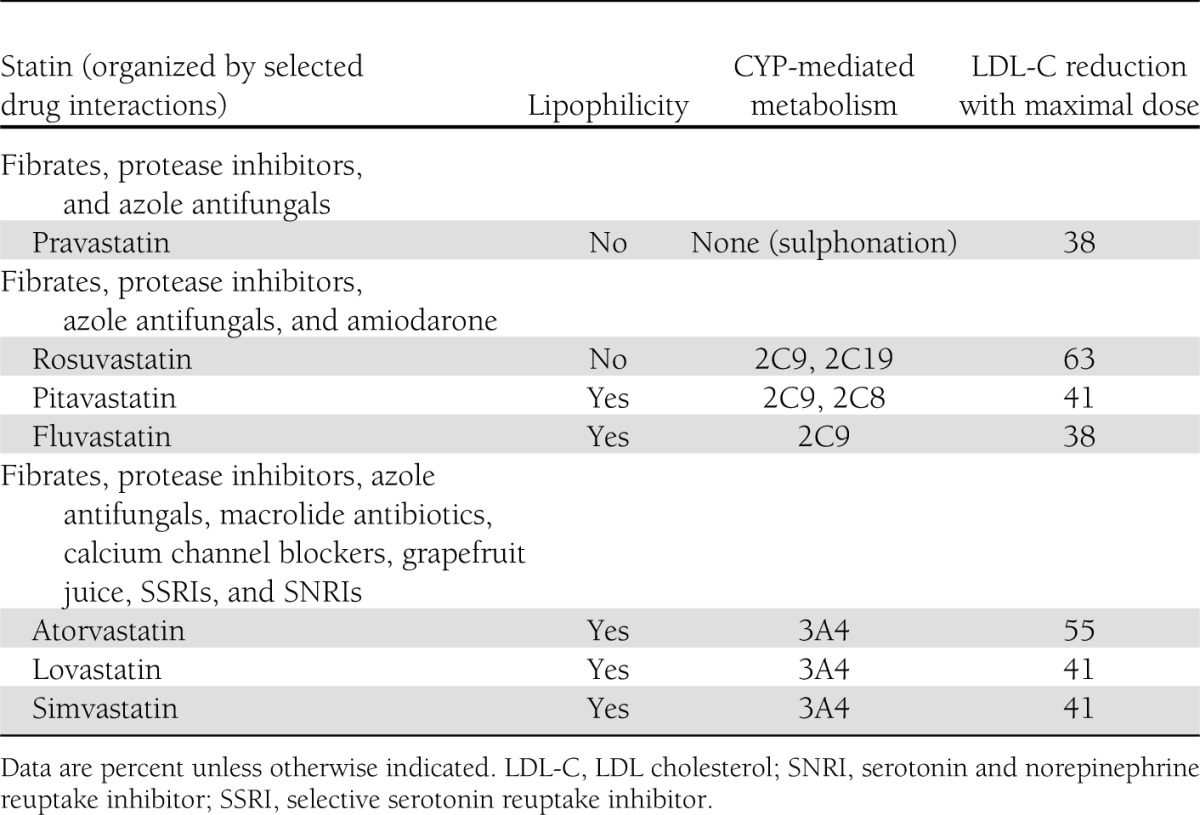
Other genetic factors implicated in statin-induced myopathy include sequence alterations (polymorphisms or mutations) in genes encoding various cytochrome P450 isoenzymes, coenzyme Q, myophosphorlyase, carnitine palmitoyl transferase 2, myoadenylate deaminase, ATP-binding cassette sub-family B (MDR/TAP), member 1 (ABCB1), ATP-binding cassette sub-family G member 2 (ABCG2), the efflux transporter multidrug resistance protein 1, and others, although data concerning these genes are not as compelling as data concerning SLCO1B1 (12,13).
Metabolic effects
Mitochondrial dysfunction and depletion of coenzyme Q10.
There are several reports of statins unmasking a previously undiagnosed mitochondrial pathology (14). However, in most cases no such preexisting pathology exists.
Muscle biopsies from four patients with statin-associated myopathy and normal CK levels demonstrated findings consistent with mitochondrial dysfunction, including increased intramuscular lipid, diminished cytochrome oxidase staining, and ragged red fibers. Three of these patients had repeat biopsies performed after discontinuation of the statin, which showed resolution of the pathologic abnormalities (15). In another study, significantly decreased mitochondrial DNA levels were found in skeletal muscle biopsies taken from patients treated with 80 mg/day simvastatin for 8 weeks but not in those treated with 40 mg/day atorvastatin (16). However, another study demonstrated that muscle structure was essentially normal in 14 of 18 patients with statin-induced increased CK levels (17).
Using respiratory exchange ratios during exercise as an indirect measure of mitochondrial function, several small studies have suggested the possibility of statin-induced mitochondrial dysfunction during exercise (18). Other studies, testing mitochondrial function by the activity of complex III of the mitochondrial respiratory chain (17) or by directly measuring concentrations of high-energy phosphates (19), found no changes in statin-treated patients, suggesting that mitochondrial function was not compromised. These studies were performed at rest, however, and may not reflect mitochondrial function during exercise.
These and other studies have raised the concern that statins may also impair mitochondrial function in cardiomyocytes and thus be cardiotoxic. However, one study demonstrated a differential effect of statins on cardiac and skeletal muscle. In human atrial cells, statins triggered transcriptional activation of mitochondrial biogenesis, probably by enhancing antioxidant capacity, whereas in skeletal muscle statins induced high oxidative stress responsible for transcriptional deactivation of mitochondrial biogenesis as well as mitochondrial dysfunction (20). Thus, statins may have beneficial effects on cardiomyocytes while having a deleterious effect on skeletal muscle.
If indeed statins cause mitochondrial dysfunction, a plausible mediator of this effect is CoQ10. CoQ10, or ubiquinone, is an end product of the mevalonate pathway, which is blocked by statins. CoQ10 is a component of the mitochondrial electron transport system. It participates in electron transport during oxidative phosphorylation in mitochondria. A reduction in CoQ10 could cause an abnormal mitochondrial respiratory function and result in mitochondrial dysfunction and myopathy.
Most studies show that statins lower serum CoQ10 levels (21). However, since CoQ10 in the serum is mainly carried by lipoproteins, mostly LDL, most of the serum reduction of CoQ10 by statins is caused simply by lowering LDL. There are few small studies of the effect of statins on CoQ10 levels in muscle. These studies mostly show that statin treatment does not appear to reduce intramuscular CoQ10 concentrations, including in subjects with symptomatic statin-induced myopathy (18).
Two small studies have tested the effect of CoQ10 supplementation on statin-induced myopathy with conflicting results. In one study, 44 simvastatin-treated patients were randomized to 200 mg/day CoQ10 or placebo for 12 weeks. No difference in myalgia score was found between the two groups (22). In another study, 32 statin-treated patients with myopathic symptoms were treated with either 100 mg/day CoQ10 or 400 IU/day vitamin E. A decrease of 40% in pain severity and 38% in pain interference with daily activities was found in the CoQ10 group, while there was no difference in the vitamin E group (23).
Lowering of farnesyl pyrophosphate and geranylgeranyl pyrophosphate.
Farnesyl pyrophosphate and geranylgeranyl pyrophosphate are other end products of the mevalonate pathway. They normally activate regulatory guanosine 5′-triphosphate–binding proteins, which promote cell maintenance and growth and diminish apoptosis. Reduction in their level was suggested as another possible mechanism for statin-induced myopathy.
Application of statins to human muscle cells has been shown to trigger apoptosis. This effect was not found when squalene epoxidase or squalene synthase (more downstream steps in cholesterol synthesis) were specifically inhibited, suggesting that this effect was not the result of a reduction in cholesterol content but was, rather, the result of the reduction in isoprenoid compounds (24).
In one in vitro study, statins caused autophagy in human rhabdomyosarcoma A204 cells. This effect paralleled inhibition of hydroxymethylglutaryl (HMG)-CoA reductase and was prevented by mevalonate and polyisoprenoid diphosphates, suggesting that statins induce autophagy by depleting cellular levels of geranylgeranyl diphosphate (25). Again, this effect was observed only with lipophilic statins, probably owing to a different transport system as previously mentioned. Another study found statin-induced morphologic changes and inhibition of protein synthesis in cultured rat skeletal muscle cells that were significantly ameliorated by supplementation with farnesol and geranylgeraniol. A squalene synthase inhibitor, blocking the synthesis of cholesterol but not of other end products of the mevalonate pathway, had no significant toxic effect, thus suggesting that depletion of metabolites of geranylgeranyl pyrophosphate, and not inhibition of cholesterol synthesis, was the primary cause of statin-induced myopathy (26).
Reduction of cholesterol content of skeletal muscle membranes.
Cholesterol is a key component of the structure and function of cell membranes. Statins seem to modulate membrane cholesterol in different tissues, including skeletal muscle (27).
Reduction of the cholesterol content of skeletal muscle cell membranes might make them unstable (28). A change in membrane fluidity may affect different ion channels, such as sodium, potassium, and chloride channels, and thus modify muscle membrane excitability. For example, a dose-dependent reduction of membrane chloride conductance was recorded in muscle fibers of simvastatin-treated rats. Again, the hydrophilic pravastatin had no such effect (29). However, since, as previously mentioned, myotoxicity does not occur in vitro when cholesterol is lowered by inhibiting squalene synthetase, this mechanism seems less plausible.
Impaired calcium signaling.
Statins may cause myopathy via impairing calcium signaling. Simvastatin has been shown to trigger mitochondrial depolarization and calcium efflux (through the permeability transition pore and sodium-calcium exchanger). Statins have been shown to trigger a massive calcium release from the sarcoplamsic reticulum via ryanodine receptors. In addition, simvastatin induced long-lasting fura-2 calcium transients in human skeletal muscle that led to activation of calpain and caspaces 3 and 9. Calcium chelation and ryanodine, via inhibition of calcium-induced calcium release, have been shown to abrogate these effects. This may be caused by impairment in the mitochondrial respiratory chain resulting in an inner-membrane depolarization and calcium extrusion by permeability transient pore and Na+/Ca2+ exchanger. These may lead first to an elevated cytoplasmic calcium concentration and consequently to sarcoplasmic reticulum calcium overload resulting in calcium waves (24).
IMMUNE MYOPATHY
Inflammatory myopathy
There are several case reports of induction of inflammatory myopathies (i.e., polymyositis and dermatomyositis) by statins (30). These cases are characterized by large elevations of CK levels, a myopathic pattern on electromyogram, and a muscle biopsy showing inflammatory infiltrates. Discontinuation of statin therapy and immunosuppressive therapy (e.g., glucocorticoids) can lead to resolution of the myopathy in these patients. Such cases, however, are rare, and in most myopathies there is no evidence of an inflammatory component.
Noninflammatory myopathy
More recently, 25 cases of a histologically distinct statin myopathy have been described, in which muscle biopsy demonstrated necrotizing myopathy without significant inflammation. These cases presented with proximal muscle weakness and elevated CK levels that persisted despite discontinuation of the statin and responded to immunosuppressive agents (31). Major histocompatibility complex-I staining was upregulated in nonnecrotic fibers in eight patients with such necrotizing myopathy (32). It is postulated that previously restricted epitopes are exposed by statins and this may trigger an autoimmune myopathy. Indeed, there is a report of autoantibodies against HMG-CoA reductase in patients with this kind of myopathy (33).
CLINICAL IMPLICATIONS FOR THE PREVENTION AND MANAGEMENT OF STATIN-INDUCED MYOPATHY
Can we identify patients at high risk of developing statin-induced myopathy?
In the PRIMO study, the strongest independent risk factor for muscular symptoms in multivariate analysis was a history of myopathy while receiving another lipid-lowering therapy (odds ratio 10.12), followed by a personal history of unexplained cramps (4.14), a history of CK elevation (2.04), a family history of muscular symptoms (1.93) or myalgia while receiving lipid-lowering therapy (1.89), and untreated hypothyroidism (1.71). Interestingly, although depression (current or in the past) was not associated with a risk of myopathy, the use of antidepressant medications was associated with a significantly lower prevalence of muscular symptoms (0.51) (4).
Although there was no effect of sex on the prevalence of myopathy in the PRIMO study, female sex is sometimes considered a risk factor (6). Women are also considered to be at lower risk of developing coronary heart disease, at least before menopause. However, all risk-assessment models take sex into consideration, and meta-analyses of statin trials show that women derive benefit from treatment similar to benefit in men (34). Therefore, the same principles regarding treatment initiation and management should be used in both sexes.
Other risk factors for developing statin-induced myopathy may be old age (>80 years), small body frame and frailty, multisystem diseases (particularly involving the liver, kidney, or both), alcoholism, consumption of large quantities of grapefruit juice (>1 quart/day), major surgery (in the perioperative period), and excessive physical activity (6).
Although the role of the aforementioned genetic polymorphisms in predisposing to statin myopathy is a subject of interest, at present there are insufficient data to warrant pharmacogenetic testing of patients to determine such risk (6).
How do we manage the patient who develops myalgia while on statin therapy?
Initial evaluation.
When a statin-treated patient develops muscle-related symptoms, CK level should be measured to rule out rhabdomyolysis (CK levels >10 times the upper limit of normal values or elevation of serum creatinine levels), which mandates immediate stopping of the statin and prompt hydration. In the vast majority of cases, CK levels will be normal or only mildly elevated
The presence of contributing factors, such as strenuous exercise and consumption of grapefruit juice, should be assessed. Depending on the statin used, the use of medications that inhibit CYP3A4 (such as azole antifungals, macrolide antibiotic, fibrates, and calcium channel blockers), or CYP2C9 (such as amiodarone) should be ruled out.
Thyroid function should be assessed, as subclinical hypothyroidism may contribute to statin-associated myopathy. Vitamin D deficiency should also be ruled out and treated if found, as it may cause myalgia. Several small studies have found that correction of vitamin D deficiency may enable patients with a history of statin-associated myopathy to tolerate statins (35).
The importance of lifestyle measures, including diet and exercise, should be stressed. Adding phytosterols to a heart-healthy diet may produce ~10% decrease in LDL cholesterol (36). Supplements such as CoQ10, vitamin E, and magnesium are often tried, but as previously mentioned, there are very little data to support their use.
Statin-based approaches.
Since statins are the only agents with a robust body of evidence proving reduction in clinical end points, every effort should be made to keep high-risk patients on a statin-based regimen.
Switching statins may be efficacious. In one small study, 43% of 37 patients who received another statin after an episode of statin-associated myopathy tolerated other statins without recurrent symptoms (37). Considering the results of the PRIMO study, the use of statins associated with a lower risk of myopathy, such as fluvastatin or pravastatin, may be considered (4), although these statins may not be potent enough if a large reduction of LDL cholesterol is needed to reach target values. In one study, 80 mg fluvastatin XL daily (as a slow-release preparation) was tolerated in 97% of patients with prior statin intolerance owing to muscle-related symptoms, and LDL cholesterol was reduced by 32.8% (38).
Another approach evaluated in several studies involves the use of long-acting statins, mainly rosuvastatin, in low doses or at a reduced frequency (1–3 times a week). For example, in a retrospective analysis of 51 patients with a statin, alternate-day rosuvastatin at a mean dose of 5.6 mg was tolerated in 72.5% of patients, with an LDL cholesterol reduction of 34.5% (39).
Nonstatin medications.
When no statin is tolerated or the maximal tolerable dose of statins fails to reduce LDL cholesterol to target levels, nonstatin therapies should be used.
The most commonly used drug is ezetimibe. Ezetimibe as monotherapy or added to 80 mg fluvastatin XL daily in patients with prior statin intolerance was well tolerated and produced a 15% reduction in LDL cholesterol (38). It should be noted that currently there are no studies that demonstrate ezetimibe's efficacy in reducing cardiovascular morbidity and mortality.
Another option to reduce cholesterol absorption is the use of bile acid sequestrants (BAS), which were proven to reduce cardiovascular events in the Lipid Research Clinics Study Coronary Primary Prevention Trial (LRC-CPPT) (40). BAS can be combined with ezetimibe to produce a greater reduction in LDL cholesterol (41). The use of BAS is associated with a high rate of gastrointestinal side effects, leading to discontinuation rates as high as 40–60%. Colesevelam has a better side effects profile and may lead to better patient compliance (42).
Niacin at daily doses from 500 to 2,000 mg lowers LDL cholesterol by ~20%. Given as monotherapy in the Coronary Drug Project study, niacin reduced cardiovascular morbidity and mortality (43). Niacin can also be used in combination with BAS and ezetimibe in statin-intolerant patients requiring a large reduction in LDL cholesterol (42). The use of niacin is limited by its side effects, mainly flushing, which can lead to the drug being discontinued in up to 25% of patients (44).
In extreme cases, when both cardiovascular risk and LDL cholesterol are high despite maximal tolerable drug therapy, LDL apheresis can be used (45). A proposed algorithm for the management of a statin-intolerant patient is provided in Fig. 1.
Figure 1.

Proposed algorithm for the management of statin-associated myopathy. ULN, upper limit of normal.
CONCLUSIONS
The exact cause of statin-induced myopathy remains elusive. This effect seems to be multifactorial, yet some conclusions can be drawn. First, myopathy is dose related, and conditions that increase serum (and possibly muscle) levels of statins, such as concomitant medications, comorbidity (i.e., hypothyroidism), smaller body size, female sex, old age, Asian ethnicity, etc., are associated with a greater incidence of myopathy. Second, myopathy seems to be directly caused by inhibition of HMG-CoA reductase. Third, myopathy is probably related not to cholesterol reduction but, rather, to the inhibition of the production of other end products of the mevalonate pathway, such as ubiquinone and other isoprenoids. Lapaquistat, a squalene synthase inhibitor, whose development was halted at an advanced stage as a result of potential hepatic safety issues, was associated with a very low frequency of muscular adverse events in >6,000 patients in phase 2 and 3 trials (46). Fourth, lipophilic statins are more prone to cause myopathy than hydrophilic ones, probably owing to different transport systems leading to greater intramuscular concentrations of the former. Finally, there seems to be a genetic susceptibility to statin-induced myopathy. Further studies may help us identify patients at increased risk of myopathy and enable better tailoring of lipid-modifying therapy.
In statin-intolerant patients at high risk of cardiovascular events, all efforts should be made to reduce LDL cholesterol to as close as possible to target levels, using lifestyle measures and combinations of nonstatin drugs. Newer therapies currently under development, such as mipomersen (an antisense inhibitor of apolipoprotein B), lomitapide (a microsomal transfer protein inhibitor), and proprotein convertase subtilisin/kexin (PCSK) 9 inhibitors, may prove useful in statin-intolerant patients.
Acknowledgments
No potential conflicts of interest relevant to this article were reported.
R.B., H.C., Y.K., and D.H. each contributed to the writing, reviewing, and editing of the manuscript. R.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This publication is based on the presentations from the 4th World Congress on Controversies to Consensus in Diabetes, Obesity and Hypertension (CODHy). The Congress and the publication of this supplement were made possible in part by unrestricted educational grants from Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Ethicon Endo-Surgery, Janssen, Medtronic, Novo Nordisk, Sanofi, and Takeda.
References
- 1.Shalev V, Chodick G, Silber H, Kokia E, Jan J, Heymann AD. Continuation of statin treatment and all-cause mortality: a population-based cohort study. Arch Intern Med 2009;169:260–268 [DOI] [PubMed] [Google Scholar]
- 2.Chodick G, Shalev V, Gerber Y, et al. Long-term persistence with statin treatment in a not-for-profit health maintenance organization: a population-based retrospective cohort study in Israel. Clin Ther 2008;30:2167–2179 [DOI] [PubMed] [Google Scholar]
- 3.Bays H. Statin safety: an overview and assessment of the data—2005. Am J Cardiol 2006;97:6C–26C [DOI] [PubMed] [Google Scholar]
- 4.Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005;19:403–414 [DOI] [PubMed] [Google Scholar]
- 5.Buettner C, Rippberger MJ, Smith JK, Leveille SG, Davis RB, Mittleman MA. Statin use and musculoskeletal pain among adults with and without arthritis. Am J Med 2012;125:176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mancini GB, Baker S, Bergeron J, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: proceedings of a Canadian Working Group Consensus Conference. Can J Cardiol 2011;27:635–662 [DOI] [PubMed] [Google Scholar]
- 7.Thompson PD, Clarkson PM, Rosenson RS, National Lipid Association Statin Safety Task Force Muscle Safety Expert Panel An assessment of statin safety by muscle experts. Am J Cardiol 2006;97:69C–76C [DOI] [PubMed] [Google Scholar]
- 8.Link E, Parish S, Armitage J, et al. SEARCH Collaborative Group SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008;359:789–799 [DOI] [PubMed] [Google Scholar]
- 9.Santos PC, Gagliardi AC, Miname MH, et al. SLCO1B1 haplotypes are not associated with atorvastatin-induced myalgia in Brazilian patients with familial hypercholesterolemia. Eur J Clin Pharmacol 2012;68:273–279 [DOI] [PubMed] [Google Scholar]
- 10.Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther 2006;80:565–581 [DOI] [PubMed] [Google Scholar]
- 11.Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res 2010;106:297–306 [DOI] [PubMed] [Google Scholar]
- 12.Sirtori CR, Mombelli G, Triolo M, Laaksonen R. Clinical response to statins: mechanism(s) of variable activity and adverse effects. Ann Med 2012;44:419–432 [DOI] [PubMed] [Google Scholar]
- 13.Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheumatol 2010;22:644–650 [DOI] [PubMed] [Google Scholar]
- 14.Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006;34:153–162 [DOI] [PubMed] [Google Scholar]
- 15.Phillips PS, Haas RH, Bannykh S, et al. Scripps Mercy Clinical Research Center Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 2002;137:581–585 [DOI] [PubMed] [Google Scholar]
- 16.Schick BA, Laaksonen R, Frohlich JJ, et al. Decreased skeletal muscle mitochondrial DNA in patients treated with high-dose simvastatin. Clin Pharmacol Ther 2007;81:650–653 [DOI] [PubMed] [Google Scholar]
- 17.Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005;62:1709–1712 [DOI] [PubMed] [Google Scholar]
- 18.Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007;49:2231–2237 [DOI] [PubMed] [Google Scholar]
- 19.Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996;77:851–854 [DOI] [PubMed] [Google Scholar]
- 20.Bouitbir J, Charles AL, Echaniz-Laguna A, et al. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J 2012;33:1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaars CF, Stalenhoef AF. Effects of ubiquinone (coenzyme Q10) on myopathy in statin users. Curr Opin Lipidol 2008;19:553–557 [DOI] [PubMed] [Google Scholar]
- 22.Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007;100:1400–1403 [DOI] [PubMed] [Google Scholar]
- 23.Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007;99:1409–1412 [DOI] [PubMed] [Google Scholar]
- 24.Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008;8:333–338 [DOI] [PubMed] [Google Scholar]
- 25.Araki M, Maeda M, Motojima K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur J Pharmacol 2012;674:95–103 [DOI] [PubMed] [Google Scholar]
- 26.Flint OP, Masters BA, Gregg RE, Durham SK. Inhibition of cholesterol synthesis by squalene synthase inhibitors does not induce myotoxicity in vitro. Toxicol Appl Pharmacol 1997;145:91–98 [DOI] [PubMed] [Google Scholar]
- 27.Nishimoto T, Tozawa R, Amano Y, Wada T, Imura Y, Sugiyama Y. Comparing myotoxic effects of squalene synthase inhibitor, T-91485, and 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors in human myocytes. Biochem Pharmacol 2003;66:2133–2139 [DOI] [PubMed] [Google Scholar]
- 28.Westwood FR, Bigley A, Randall K, Marsden AM, Scott RC. Statin-induced muscle necrosis in the rat: distribution, development, and fibre selectivity. Toxicol Pathol 2005;33:246–257 [DOI] [PubMed] [Google Scholar]
- 29.Pierno S, De Luca A, Tricarico D, et al. Potential risk of myopathy by HMG-CoA reductase inhibitors: a comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibers. J Pharmacol Exp Ther 1995;275:1490–1496 [PubMed] [Google Scholar]
- 30.Schalke BB, Schmidt B, Toyka K, Hartung HP. Pravastatin-associated inflammatory myopathy. N Engl J Med 1992;327:649–650 [DOI] [PubMed] [Google Scholar]
- 31.Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010;41:185–190 [DOI] [PubMed] [Google Scholar]
- 32.Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007;17:194–200 [DOI] [PubMed] [Google Scholar]
- 33.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011;63:713–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baigent C, Blackwell L, Emberson J, et al. Cholesterol Treatment Trialists’ (CTT) Collaboration Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee P, Greenfield JR, Campbell LV, Vitamin D insufficiency--a novel mechanism of statin-induced myalgia? Clin Endocrinol (Oxf) 2009;71:154–155 [DOI] [PubMed] [Google Scholar]
- 36.Law M. Plant sterol and stanol margarines and health. BMJ 2000;320:861–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen KE, Hildebrand JP, Ferguson EE, Stein JH. Outcomes in 45 patients with statin-associated myopathy. Arch Intern Med 2005;165:2671–2676 [DOI] [PubMed] [Google Scholar]
- 38.Stein EA, Ballantyne CM, Windler E, et al. Efficacy and tolerability of fluvastatin XL 80 mg alone, ezetimibe alone, and the combination of fluvastatin XL 80 mg with ezetimibe in patients with a history of muscle-related side effects with other statins. Am J Cardiol 2008;101:490–496 [DOI] [PubMed] [Google Scholar]
- 39.Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother 2008;42:341–346 [DOI] [PubMed] [Google Scholar]
- 40.The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA 1984;251:351–364 [DOI] [PubMed] [Google Scholar]
- 41.Zema MJ. Colesevelam HCl and ezetimibe combination therapy provides effective lipid-lowering in difficult-to-treat patients with hypercholesterolemia. Am J Ther 2005;12:306–310 [DOI] [PubMed] [Google Scholar]
- 42.Insull W., Jr Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J 2006;99:257–273 [DOI] [PubMed] [Google Scholar]
- 43.Berge KG, Canner PL, Coronary Drug Project Research Group Coronary drug project: experience with niacin. Eur J Clin Pharmacol 1991;40(Suppl 1):S49–S51 [PubMed] [Google Scholar]
- 44.AIM-HIGH Investigators Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011;365:2255–2267 [DOI] [PubMed] [Google Scholar]
- 45.Thompson GR, HEART-UK LDL Apheresis Working Group Recommendations for the use of LDL apheresis. Atherosclerosis 2008;198:247–255 [DOI] [PubMed] [Google Scholar]
- 46.Stein EA, Bays H, O’Brien D, Pedicano J, Piper E, Spezzi A. Lapaquistat acetate: development of a squalene synthase inhibitor for the treatment of hypercholesterolemia. Circulation 2011;123:1974–1985 [DOI] [PubMed] [Google Scholar]