

The prevalence and incidence of type 2 diabetes, representing >90% of all cases of diabetes, are increasing rapidly throughout the world. The International Diabetes Federation has estimated that the number of people with diabetes is expected to rise from 366 million in 2011 to 552 million by 2030 if no urgent action is taken. Furthermore, as many as 183 million people are unaware that they have diabetes (www.idf.org). Therefore, the identification of individuals at high risk of developing diabetes is of great importance and interest for investigators and health care providers.
Type 2 diabetes is a complex disorder resulting from an interaction between genes and environment. Several risk factors for type 2 diabetes have been identified, including age, sex, obesity and central obesity, low physical activity, smoking, diet including low amount of fiber and high amount of saturated fat, ethnicity, family history, history of gestational diabetes mellitus, history of the nondiabetic elevation of fasting or 2-h glucose, elevated blood pressure, dyslipidemia, and different drug treatments (diuretics, unselected β-blockers, etc.) (1–3).
There is also ample evidence that type 2 diabetes has a strong genetic basis. The concordance of type 2 diabetes in monozygotic twins is ~70% compared with 20–30% in dizygotic twins (4). The lifetime risk of developing the disease is ~40% in offspring of one parent with type 2 diabetes, greater if the mother is affected (5), and approaching 70% if both parents have diabetes. In prospective studies, we have demonstrated that first-degree family history is associated with twofold increased risk of future type 2 diabetes (1,6). The challenge has been to find genetic markers that explain the excess risk associated with family history of diabetes.
Advances in genotyping technology during the last 5 years have facilitated rapid progress in large-scale genetic studies. Since 2007, genome-wide association studies (GWAS) have identified >65 genetic variants that increase the risk of type 2 diabetes by 10–30% (7,8). Most of these variants are noncoding variants, and therefore their functional consequences are challenging to investigate. Many of the variants identified to date regulate insulin secretion and not insulin action in insulin-sensitive tissues.
In a review by Noble et al. (3), a total of 43 different studies were presented where nongenetic prediction models for type 2 diabetes, including known risk factors for type 2 diabetes with different combinations, had been analyzed. Heterogeneity of data and highly variable methodology of primary studies precluded meta-analysis. Altogether, 84 different risk prediction models were presented in 43 studies. C statistics varied from 0.60 to 0.91 (from 0.60 to 0.69 in 5 models, from 0.70 to 0.79 in 44 models, from 0.80 to 0.89 in 32 models, and ≥0.90 in 3 models). These results indicate that clinical, laboratory, and other easily collected information by interview constitutes in most cases a solid basis for nongenetic prediction models in type 2 diabetes.
Identification of a large number of novel genetic variants increasing susceptibility to type 2 diabetes and related traits opened up opportunity, not existing thus far, to translate this genetic information to the clinical practice and possibly improve risk prediction. However, available data to date do not yet provide convincing evidence to support use of genetic screening for the prediction of type 2 diabetes.
In this review, we summarize the current evidence on the role of genetic variants to predict type 2 diabetes above and beyond nongenetic factors and discuss the limitations and future potential of genetic studies.
Genetic prediction models for type 2 diabetes: evidence from cross-sectional and longitudinal studies
Several studies have indicated that different genetic variants (single nucleotide polymorphisms [SNPs]) are associated with type 2 diabetes. Genetic risk models for type 2 diabetes, based on both cross-sectional (9–17) and longitudinal (1,17–24) studies, are summarized in Table 1.
Table 1.
Comparison of clinical and genetic prediction models for type 2 diabetes
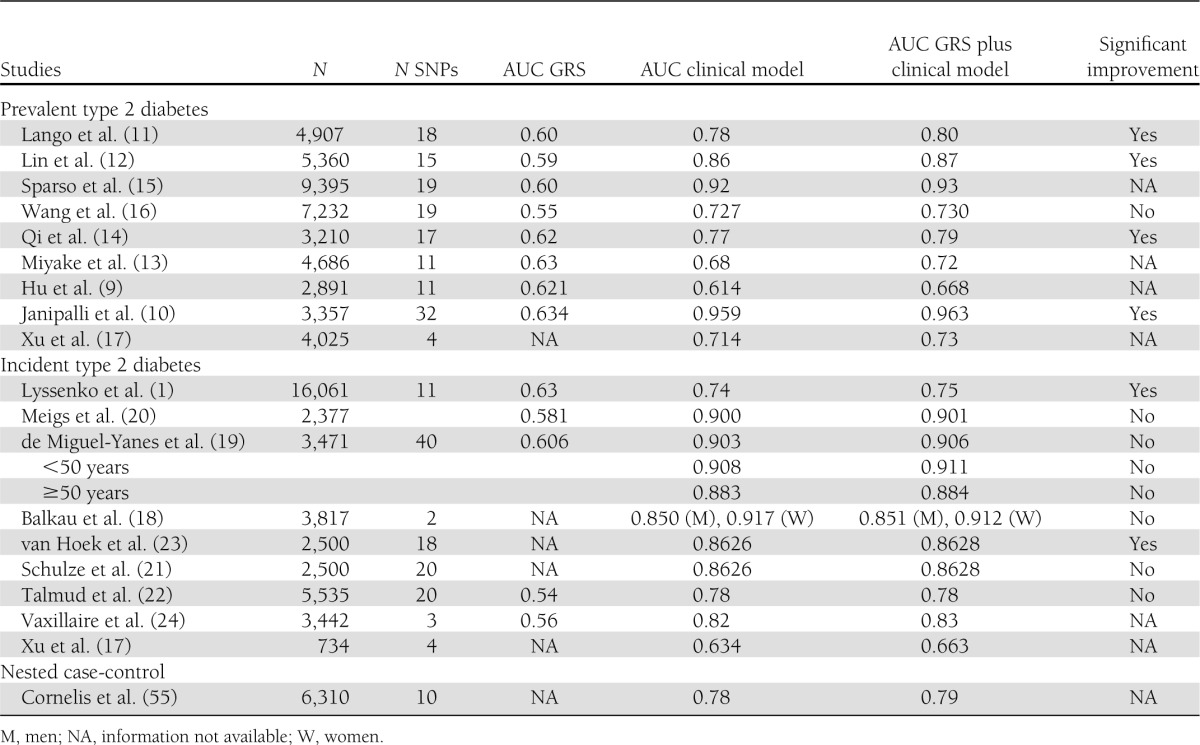
Cross-sectional studies.
In cross-sectional studies including 3,000–9,000 individuals with and without type 2 diabetes, the discriminatory ability of the combined SNP information has been assessed by grouping individuals based on the number of risk alleles and determining relative odds of type 2 diabetes, as well as by calculating the area under the receiver operating characteristic curve (AUC). As shown in Table 1, the AUC of the genetic risk score (GRS), which combines the information from all risk variants included in the study, has ranged from 0.54 to 0.63, indicating that genetic factors have limited use in predicting an individual’s risk of the disease. In contrast, the AUC has been considerably larger (from 0.61 to 0.95) for clinical models including different combinations of clinical and laboratory parameters (age, sex, and BMI in all models and family history of diabetes and fasting glucose in most of the models) predicting the risk of type 2 diabetes. Adding the GRS in the same model shows that in addition to clinical and laboratory parameters, risk variants increase only minimally the predictive value at the population level, although the model improvement could be statistically significant (P < 0.05) in some cases.
Perhaps the most important clinical question in cross-sectional studies is trying to identify undiagnosed individuals with type 2 diabetes. We addressed this question in our large population-based Metabolic Syndrome in Men (METSIM) Study (16). We identified undiagnosed type 2 diabetic patients using the Finnish Diabetes Risk Score alone (25), which was the best single indicator of prevalent undiagnosed diabetes among all variables tested in our study. The AUC based on logistic regression models for the identification of previously undiagnosed type 2 diabetic subjects with the Finnish Diabetes Risk Score alone was 0.727, and it was 0.772 after adding total triglycerides, HDL cholesterol, adiponectin, and alanine transaminase in the model. Adding type 2 diabetes risk alleles (20 SNPs) did not further improve the model (0.772) (16). Therefore, in our study common genetic variants did not seem to add any information on the identification of people having undiagnosed diabetes.
Longitudinal studies.
Longitudinal studies can address the question of what the nongenetic and genetic risk factors predicting incident type 2 diabetes are. Several large population-based follow-up studies have been published aiming to investigate the predictive power of common genetic variants on the risk of incident type 2 diabetes (Table 1). These studies, including genetic information from 2 to 40 SNPs, reported results surprisingly similar to those from cross-sectional case-control studies. Estimates of C statistics have ranged from 0.54 to 0.63. Different clinical predicting models gave/provided more significant C statistic values from 0.63 to 0.917, which are also quite similar to those based on cross-sectional studies. Risk variants did not essentially increase the AUC to predict type 2 diabetes when combined with clinical risk factors. In one study, type 2 diabetes risk prediction of a combined clinical and genetic model was somewhat better in younger (<50 years) than in older (≥50 years) individuals (19) and in women than in men (18). Most of these prospective studies were performed in Caucasian populations, with only one in Chinese (17).
Are genetic prediction models for type 2 diabetes worthless?
Both cross-sectional and longitudinal studies published thus far (Table 1) demonstrate that genetic screening for the prediction of type 2 diabetes in high-risk individuals is currently of little value in clinical practice. Table 2 lists several limitations of GRSs published (Table 2).
Table 2.
Limitations and potential of GRS studies

Small effect size of genetic loci.
Effect sizes of common genetic variants for type 2 diabetes identified to date are rather modest, ranging from 10 to 35% (7,8). An attempt to compose a GRS combining several genetic variants has shown only a 10–12% increased risk of disease with increasing number of the risk alleles. In the Malmö Preventive Project study (1), the effect was approximately twofold increased when carriers of the highest and the lowest number of risk alleles were compared (top 20% ≥12 vs. bottom 20% ≤8 risk alleles). Increasing the number of novel genetic variants up to 40 did not seem to largely improve the risk prediction (19). The observed modest effect sizes could be partially attributed to the fact that low frequency or rare variants have not yet been reported. Also, it is worth mentioning that the majority of the identified loci from GWAS are not, in fact, genes. The type 2 diabetes–associated loci represent an associated SNP, and there are still no data on whether the top associated signal represents the “causal gene”—much less the “causal variant.”
Low discriminative ability of the GRS.
A good diagnostic test in clinical practice has high sensitivity and specificity. Consistently across all studies, the C statistics of the AUC for genetic models are typically ~0.60 suggesting that a genetic test performs just a little better than flipping a coin. These results demonstrate that the performance of a genetic test remains rather poor even after adding all recently identified genetic variants in the model (19). This may not be very surprising, since these variants explain only ~10–15% of the heritability of type 2 diabetes (7). The application of the genome-wide effects taking into account all SNPs and not just those that reach a Bonferroni level of significance has recently been used in studies on height (26). The results suggested that this approach can reduce the amount of missing heritability and may permit a better GRS. However, the clinical utility of this application for the prediction of type 2 diabetes needs to be tested and validated. Finally, type 2 diabetes represents a heterogeneous condition defined by hyperglycemia, and there may be several subtypes of diabetes yet to be defined. Genetic variants operating through different pathways in the disease pathogenesis, such as obesity, and contributing to variation in glycemic traits together may have greater predictive value for diabetes and its different subtypes. Therefore, the analyses evaluating prediction models based on all reported variants associated with type 2 diabetes (8) and glycemic traits including glucose and insulin levels during an oral glucose tolerance test (OGTT) (27) but also obesity (28) should be performed.
Small added value of GRS compared with clinical risk factors.
Another question that rises about the usefulness of a genetic screening in clinical practice is whether genetic information improves the discriminative accuracy of a test using traditional routine clinical risk factors alone. Both prospective and cross-sectional studies have reported somewhat different discriminatory values across different studies depending on study ascertainment (inclusion and exclusion criteria of different metabolic risk factors), the length of the follow-up period in the prospective cohorts, obesity, and the presence of family history of diabetes. A consistent finding in all of these has been that GRS has added very little to the information provided by clinical risk factors alone. Thus, the addition of data from genotyped genetic variants to the clinical model only slightly improved the discriminative power of the AUC in the largest prospective studies from 0.74 to 0.75 in the Swedish Malmö Preventive Project study (1), from 0.900 to 0.901 in the Framingham Offspring study (20), and from 0.66 to 0.68 in the Rotterdam study (23). One explanation for these findings could be that clinical risk factors themselves, such as obesity and elevated glucose levels, harbor a substantial genetic component, and therefore different GRS models underestimate the true significance of genetic variation as a predictor for type 2 diabetes.
Questionable clinical relevance of some genetic variants in disease prediction.
Once genetic loci are identified in the case-control studies, it is very important to validate their ability to predict disease in prospective studies. Prospective studies represent a more controlled setting where both case and control subjects are ascertained in the same way and have similar environmental exposure and therefore give the true incidence of the disease in a population. In the Malmö Preventive Project study (1), 11 of 16 genetic loci studied, in the Framingham Offspring study (20) 2 of 18, and in the Rotterdam study (12) 9 of 18 were associated with the risk of developing future type 2 diabetes. These results may suggest that not all genetic variants that were significantly associated with type 2 diabetes in case-control studies are clinically relevant in the processes responsible for the conversion to type 2 diabetes. However, we could not rule out a lack of power, since similar observations have also been made in case-control studies. We are currently conducting the largest to date meta-analysis of prospective cohorts in European consortia (ENGAGE) including a total of ~55,000 individuals, followed for >15 years, to increase sample size and, thus, improve statistical power. Our preliminary findings support the notion that the validation and characterization of genetic variants identified in case-control studies should be performed before any claims of their clinical relevance are made.
Lack of appropriate models for studies of gene-gene and gene-environment interactions in risk prediction.
There is very little information on how much gene-gene and gene-environment interactions contribute to the prediction of type 2 diabetes. The success in the application of the methodological techniques to study epistatic effects in different populations has been limited. Given the excessive calculation and power capacity required for running these tests, researchers have mainly studied interaction between genomic loci that have already been found (29). However, studies in plants and animals clearly demonstrate that epistatic/interactions effects are often detected in the absence of main effects (30). Our recent studies demonstrate that the risk of disease conferred by genetic variants might be neutralized by their concomitant beneficial effects in other key organs and tissues involved in the pathogenesis of type 2 diabetes or having different responses to nutrition—so-called pleiotropic effects (31,32). For example, insulin secretion reducing effect of a genetic variant in GIPR is ameliorated by its beneficial effects on body composition, including BMI, waist, and fat mass (31). Furthermore, the carriers of the GIPR variant seem to respond differently to food rich in carbohydrates and fat (32,33). Similar observations have been reported for an interaction between a variant in the FTO gene and physical activity on the risk of obesity and cardiovascular diseases (34,35). Carriers of the obesity-associated allele in FTO have a higher risk for cardiovascular risk only in women who are physically inactive but not in those who are physically active, suggesting that the risk for developing cardiovascular disease can be prevented or delayed in the risk allele carriers if they are physically active. Thus, defining the nature of the gene-gene and gene-environment interactions can clearly help to improve prediction and identify persons at increased risk of type 2 diabetes (36).
Genetic prediction models for type 2 diabetes can be valuable in the future
Previously published genetic studies have severe limitations that underestimate the true significance of genetic variants in predicting type 2 diabetes (Table 2). Genetic prediction models can be improved by increasing the precision of the diagnosis of type 2 diabetes, by identification of low-frequency and rare genetic variants, by identification of risk variants for type 2 diabetes in non–European ancestry populations, by increasing knowledge on structural variation and epigenetics, and by developing statistical techniques to evaluate gene-gene and gene-environment interactions.
Necessity of improving the precision of the diagnosis of individuals with diabetes.
Type 2 diabetes is a chronic hyperglycemic condition that is not type 1 diabetes or other subtypes of diabetes, which include genetic defects of insulin secretion and action, diseases of exocrine pancreas, endocrinopathies, drug- or chemically induced diabetes, diabetes in connection with infections, uncommon forms of immunomediated diabetes, other genetic syndromes sometimes associated with diabetes, or gestational diabetes mellitus (37). In other words, there is no precise definition of type 2 diabetes. In fact, this main subtype of diabetes is defined by excluding all other conditions leading to chronic hyperglycemia.
Differential diagnosis between different subtypes of diabetes is challenging, especially between type 2 diabetes and late-onset and slowly developing type 1 diabetes. Patients having this subtype of diabetes, also called latent autoimmune diabetes in adults, have a progressive insulin secretion defect, share a genetic predisposition with both type 1 and type 2 diabetic patients, and are often diagnosed erroneously as type 2 patients (38). These patients, positive for GAD antibodies, may include ~10% of all diabetic patients (39) and are the most important subtype of diabetes leading to misclassification of diabetic patients. Additionally, recent exome sequencing studies have demonstrated that there is a continuously increasing number of monogenic forms of diabetes, which implies that the definition of type 2 diabetes in previous genetic studies may have been imprecise (40,41). Thus, it is very likely that every study population includes a varying number of individuals who have monogenic diabetes and who have been misclassified as having type 2 diabetes. Finally, it is important to note that several large-scale case-control or cohort studies have not applied an OGTT, which implies that their nondiabetic control group includes a varying number of individuals having type 2 diabetes. Imprecise classification of individuals with diabetes into subtypes and poor diagnostic procedures to find or exclude individuals with diabetes have considerably weakened the power of previous genetic prediction models. More careful phenotyping and classification of participants into different subtypes of diabetes are needed in future studies aiming to improve genetic prediction models. Dynamic measures of β-cell function (i.e., glucose-stimulated insulin secretion during an OGTT) and insulin resistance (i.e., during clamp) among nondiabetic individuals will be largely insightful for the design of future studies.
New sequencing techniques will identify low-frequency and rare variants with large effect sizes.
Genome-wide association studies are based on the “common disease, common variant” hypothesis, assuming that common diseases are attributable in part to allelic variants present in >5% of the population (42). These studies have been able to identify only relatively common variants that essentially contributed to the generation of different genetic risk models for complex diseases, including type 2 diabetes. Therefore, new technologies (exome sequencing, custom-made exome chips) are needed to identify low frequency (<5%) or rare (<0.5%) variants having larger effect sizes that could potentially explain a part of the “missing heritability” (43). Importantly, as previously mentioned, the GRS that emerges from GWAS may not, in fact, be using the “true” causal variant (or may not even be in the true causal gene). As a result, through fine-mapping and sequencing, perhaps the true genes/variants can be identified and, with use of these in a GRS, the prediction ability might increase. It has been estimated that 20 variants with risk allele frequency of 1% and allelic odds ratio of 3.0 could account for most familial aggregation of type 2 diabetes (43). Results from exome sequencing and custom-made exome chip studies soon to be published will clarify the role of variants with a population frequency <5% in chronic diseases, including type 2 diabetes. This work will be facilitated by the comprehensive catalog of variants with the minor allele frequency >1% generated by the 1000 Genomes Project (http://www.1000genomes.org/page.php). Identification of low frequency and rare variants makes it possible to search for causal variants in gene regions having simultaneously common variants associated with the disease.
Studies on monogenic forms of diabetes have clarified the relative importance of rare mutations having large effects sizes versus common SNPs having small effect sizes. Lango et al. (44) included a total of 410 individuals having causal mutations in the hepatic nuclear receptor 1α (maturity-onset diabetes of the young 3) in their study. They generated a single GRS representing the combined genetic susceptibility for type 2 diabetes, based on 17 SNPs known to influence the risk of type 2 diabetes. Each additional type 2 diabetes risk allele was associated with a 0.35-year reduction in age at diagnosis (P = 0.005) in all individuals and with a 0.28-year reduction in unrelated probands (P = 0.094). These results imply that the age of onset of monogenic diabetes caused by rare mutations having large effect sizes is not substantially modified by common polygenic variants. This example emphasizes the potential significance of rare variants having large effect sizes over common variants having small effect sizes in the risk prediction of diabetes.
Studies on non–European ancestry populations will help identify new variants relevant to type 2 diabetes prediction.
Most of the GWA studies have been performed in European ancestry populations, and therefore current type 2 diabetes genetic risk models are not likely to be applicable to all populations. Genetic variation is greatest in recent African ancestry populations (45), but there are no large GWAS where risk variants for type 2 diabetes in African populations have been investigated in detail. This information could greatly facilitate the identification of trait-defining variants as shown by a recent study in an African American type 2 diabetic case-control population (46). The investigators resequenced the critical chromosomal region for association and by haplotype analysis showed that rs7903146, originally found in Caucasian populations, was indeed a causal variant and sufficient to explain the haplotype association. The identification of causal variants, instead of their originally identified proxy SNPs, can potentially improve type 2 diabetes prediction models (47). Furthermore, the differences in genetic architecture among the populations could help to identify variants that are relatively rare in the Europeans but are more common in other ethnic groups. Thus, for example, the KCNQ1 gene was first identified in Asians where the minor allele frequency of the associated variants ranged between 30 and 40%, which was much higher than in Europeans with a frequency 10% (48,49).
Studies of structural variation and epigenetics may help identify new variants relevant to type 2 diabetes prediction.
The contribution of structural variation, including copy no. variants (CNVs) (insertions and deletions) and copy neutral variants (inversions and translocations), to the risk of type 2 diabetes is poorly known. To date, robustly replicated findings of CNVs associated with type 2 diabetes have not been reported. The reason for this is, in part, that most of the CNV analysis has been based on CNVs that are “tagged” by GWAS SNPs and thus covering only a small well-behaved genomic regions (in Hardy-Weinberg equilibrium), whereas the amount of “structural dark matter” remains relatively untouched by arrays or sequencing of those genomic regions that are not amenable to GWAS arrays (48). Next-generation sequencing has a considerably better potential than conventional sequencing to find structural variation, which could contribute to the understanding of the genetics of type 2 diabetes. However, it is not very likely that CNVs play a major role in the genetics of type 2 diabetes, given the fact that CNVs have been estimated to affect up to 5% of the human genome (43).
Epigenetics means heritable changes in gene function attributable to chemical modifications of DNA and its associated proteins, independent of the DNA sequence. The most investigated epigenetic modifications are methylation of cytosine residues in DNA and histone modifications (50). Changes in DNA methylation have been shown to be linked with some variants increasing the risk of type 2 diabetes. Hypermethylation of the maternal allele of KCNQ1 results in monoallelic activity of the neighboring maternally expressed protein-coding genes and is associated with the risk of type 2 diabetes (51). Similarly, maternally expressed KLF14 only increases the risk when carried on the maternal chromosome and acts as a master trans regulator of adipose tissue expression (52). These examples demonstrate the possibility that several other genes, yet to be discovered, can contribute to the risk of type 2 diabetes via epigenetic mechanisms. Combining the advantages of GWAS and epigenome analyses might pave the way to better understanding of the pathogenesis of type 2 diabetes and improve genetic risk models. Unfortunately, methods to estimate whole-genome methylation are still under development and catch only a minor fraction of all methylation sites. Technical improvements in near future might make genome-wide methylation scans more extensive and reliable.
Large population-based studies and development of statistical methods will improve analyses of gene-gene and gene-environment interactions.
Most previous studies on the genetics of type 2 diabetes, especially before the era of GWAS, applied a single-locus analysis strategy and thus ignored interactions. Recent advances in genotyping have considerably improved the opportunity to investigate the genetic architecture of type 2 diabetes and have made it possible to perform meta-analyses of several population-based studies often including >100,000 participants. Although these studies exhibit considerable heterogeneity, which weakens their power, they have paved the way to studies of gene-gene and gene-environment interactions. Recent advances include Metabochip, a custom-made Illumina array (Illumina, San Diego, CA), including 217,000 SNPs, and Illumina Human Exome BeadChip including >250,000 putative functional exonic variants that are especially suited for genetic studies of type 2 diabetes. These large populations allow meta-analyses based on identical genetic platforms, which minimize the heterogeneity of genotyping results.
Gene-gene and gene-environment interaction analyses based on large populations increase the power to detect novel variants and more accurately characterize the genetic effects. They also may help to elucidate the biological and biochemical pathways responsible for complex diseases, e.g., type 2 diabetes, and identify the environmental effects. Risk prediction models including significant interactions also improve disease risk prediction. Interaction analyses require sophisticated statistical methods to analyze genetic interactions. For example, exhaustive evaluation of all two-marker models in GWAS data are already challenging, given the fact that 5 × 10−11 possible models from a set of 1 million SNPs need to be calculated (53). The ultimate goal is to integrate modern statistical methods with genetic data and biological knowledge, which will further improve the power to detect complex interactions (54).
Conclusions
Genetic testing for the prediction of type 2 diabetes in high risk individuals is currently of little value in clinical practice.
The limitations of genetic risk models are small effect size of genetic loci, low discriminative ability of the genetic test, small added value of genetic information compared with the clinical risk factors, questionable clinical relevance of some genetic variants in disease prediction, and the lack of appropriate models for studies of gene-gene and gene-environment interactions in the risk prediction.
For improvement of the genetic risk models in the future, the definition of type 2 diabetes and classification of subtypes of diabetes should be more precise, new sequencing techniques should be applied to identify low-frequency and rare variants having a large effect size, non–European ancestry populations should be investigated to identify new variants relevant to type 2 diabetes prediction, studies of structural variation and epigenetics should be performed to identify new variants relevant to type 2 diabetes prediction, and modern statistical methods should be developed and applied in studies of gene-gene and gene-environment interaction in large populations.
Acknowledgments
Studies at LUDC, Malmö, were supported by grants from the Swedish Research Council, including a Linné grant (31475113580) and Strategic Research Grant EXODIAB (Dnr 2009-1039); the Heart and Lung Foundation; the Swedish Diabetes Research Society; the European Community's Seventh Framework Programme grant CEED3 (223211); the Diabetes Programme at Lund University; the Påhlsson Foundation; the Crafoord Foundation; the Knut and Alice Wallenberg Foundation; Novo Nordisk Foundation; and the European Foundation for the Study of Diabetes.
No potential conflicts of interest relevant to this article were reported.
V.L. and M.L. wrote the manuscript, researched data, and reviewed and approved the final version of the manuscript. V.L. and M.L. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This publication is based on the presentations from the 4th World Congress on Controversies to Consensus in Diabetes, Obesity and Hypertension (CODHy). The Congress and the publication of this supplement were made possible in part by unrestricted educational grants from Abbott, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Ethicon Endo-Surgery, Janssen, Medtronic, Novo Nordisk, Sanofi, and Takeda.
References
- 1.Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 2008;359:2220–2232 [DOI] [PubMed] [Google Scholar]
- 2.Mykkänen L, Kuusisto J, Pyörälä K, Laakso M. Cardiovascular disease risk factors as predictors of type 2 (non-insulin-dependent) diabetes mellitus in elderly subjects. Diabetologia 1993;36:553–559 [DOI] [PubMed] [Google Scholar]
- 3.Noble D, Mathur R, Dent T, Meads C, Greenhalgh T. Risk models and scores for type 2 diabetes: systematic review. BMJ 2011;343:d7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaprio J, Tuomilehto J, Koskenvuo M, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992;35:1060–1067 [DOI] [PubMed] [Google Scholar]
- 5.Groop L, Forsblom C, Lehtovirta M, et al. Metabolic consequences of a family history of NIDDM (the Botnia study): evidence for sex-specific parental effects. Diabetes 1996;45:1585–1593 [DOI] [PubMed] [Google Scholar]
- 6.Lyssenko V, Almgren P, Anevski D, et al. Botnia study group Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes 2005;54:166–174 [DOI] [PubMed] [Google Scholar]
- 7.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med 2010;363:2339–2350 [DOI] [PubMed] [Google Scholar]
- 8.Morris AP, Voight BF, Teslovich TM, et al. Wellcome Trust Case Control Consortium. Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators. Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium. South Asian Type 2 Diabetes (SAT2D) Consortium. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu C, Zhang R, Wang C, et al. PPARG, KCNJ11, CDKAL1, CDKN2A-CDKN2B, IDE-KIF11-HHEX, IGF2BP2 and SLC30A8 are associated with type 2 diabetes in a Chinese population. PLoS ONE 2009;4:e7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janipalli CS, Kumar MV, Vinay DG, et al. Analysis of 32 common susceptibility genetic variants and their combined effect in predicting risk of Type 2 diabetes and related traits in Indians. Diabet Med 2012;29:121–127 [DOI] [PubMed] [Google Scholar]
- 11.Lango H, Palmer CN, Morris AD, et al. UK Type 2 Diabetes Genetics Consortium Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes 2008;57:3129–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin X, Song K, Lim N, et al. Risk prediction of prevalent diabetes in a Swiss population using a weighted genetic score—the CoLaus Study. Diabetologia 2009;52:600–608 [DOI] [PubMed] [Google Scholar]
- 13.Miyake K, Yang W, Hara K, et al. Construction of a prediction model for type 2 diabetes mellitus in the Japanese population based on 11 genes with strong evidence of the association. J Hum Genet 2009;54:236–241 [DOI] [PubMed] [Google Scholar]
- 14.Qi Q, Li H, Wu Y, et al. Combined effects of 17 common genetic variants on type 2 diabetes risk in a Han Chinese population. Diabetologia 2010;53:2163–2166 [DOI] [PubMed] [Google Scholar]
- 15.Sparsø T, Grarup N, Andreasen C, et al. Combined analysis of 19 common validated type 2 diabetes susceptibility gene variants shows moderate discriminative value and no evidence of gene-gene interaction. Diabetologia 2009;52:1308–1314 [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Stancáková A, Kuusisto J, Laakso M. Identification of undiagnosed type 2 diabetic individuals by the finnish diabetes risk score and biochemical and genetic markers: a population-based study of 7232 Finnish men. J Clin Endocrinol Metab 2010;95:3858–3862 [DOI] [PubMed] [Google Scholar]
- 17.Xu M, Bi Y, Xu Y, et al. Combined effects of 19 common variations on type 2 diabetes in Chinese: results from two community-based studies. PLoS ONE 2010;5:e14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balkau B, Lange C, Fezeu L, et al. Predicting diabetes: clinical, biological, and genetic approaches: data from the Epidemiological Study on the Insulin Resistance Syndrome (DESIR). Diabetes Care 2008;31:2056–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Miguel-Yanes JM, Shrader P, Pencina MJ, et al. MAGIC Investigators. DIAGRAM+ Investigators Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care 2011;34:121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med 2008;359:2208–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulze MB, Weikert C, Pischon T, et al. Use of multiple metabolic and genetic markers to improve the prediction of type 2 diabetes: the EPIC-Potsdam Study. Diabetes Care 2009;32:2116–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talmud PJ, Hingorani AD, Cooper JA, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ 2010;340:b4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Hoek M, Dehghan A, Witteman JC, et al. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes 2008;57:3122–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaxillaire M, Veslot J, Dina C, et al. DESIR Study Group Impact of common type 2 diabetes risk polymorphisms in the DESIR prospective study. Diabetes 2008;57:244–254 [DOI] [PubMed] [Google Scholar]
- 25.Lindström J, Tuomilehto J. The diabetes risk score: a practical tool to predict type 2 diabetes risk. Diabetes Care 2003;26:725–731 [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010;42:565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott RA, Lagou V, Welch RP, et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manning AK, Hivert MF, Scott RA, et al. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Multiple Tissue Human Expression Resource (MUTHER) Consortium A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dempfle A, Scherag A, Hein R, Beckmann L, Chang-Claude J, Schäfer H. Gene-environment interactions for complex traits: definitions, methodological requirements and challenges. Eur J Hum Genet 2008;16:1164–1172 [DOI] [PubMed] [Google Scholar]
- 30.Carlborg O, Jacobsson L, Ahgren P, Siegel P, Andersson L. Epistasis and the release of genetic variation during long-term selection. Nat Genet 2006;38:418–420 [DOI] [PubMed] [Google Scholar]
- 31.Lyssenko V, Eliasson L, Kotova O, et al. Pleiotropic effects of GIP on islet function involve osteopontin. Diabetes 2011;60:2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonestedt E, Lyssenko V, Ericson U, et al. Genetic variation in the glucose-dependent insulinotropic polypeptide receptor modifies the association between carbohydrate and fat intake and risk of type 2 diabetes in the Malmo Diet and Cancer cohort. J Clin Endocrinol Metab 2012;97:E810–E818 [DOI] [PubMed] [Google Scholar]
- 33.Qi Q, Bray GA, Hu FB, Sacks FM, Qi L. Weight-loss diets modify glucose-dependent insulinotropic polypeptide receptor rs2287019 genotype effects on changes in body weight, fasting glucose, and insulin resistance: the Preventing Overweight Using Novel Dietary Strategies trial. Am J Clin Nutr 2012;95:506–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmad T, Chasman DI, Mora S, et al. The fat-mass and obesity-associated (FTO) gene, physical activity, and risk of incident cardiovascular events in white women. Am Heart J 2010;160:1163–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilpeläinen TO, Qi L, Brage S, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med 2011;8:e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank P, Pearson E, Florez JC. Gene-environment and gene-treatment interactions in type 2 diabetes: progress, pitfalls, and prospects. Diabetes Care 2013;36:1413–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care 2012;35(Suppl. 1):S64–S71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersen MK, Lundgren V, Turunen JA, et al. Latent autoimmune diabetes in adults differs genetically from classical type 1 diabetes diagnosed after the age of 35 years. Diabetes Care 2010;33:2062–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isomaa B, Almgren P, Henricsson M, et al. Chronic complications in patients with slowly progressing autoimmune type 1 diabetes (LADA). Diabetes Care 1999;22:1347–1353 [DOI] [PubMed] [Google Scholar]
- 40.Bonnefond A, Durand E, Sand O, et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS ONE 2010;5:e13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johansson S, Irgens H, Chudasama KK, et al. Exome sequencing and genetic testing for MODY. PLoS ONE 2012;7:e38050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet 2001;17:502–510 [DOI] [PubMed] [Google Scholar]
- 43.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lango Allen H, Johansson S, Ellard S, et al. Polygenic risk variants for type 2 diabetes susceptibility modify age at diagnosis in monogenic HNF1A diabetes. Diabetes 2010;59:266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009;106:9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer ND, Hester JM, An SS, et al. Resequencing and analysis of variation in the TCF7L2 gene in African Americans suggests that SNP rs7903146 is the causal diabetes susceptibility variant. Diabetes 2011;60:662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vassy JL, Meigs JB. Is genetic testing useful to predict type 2 diabetes? Best Pract Res Clin Endocrinol Metab 2012;26:189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girirajan S, Brkanac Z, Coe BP, et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011;7:e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yasuda K, Miyake K, Horikawa Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008;40:1092–1097 [DOI] [PubMed] [Google Scholar]
- 50.Schnabel RB, Baccarelli A, Lin H, Ellinor PT, Benjamin EJ. Next steps in cardiovascular disease genomic research—sequencing, epigenetics, and transcriptomics. Clin Chem 2012;58:113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong A, Steinthorsdottir V, Masson G, et al. DIAGRAM Consortium Parental origin of sequence variants associated with complex diseases. Nature 2009;462:868–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Small KS, Hedman AK, Grundberg E, et al. GIANT Consortium. MAGIC Investigators. DIAGRAM Consortium. MuTHER Consortium Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet 2011;43:561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie MD. Using biological knowledge to uncover the mystery in the search for epistasis in genome-wide association studies. Ann Hum Genet 2011;75:172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yi N. Statistical analysis of genetic interactions. Genet Res (Camb) 2010;92:443–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cornelis MC, Qi L, Zhang C, et al. Joint effects of common genetic variants on the risk for type 2 diabetes in U.S. men and women of European ancestry. Ann Intern Med 2009;150:541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]