

Abstract
Purpose.
To determine whether knockdown of Müller cell–derived VEGFA-splice variant, VEGF164, which is upregulated in the rat retinopathy of prematurity (ROP) model, safely inhibits intravitreal neovascularization (IVNV).
Methods.
Short hairpin RNAs for VEGF164 (VEGF164.shRNAs) or luciferase.shRNA control were cloned into lentivectors with CD44 promoters that specifically target Müller cells. Knockdown efficiency, off-target effects, and specificity were tested in HEK reporter cell lines that expressed green fluorescent protein (GFP)-tagged VEGF164 or VEGF120 with flow cytometry or in rat Müller cells (rMC-1) by real-time PCR. In the rat oxygen-induced retinopathy (OIR) ROP model, pups received 1 μL subretinal lentivector-driven luciferase.shRNA, VEGFA.shRNA, or VEGF164.shRNA at postnatal day 8 (P8). Analyses at P18 and P25 included: IVNV and avascular retina (AVA); retinal and serum VEGF (ELISA); density of phosphorylated VEGFR2 (p-VEGFR2) in lectin-labeled retinal endothelial cells (ECs; immunohistochemistry); TUNEL staining and thickness of inner nuclear (INL) and outer nuclear layers (ONL) in retinal cryosections; and pup weight gain.
Results.
In HEK reporter and in rMC-1 cells and in comparison to lucifferase.shRNA, VEGFA.shRNA reduced both VEGF120 and VEGF164, but VEGF164.shRNA only reduced VEGF164 and not VEGF120. Compared with luciferase.shRNA, VEGFA.shRNA and VEGF164.shRNA reduced retinal VEGF and IVNV without affecting AVA at P18 and P25. At P25, VEGF164.shRNA more effectively maintained IVNV inhibition than VEGFA.shRNA. VEGFA.shRNA and VEGF164.shRNA reduced pVEGFR2 in retinal ECs at P18, but VEGFA.shRNA increased it at P25. VEGFA.shRNA increased TUNEL+ cells at P18 and decreased ONL thickness at P18 and P25. VEGFA.shRNA and VEGF164.shRNA did not affect pup weight gain and serum VEGF.
Conclusions.
Short hairpin RNA to Müller cell VEGF164 maintained long-term inhibition of IVNV and limited cell death compared with shRNA to VEGFA.
Keywords: vascular endothelial growth factor, lentivector, short hairpin RNA, intravitreal neovascularization, Müller cells
Using a relevant model of current ROP, the rat 50/10 oxygen-induced retinopathy model, we determined that targeting Müller cell VEGF164 by lentivector-delivered shRNA achieved better inhibition of intravitreal neovascularization without causing retinal cell death compared to shRNA to VEGFA.
Introduction
Vascular endothelial growth factor is important in several angiogenic eye diseases, including AMD,1,2 diabetic retinopathy,3,4 retinal vein occlusion,3 and retinopathy of prematurity (ROP).5,6 We previously found that overactivation of VEGF receptor 2 (VEGFR2) led to disordered developmental angiogenesis7 in a similar pattern as seen in intravitreal neovascularization (IVNV) in severe ROP.8 Broad inhibition of VEGF with an intravitreal neutralizing antibody reduced IVNV in a rat model of ROP, but also reduced pup growth and serum VEGF levels.9 To target pathologic effects of VEGFA without affecting physiologic ones, we localized VEGFA splice variant mRNAs to cellular retinaldehyde-binding protein (CRALBP)-labeled Müller cells, RPE, and cells in the ganglion cell layer in the retina of a rat model of ROP at a time point when total retinal VEGFA expression was significantly increased compared with room air-raised pups.10 We created lentivectors that used CD44 promoters and efficiently drove VEGFA shRNA specifically in Müller cells when delivered into the subretinal space.11,12 In the short-term, IVNV was inhibited without affecting serum VEGFA, pup growth, or retinal apoptosis.12 However, Müller cells also depend on VEGFA for survival13 and this strategy may potentially have adverse effects. Therefore, in this study, we created lentivectors with CD44 promoters that specifically targeted Müller cell-VEGF164, which leads to pathologic angiogenesis14,15 and which we found to be the most prevalent splice variant in the ROP model.10 We tested the hypothesis that the lentivector shRNA to rat VEGF164 in Müller cells would safely and effectively reduce IVNV compared with knockdown of VEGFA or control and also tested efficacy and retinal cell survival at a later time point in the ROP model. We found that targeted lentivector delivery of shRNAs to VEGFA or VEGF164 reduced IVNV, but that shRNA to VEGF164 maintained long-term inhibition of IVNV and limited cell death compared with shRNA to VEGFA.
Methods
Rat Model of ROP (Rat 50/10 OIR Model)
All animal care was in accordance with the Use of Laboratory Animals and the ARVO Statement for the Use of Animals in Ophthalmology and Vision Research and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Utah. In the well-described rat oxygen-induced retinopathy (OIR) model (rat model of ROP),16,17 Sprague-Dawley (Charles River Laboratories, Inc., Wilmington, MA) dams and pups are placed into a dual channel/dual gas controller (OxyCycler; BioSpherix, Ltd., Lacona, NY) within 6 hours of birth, where oxygen is cycled between 50% and 10% every 24 hours. At postnatal day (P)14, litters were placed into room air until P18 or P25. Litter size was maintained at 12 to 16 pups. Pup weight was obtained at P8, P18, and P25. Pups were euthanized by intraperitoneal injection (IP) of ketamine (60 mg/kg) and xylazine (18 mg/kg) followed by IP pentobarbital (80 mg/kg). For all pups, one eye was processed for flatmount analysis and the fellow eye for protein or immunohistochemistry (IHC).
Construction of Lentivector-Driven shRNA
Short hairpin RNAs targeting rat VEGFA (NM_031836, VEGFA.shRNA); rat VEGF164 (AF260425, VEGF164.shRNA); or nonmammalian gene luciferase (M15077, luciferase.shRNA) were developed. shRNAs were embedded within a microRNA (miR-30) context as an efficient method to knock down a gene.18,19 The shRNA-microRNA30 was each cloned into a lentiviral transfer vector driven by a CD44 promoter specific to Müller cells (pFmCD44.1GW) and with multicistronic cotranscription of green fluorescent protein (GFP)20 or red fluorescent protein (RFP).11 The VEGFA.shRNAs were previously developed and tested for off-target effects and efficiency.12 Here, we describe methods for lentivectors with VEGF164.shRNAs. Knockdown and off-target efficiency was tested in rat Müller cells (rMC-1; line provided by Vijay Sarthy, Northwestern University, Evanston, IL) and VEGF120 and VEGF164 HEK reporter cell lines,12 with the goal to choose shRNAs with better knockdown efficiency in VEGF164 but not VEGF120.
Cell Culture and Assay for In Vitro–Knockdown Efficiency
Rat Müller cells, VEGF120, and VEGF164 HEK reporter cell lines were maintained in DMEM/high glucose (Gibco/Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Both VEGF120 and VEGF164 HEK reporter cell lines were transfected with plasmid DNA-pFmCD44.1GW containing VEGF164.shRNA-1 or VEGF164.shRNA-2 expressed with RFP or an empty vector without shRNA as control. Forty-eight hours after transfection, the knockdown efficiency of VEGF164.shRNAs was determined by flow cytometry of GFP fluorescence in RFP positive cells. Silencing was calculated as a percentage of GFP to control vector transfected-cells. The shRNA with better knockdown efficiency in VEGF164 but not VEGF120 was chosen for all following experiments and designated VEGF164.shRNA. rMC-1 cells in 6-well plates (Corning, Inc., Corning, NY) with 80% confluence were infected with lentivirus (5.0 × 106 viral particles/mL) containing VEGFA.shRNA, VEGF164.shRNA, luciferase.shRNA, or vehicle without viral infection. After 48 hours, cells were extracted and real-time PCR was performed. Each condition was performed in triplicate.
Subretinal Injection
At the beginning of P8 (50% oxygen cycle), pups were anesthetized by IP ketamine (20 mg/kg) and xylazine (2.5 mg/kg). Subretinal injections were performed by creating an initial opening beneath the limbus with a 30-gauge needle. One μL of 1 × 109 viral particles/mL (VP/mL) of VEGFA.shRNA, VEGF164.shRNA, or control luciferase.shRNA was delivered into the subretinal space using a 33-gauge needle attached to a syringe (Hamilton Company, Reno, NY). In some pups, 1 μL of subretinal PBS was used as an additional control. The created retinal detachments resolved within 24 hours. The same virus and dose were used in each eye of the same pup. Attempts were made to represent all lentivector types in each litter and to inject the same number of pups with each lentivector preparation in each litter. Pups weighing less than 7 g were not used and all pups analyzed were within ±2 g of mean pup weight based on overall growth chart.21,22
In Vivo Retinal Imaging
Pupil dilation was achieved with tropicamide (1% solution; Bausch & Lomb Pharmaceuticals, Inc., Rochester, NY). Genteal gel (Novartis Pharmaceuticals Corp., East Hanover, NJ) was the coupling agent for retinal imaging with a commercial applanation imaging system (Micron III; Phoenix Research Laboratories, Inc., Pleasanton, CA) and multiple camera recording software (StreamPix 5; Norpix, Inc., Montreal, Quebec, Canada). Both GFP and bright field were used for imaging.
Retinal Flatmount Preparation, Imaging, and Analysis
After euthanasia, eyes were enucleated, pierced through the cornea with a 30-gauge needle, placed into freshly made 4% paraformaldehyde (PFA) containing 10 mM sodium orthovanadate for 2 hours on ice, and then transferred to PBS. Corneas, lenses, and vitreous were removed, and retinas were dissected from the RPE/choroid/sclera. Retinal flatmounts were labeled using 5 μg/mL AlexaFluor 568 conjugated isolectin GS-IB4 from Griffonia simplicifolia (Bandeiraea; Molecular Probes, Eugene, OR) and imaged using an inverted fluorescence microscope (Olympus IX81; Olympus Corp., Tokyo, Japan).12 Whole retinal flatmount images were stitched using the scan-slide stitching function of imaging software (Metamorph version 7.0; Molecular Devices, Inc., Sunnyvale, CA). The avascular retina (AVA) and IVNV areas were analyzed by two masked reviewers and calculated as a percentage of total retinal area for each flatmount using Java-based imaging software (ImageJ version 1.46; National Institutes of Health, Bethesda, MD).
Cryosection Preparation and Immunofluorescence Staining and Quantification
Whole eye globes were fixed in 4% PFA containing 10 mM sodium orthovanadate for 10 minutes. Corneas and lenses were removed, and posterior eyecups were fixed for another 15 minutes in 4% PFA, then incubated in 30% sucrose/PBS at 4°C overnight, and mounted in optimal cutting temperature compound (Tissue-Tek; Electron Microscopy Sciences, Hatfield, PA). Cryosections (12 μm) were cut sequentially and stained for immunofluorescence analysis. Cryosections were incubated with rabbit anti-phosphorylated VEGFR2 (p-VEGFR2 at Y951; Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. After washes, sections were incubated with AlexaFluor 405 conjugated goat anti-rabbit second antibody for p-VEGFR2 and lectin for 1 hour. Sections stained with only secondary antibody and DAPI were controls. TUNEL staining was performed per instructions in the cell death detection kit (In Situ Cell Death Detection Kit, TMR red; Roche Diagnostics, Indianapolis, IN). DNase-treated sections were used as positive controls. Images were captured with confocal microscopy (Olympus IX81; Olympus Corp.). To determine the effects of knockdown on retinal VEGFR2 activation in captured images, semiquantitative assessment of the density of p-VEGFR2 was performed in sections of retina extending from the ganglion cell layer to lectin stained choroidal vessels depicting the RPE/choroid layer using Java-based imaging software (NIH). For p-VEGFR2 in retinal vessels, the density of p-VEGFR2 colabeling with lectin-stained ECs of the primary vascular plexus at the junctions between avascular retina and vascular retina was measured with the threshold function of the Java-based imaging software (NIH). TUNEL-positive cells colabeled with tetramethylrhodamine red (TMR red) and diamino-2-phenylindole (DAPI) were counted in retinal sections imaged at ×4 magnification. Retinal thickness was measured from ganglion cell to ONL in DAPI-stained sections captured at ×40 magnification using Java-based imaging software (NIH). In total, six sections taken at 60-μm intervals from three eyes of three pups in three litters were used for immunohistochemical analyses.
Retinal Protein Preparation and VEGF ELISA
Retinas were homogenized in modified radio-immunoprecipitation assay buffer containing 2 mM orthovanadate and protease inhibitors (Roche Diagnostics). Protein concentration was determined by bicinchocinic acid (BCA) protein assay (Pierce Biotechnology, Inc., Rockford, IL). Total retinal and serum VEGF concentration was measured using a commercial ELISA kit (Quantikine Rat VEGFA RRV00; R&D Systems, Minneapolis, MN) following manufacturer's instructions. Serum (50 μL) or 50 μg protein of retinal lysates was used for each sample, and samples were in duplicate.
Statistical Analysis
Significant differences between treatment groups were determined with ANOVA and Newman-Keuls multiple comparison test. For each test, a minimum value of P < 0.05 was considered statistically significant. Except where indicated otherwise, at least eight flat mounts, six samples for Western blot, and four samples for ELISA were analyzed. All samples were taken from different pups from at least three different litters. Results are mean ± SD.
Results
Generation and Knockdown Efficiency of Lentivector-Driven VEGF164-shRNA
We designed two different shRNAs for the rat VEGF164 coding sequence (GenBank: AF260425) and cloned each into the lentivector pFmCD44.1 GW, which contains a CD44 promoter specific to Müller cells and drives a microRNA30 (miR-30)–based shRNA cassette and either RFP or GFP. To test knockdown efficiency of the designed shRNAs, lentivector plasmids containing either of the two shRNAs and an RFP tag or an empty vector were transfected into one of two HEK293 reporter cell lines that expressed either GFP-tagged rat VEGF120 or VEGF164. Green fluorescent protein fluorescence intensity in RFP-expressed cells containing the shRNAs or empty vector was read using flow cytometry (Fig. 1A). Neither VEGF164.shRNA-1 nor -2 had an effect on knockdown of VEGF120 (Fig. 1B). Compared with cells transfected with empty vector, VEGF164.shRNA-2 caused 35% knockdown of VEGF164, whereas VEGF164.shRNA-1 only caused 15% knockdown of VEGF164. Therefore, VEGF164.shRNA-2 (designated hereafter as VEGF164.shRNA) was chosen for later experiments. The sequence of VEGF164.shRNA was: 5′-TGCTGTTGACAGTGAGCGCAGCCAGAAAATCACTGTGAGCTAGTGAAGCCACAGATGTAGCTCACAGTGATTTTCTGGCTTTGCCTACTGCCTCGGA-3′. We previously reported how lentivectors targeting rat Müller cell VEGFA were designed and tested and chose a VEGFA.shRNA that caused 35% reduction in VEGF120 and 50% of VEGF164 in HEK reporter cell lines.12
Figure 1.

Generation of lentivector-delivered shRNA for specific knockdown of VEGF164 in Müller cells. HEK reporter cell lines expressed GFP-tagged VEGF120 or VEGF164 were transfected with RFP-expressed lentivector VEGF164.shRNA plasmids or empty vector without shRNA. (A) Flow cytometry (fluorescence activated cell sorting [FACS]) of GFP fluorescence. (B) Quantification of percent silencing of VEGF120 and VEGF164 by VEGF164 shRNAs from FACS analysis. (C) Real-time PCR of mRNA of VEGF120 and VEGF164 in rMC-1 infected without lentivirus (uninfected) or with lentivector-driven shRNA to luciferase (luc.shRNA), VEGFA (VEGFA.shRNA), or VEGF164 (VEGF164.shRNA). *P < 0.05 and **P < 0.01 versus luc.shRNA).
To test knockdown efficiency and specificity of lentivector-driven VEGF164.shRNA and VEGFA.shRNA in Müller cells, we infected rMC-1s, a rat Müller cell line, with lentivectors containing the CD44 promoter driving GFP and one of the three different shRNAs (luciferase.shRNA, VEGFA.shRNA, VEGF164.shRNA) or an uninfected control. After 48 hours, rMC-1s were analyzed with real-time PCR for VEGF120 or VEGF164 mRNAs and expressed as fold difference compared with the mRNA from the uninfected control. There was no difference in mRNA level of VEGF120 and VEGF164 splice variants between uninfected and luciferase.shRNA. Compared with luciferase.shRNA, VEGFA.shRNA reduced expression of both VEGF120 and VEGF164 (Fig. 1C), whereas only VEGF164 mRNA was reduced in the VEGF164.shRNA group. These results provide evidence that VEGF164.shRNA specifically knocked down VEGF164 mRNA and not VEGF120.
Knockdown of VEGF164 Reduced IVNV at P18 and P25
Images were taken before euthanasia using an applanation imaging system (Phoenix Research Laboratories, Inc.) to assess transduction of each of the three lentivectors that had been delivered as subretinal injections to P8 pup eyes. At P25, GFP fluorescence demonstrated that approximately 33% of the retina was transduced by each subretinal injection of lentivector shRNA and not with the PBS injection (Fig. 2A). Müller cell specificity was determined by colabeling of GFP and CRALBP in retinal cryosections (Fig. 2B). To determine knockdown efficiency of VEGF in vivo, total retina lysates were analyzed for VEGF by ELISA. Compared with respective luciferase.shRNA controls at P18 and P25, retinal VEGF was significantly decreased by treatment with lentivector VEGFA.shRNA or VEGF164.shRNA at both time points (Fig. 2C).
Figure 2.
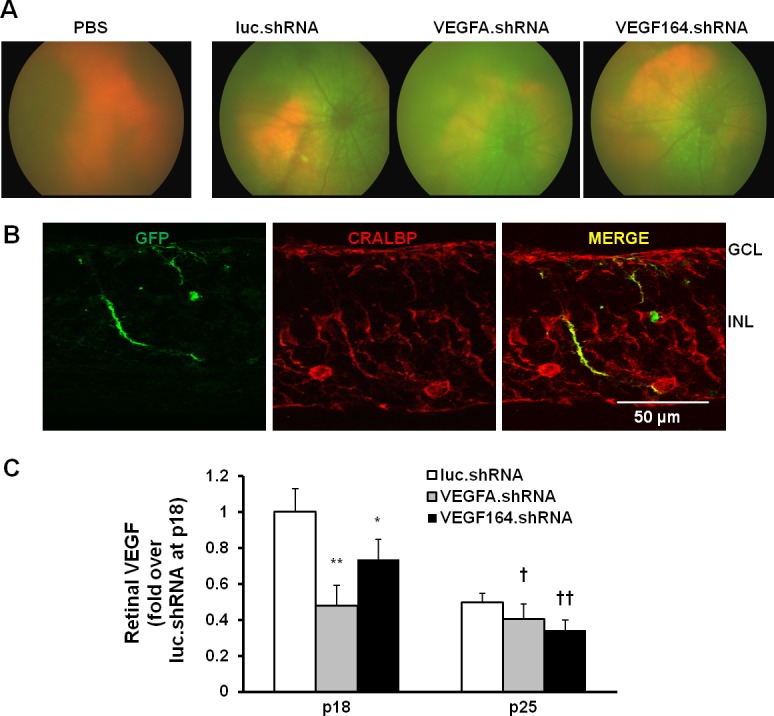
In vivo analysis of lentivector-delivered shRNA transduction in retina of pups raised in the rat ROP model at P18 and P25 following subretinal injection at P8. (A) Micron III images show GFP expression in retina of pups following lentivirus injection at P25. (B) GFP expression is localized with CRALBP-labeled Müller cells in retinal cryosections at P25. (C) ELISA of retinal VEGFA protein at P18 and P25. *P < 0.05. **P < 0.001 versus luc.shRNA at P18. †P < 0.05. ††P < 0.01 versus luc.shRNA at P25.
VEGFA.shRNA and VEGF164.shRNA each significantly inhibited IVNV at P18 compared with luciferase.shRNA (Figs. 3A, 3B). At P25, IVNV was significantly reduced in pup eyes treated with the luciferase.shRNA compared with those treated at P18. This was anticipated, because IVNV naturally regresses in the ROP model. At P25, VEGF164.shRNA significantly inhibited IVNV compared with VEGFA.shRNA, whereas VEGFA.shRNA increased IVNV at P25 compared with P18. These findings suggest that VEGF164.shRNA, but not VEGFA.shRNA, was superior in maintaining inhibition of IVNV (Figs. 3A, 3B), and that VEGFA.shRNA-treated eyes had recurrent IVNV. Neither VEGFA.shRNA nor VEGF164.shRNA had an effect on AVA at either P18 or P25, compared with luciferase.shRNA (Figs. 3A, 3C).
Figure 3.

Lentivector-derived shRNA to VEGFA or VEGF164 reduces IVNV without affecting physiological retinal vascular development (AVA) in the rat ROP model. Images of retinal flatmounts at P18 and P25 following subretinal injections in each group. (A) luc.shRNA, VEGFA.shRNA, and VEGF164.shRNA. *Points to avascular retinal area. A white arrow points to IVNV. (B) Quantification of IVNV. *P < 0.05. ***P < 0.001 versus luc.shRNA at P8. †P < 0.01 versus VEGFA.shRNA at P25. ###P < 0.001 versus VEGFA.shRNA at P18. (C) Avascular retina.
VEGFR2 Activation in Retinal ECs After Targeted VEGFA.shRNA or VEGF164.shRNA
To determine if targeted treatment with VEGFA.shRNA or VEGF164.shRNA inhibited total retinal VEGFR2 signaling, densitometry of p-VEGFR2 was measured from the ganglion cell layer to the RPE/choroid layer in cryosections colabeled with p-VEGFR2 and lectin (Figs. 4A, 4B). To determine the effect of VEGFA.shRNA or VEGF164.shRNA on VEGFR2 activation in endothelial cells, lectin and p-VEGFR2 colabeling was analyzed with densitometry in the cryosections at the junctions between avascular and vascular retina in the inner plexus (Figs. 4A, 4C). At P18, neither treatment significantly inhibited total retinal p-VEGFR2 density (Fig. 4B) compared with luciferase.shRNA, but VEGFA.shRNA and VEGF164.shRNA each reduced colabeling of lectin and p-VEGFR2 in the vascular/avascular junction where IVNV developed (Fig. 4C). At P25, there was increased p-VEGFR2 labeling in the retina in both VEGFA and VEGF164.shRNA sections compared with luciferase.shRNA (Fig. 4B). There was also increased p-VEGFR2 colabeling with endothelial cells in VEGFA.shRNA-treated sections but not in VEGF164.shRNA-treated ones (Fig. 4C).
Figure 4.

Analysis of VEGFR2 activation in pups treated with subretinal injections of lentivector-driven shRNAs in the rat ROP model. (A) Immunohistochemistry of p-VEGFR2 in retinal cryosections. (B) Semiquantification of p-VEGFR2 (blue) in total retina (***P < 0.001 versus luc.shRNA at P25). (C) Colabeling of p-VEGFR2 (blue) in lectin (red)-labeled ECs in the primary plexus (depicted within boxes; *P < 0.05, **P < 0.01 versus luc.shRNA at P18; ††P < 0.01 versus luc.shRNA at P25) from P18 and P25 pups treated with luc.shRNA, VEGFA.shRNA, and VEGF164.shRNA.
VEGFA.shRNA and VEGF164.shRNA Effects on Retinal Survival, Serum VEGF, and Pup Growth
VEGF is important to Müller cell health and neuroprotection.13,23,24 Therefore, we tested whether shRNAs to either VEGFA or VEGF164 affected retinal cell survival at P18 or P25 by assessing TUNEL staining (Fig. 5A). Compared with PBS, luciferase.shRNA increased TUNEL+ cells at P18 but not at P25 (Fig. 5B). Although we previously found VEGFA knockdown did not increase apoptosis in P18 retinal lysates,12 sections from VEGFA.shRNA, but not VEGF164.shRNA treated eyes had significantly increased TUNEL+ cells in both INL and ONL compared with luciferase.shRNA at P18. At P25, TUNEL+ cells were significantly reduced compared with respective groups at P18, and there were no significant differences in TUNEL+ cells among the groups at P25. Compared with PBS, luciferase.shRNA treatment did not alter the thickness of the retina (data not shown), the INL (Fig. 5C) or ONL (Fig. 5D). However, eyes treated with the VEGFA.shRNA had significantly reduced ONL thickness compared with luciferase.shRNA at both P18 and P25 (Fig. 5D). Compared with luciferase.shRNA, no differences in thickness of total retina at P18 or P25 (data not shown) or INL at P18 were detected after VEGFA.shRNA or VEGF164.shRNA injections (Fig. 5C). At P25, the INL was thicker in the VEGF164.shRNA group than the other groups (Fig. 5C). Subretinal VEGFA.shRNA or VEGF164.shRNA did not affect body weight gain (Fig. 6A) or serum VEGF (Fig. 6B) compared with luciferase.shRNA at either time point.
Figure 5.

Analysis of retinal apoptosis and retinal morphological changes in the pups treated with lentivector-driven shRNAs in the rat ROP model. Images of TUNEL staining (A) and number of TUNEL (red) positive cells (B) in retinal DAPI (blue) stained cryosections from P18 and P25 pups treated with luc.shRNA, VEGFA.shRNA, and VEGF164.shRNA (**P < 0.01, ***P < 0.001 versus PBS at P18; †††P < 0.001 versus luc.shRNA at P18). Quantification of the thickness of the INL (C) (***P < 0.001 versus PBS at P18; ††P < 0.01 versus luc.shRNA at P25) and the ONL (D) (***P < 0.001 versus luc.shRNA at P18; ††P < 0.001 versus luc.shRNA at P25) in DAPI-stained retinal cryosections from P18 and P25 OIR pups treated with luciferase.shRNA, VEGFA.shRNA, and VEGF164.shRNA.
Figure 6.
Lentivector-driven shRNAs treatment has no effect on pup growth and serum VEGF. Pup weight gains from P8 to P18 or P25 (A) and ELISA of serum VEGF in P18 or P25 pups (B) treated with luc.shRNA, VEGFA.shRNA, and VEGF164.shRNA in the rat ROP model.
Discussion
We provide evidence that targeting Müller cells with an shRNA to one splice variant of VEGF, VEGF164, was safer and more effective at inhibiting and maintaining inhibition of IVNV than targeting all splice variants of VEGFA in a rat model representative of human ROP. We chose to target Müller cells, because we found that VEGF–splice variant mRNAs localized to cells in the INL corresponding to CRALBP-labeled Müller cells and that knockdown of VEGFA in Müller cells inhibited IVNV at the early time point, P18.12 Previously, Bai et al.25 found that Müller cell-expressed VEGF was important in causing IVNV by developing and testing a Müller cell conditional VEGF knockout in the mouse OIR model. Our study not only differs by testing long-term safety and comparing knockdown of a Müller cell-expressed splice variant—VEGF164—to knockdown of Müller cell-VEGFA, but also studies the rat OIR model, which is a representative model of human ROP in places where oxygen is regulated.26
Studies in human preterm infants show that IVNV can recur following anti-VEGF treatments.27–29 Experimentally, broad inhibition of VEGF with an intravitreal neutralizing antibody led to upregulation of angiogenic compounds in association with recurrent IVNV.9 We hypothesized that inhibition of all VEGF splice variants would affect both pathologic and physiologic processes and lead to cell death or compensatory upregulation of angiogenic pathways. Therefore, we targeted one splice variant of VEGF to inhibit pathologic angiogenesis and potentially preserve physiologic effects from VEGF. We chose VEGF164 based on the finding that of the three rat splice variants of VEGFA (VEGF120, VEGF164, and VEGF188), VEGF164 was the most prevalent10,30 and unlike VEGF120 or VEGF188, was increased in association with both older developmental age and the ROP model compared with RA.10 We also found that VEGF164 was upregulated by repeated fluctuations in oxygenation, whereas VEGF120 was upregulated by hypoxia alone.30 Physiologic events can be stimulated by hypoxia alone.31 Also, evidence is accumulating that fluctuations in oxygenation are important in ROP.32,33 Others have reported VEGF164 as the splice variant most likely to cause inflammation and abnormal angiogenesis.14 For all these reasons, we compared the efficacy and safety of targeting Müller cell-VEGF164 or -VEGFA with shRNAs in the rat model of ROP. We chose a lentivector because lentivirus is incorporated into the genome and provides a robust means to study VEGF knockdown longitudinally.
Although we found that targeting VEGF164 with shRNA was more effective at maintaining inhibition of IVNV than targeting VEGFA, we cannot directly compare the two. The shRNA to VEGFA had greater knockdown in HEK164 reporter cell lines than did VEGF164-specific shRNA, which was selective to VEGF164 and not VEGF120. Therefore, the dose effect of VEGF164.shRNA in Müller cells may account for some of the outcomes we found. For example, there was greater knockdown of VEGF in P18 retinal lysates from eyes treated with VEGFA.shRNA than with VEGF164.shRNA; but after a longer time for lentiviral transduction, VEGF164.shRNA treatment reduced VEGF in P25 retinal lysates similar to VEGFA.shRNA. VEGF120, which was not knocked down by VEGF164.shRNA, has soluble properties enabling it to move to other cells within the retina, and may have accounted for the lack of cell death and increased INL thickness noted in P25 sections from eyes treated with VEGF164.shRNA.
Based on densitometry of labeled retinal sections, knockdown of VEGF164 or VEGFA had no effect on overall retinal VEGFR2 activation at P18; but at a later time point, both VEGFA.shRNA and VEGF164.shRNA each led to greater retinal p-VEGFR2. This suggests a compensatory increase in VEGFR2 activation. One explanation may be upregulation of VEGF in other cells in the retina. However, we did not find evidence of increased retinal VEGF at P25. The ELISA technique measures only unbound VEGF and may miss VEGF bound to VEGFR.34 We previously found that Müller cell-VEGF activates endothelial cell VEGFR2.12,35 Therefore, to detect differences in p-VEGFR2 among the groups, we used a semiquantitative approach with densitometry of labeled retinal sections for endothelial cell p-VEGFR2. This method showed reduced p-VEGFR2 densitometry in ECs at P18, but increased p-VEGFR2 in ECs at P25 retinas that had been treated with the VEGFA.shRNA, not VEGF164.shRNA, and in association with an increase in IVNV at P25, suggesting compensatory signaling effects.
In a previous publication12 and confirmed here, targeted knockdown of overexpressed VEGFA in Müller cells reduced IVNV at P18 without adversely affecting body weight gain or causing apoptosis measured as cleaved caspase-3 in retinal lysates.9 Here, using TUNEL, we found VEGFA knockdown caused more cell death compared with luciferase.shRNA treatment. This may be because TUNEL does not discriminate apoptosis from necrosis and that the sensitivity in detecting a small group of cells undergoing apoptosis was insufficient using Western analysis of whole retinal lysates. At P25, there was no difference in TUNEL+ cells, suggesting absence of ongoing cell death. However, VEGFA.shRNA also reduced ONL thickness at P25. Müller cells require VEGF for their own survival13 and produce VEGF and other factors important to retinal neuronal survival,28,29 including photoreceptors whose nuclei reside in the ONL. Conditional knockout of Müller cells in a mouse model led to photoreceptor degeneration.36 Future studies of the effects of long-term knockdown of Müller cell-VEGF such as through electrophysiology and spectral domain optical coherence tomography are warranted.
In summary, knockdown of splice variant, VEGF164, in Müller cells appears safer and more effective in inhibiting IVNV in the long-term than targeted knockdown of VEGFA, suggesting that maintaining the expression of some splice variants of VEGFA may improve survival of cells within the retina. Additional studies regarding potential compensatory signaling of Müller cells after VEGF inhibition and on long-term structure and function from knockdown of VEGFA or VEGF164 are warranted. Effects of VEGF knockdown on endothelial effectors are also important to determine.
Acknowledgments
Supported by National Institutes of Health (NIH)/National Eye Institute (NEI) Grants R01EY015130 (MEH), R01EY017011 (MEH), and R01-DK058702-10 (TK), and in part by an unrestricted grant from Research to Prevent Blindness, Inc., New York, New York, to the Department of Ophthalmology & Visual Sciences, University of Utah.
Disclosure: Y. Jiang, None; H. Wang, None; D. Culp, None; Z. Yang, None; L. Fotheringham, None; J. Flannery, None; S. Hammond, None; T. Kafri, None; M.E. Hartnett, None
References
- 1. Churchill AJ, Carter JG, Lovell HC, et al. VEGF polymorphisms are associated with neovascular age-related macular degeneration. Hum Mol Genet. 2006; 15: 2955–2961 [DOI] [PubMed] [Google Scholar]
- 2. Bird AC. Therapeutic targets in age-related macular disease. J Clin Invest. 2010; 120: 3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994; 331: 1480–1487 [DOI] [PubMed] [Google Scholar]
- 4. Nicholson B, Schachat AP. A review of clinical trials of anti-VEGF agents for diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2010; 248: 915–930 [DOI] [PubMed] [Google Scholar]
- 5. Cooke RWI, Drury JA, Mountford R, Clark D. Genetic Polymorphisms and Retinopathy of Prematurity. Invest Ophthalmol Vis Sci. 2004; 45: 1712–1715 [DOI] [PubMed] [Google Scholar]
- 6. Mititelu M, Chaudhary KM, Lieberman RM. An evidence-based meta-analysis of vascular endothelial growth factor inhibition in pediatric retinal diseases: Part 1. Retinopathy of prematurity. J Pediatr Ophthalmol Strabismus. 2012; 49: 332–340 [DOI] [PubMed] [Google Scholar]
- 7. Zeng G, Taylor SM, McColm JR, et al. Orientation of endothelial cell division is regulated by VEGF signaling during blood vessel formation. Blood. 2007; 109: 1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartnett ME, Martiniuk D, Byfield G, Geisen P, Zeng G, Bautch VL. Neutralizing VEGF decreases tortuosity and alters endothelial cell division orientation in arterioles and veins in a rat model of ROP: relevance to plus disease. Invest Ophthalmol Vis Sci. 2008; 49: 3107–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCloskey M, Wang H, Jiang Y, Smith GW, Strange J, Hartnett ME. Anti-VEGF antibody leads to later atypical intravitreous neovascularization and activation of angiogenic pathways in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2013; 54: 2020–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Budd SJ, Hartnett ME. Increased angiogenic factors associated with peripheral avascular retina and intravitreous neovascularization: a model of retinopathy of prematurity. Arch Ophthalmol. 2010; 128: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenberg KP, Geller SF, Schaffer DV, Flannery JG. Targeted transgene expression in Müller glia of normal and diseased retinas using lentiviral vectors. Invest Ophthalmol Vis Sci. 2007; 48: 1844–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Smith GW, Yang Z, et al. Short hairpin RNA-mediated knockdown of VEGFA in Müller cells reduces intravitreal neovascularization in a rat model of retinopathy of prematurity. Am J Pathol. 2013; 183: 964–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saint-Geniez M, Maharaj AS, Walshe TE, et al. Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PLoS One. 2008; 3: e3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003; 198: 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishida S, Usui T, Yamashiro K, et al. VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci. 2003; 44: 2155–2162 [DOI] [PubMed] [Google Scholar]
- 16. Penn JS, Henry MM, Tolman BL. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res. 1994; 36: 724–731 [DOI] [PubMed] [Google Scholar]
- 17. Wang H, Byfield G, Jiang Y, Smith GW, McCloskey M, Hartnett ME. VEGF-mediated STAT3 activation inhibits retinal vascularization by down-regulating local erythropoietin expression. Am J Pathol. 2012; 180: 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005; 102: 13212–13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giering JC, Grimm D, Storm TA, Kay MA. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol Ther. 2008; 16: 1630–1636 [DOI] [PubMed] [Google Scholar]
- 20. Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005; 102: 13212–13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu C, Vanderveen DK, Hellstrom A, Lofqvist C, Smith LE. Longitudinal postnatal weight measurements for the prediction of retinopathy of prematurity. Arch Ophthalmol. 2010; 128: 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vanhaesebrouck S, Daniels H, Moons L, Vanhole C, Carmeliet P, De Zegher F. Oxygen-induced retinopathy in mice: amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr Res. 2009; 65: 307–310 [DOI] [PubMed] [Google Scholar]
- 23. Foxton RH, Finkelstein A, Vijay S, et al. VEGF-A is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am J Pathol. 2013; 182: 1379–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yourey PA, Gohari S, Su JL, Alderson RF. Vascular endothelial cell growth factors promote the in vitro development of rat photoreceptor cells. J Neurosci. 2000; 20: 6781–6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bai Y, Ma J-X, Guo J, et al. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol. 2009; 219: 446–454 [DOI] [PubMed] [Google Scholar]
- 26. Hartnett ME, Penn JS. Mechanisms and management of retinopathy of prematurity. N Engl J Med. 2012; 367: 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel RD, Blair MP, Shapiro MJ, Lichtenstein SJ. Significant treatment failure with intravitreous bevacizumab for retinopathy of prematurity. Arch Ophthalmol. 2012; 130: 801–802 [DOI] [PubMed] [Google Scholar]
- 28. Ittiara S, Blair MP, Shapiro MJ, Lichtenstein SJ. Exudative retinopathy and detachment: a late reactivation of retinopathy of prematurity after intravitreal bevacizumab. J AAPOS. 2013; 17: 323–325 [DOI] [PubMed] [Google Scholar]
- 29. Hu J, Blair MP, Shapiro MJ, Lichtenstein SJ, Galasso JM, Kapur R. Reactivation of retinopathy of prematurity after bevacizumab injection. Arch Ophthalmol. 2012; 130: 1000–1006 [DOI] [PubMed] [Google Scholar]
- 30. McColm JR, Geisen P, Hartnett ME. VEGF isoforms and their expression after a single episode of hypoxia or repeated fluctuations between hyperoxia and hypoxia: relevance to clinical ROP. Mol Vis. 2004; 10: 512–520 [PMC free article] [PubMed] [Google Scholar]
- 31. Chan-Ling T, Gock B, Stone J. The effect of oxygen on vasoformative cell division: evidence that ‘physiological hypoxia' is the stimulus for normal retinal vasculogenesis. Invest Ophthalmol Vis Sci. 1995; 36: 1201–1214 [PubMed] [Google Scholar]
- 32. York JR, Landers S, Kirby RS, Arbogast PG, Penn JS. Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birth-weight infants. J Perinatol. 2004; 24: 82–87 [DOI] [PubMed] [Google Scholar]
- 33. Hauspurg AK, Allred EN, Vanderveen DK, et al. Blood gases and retinopathy of prematurity: the ELGAN study. Neonatology. 2011; 99: 104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geisen P, Peterson L, Martiniuk D, Uppal A, Saito Y, Hartnett M. Neutralizing antibody to VEGF reduces intravitreous neovascularization and does not interfere with vascularization of avascular retina in an ROP model. Mol Vis. 2008; 14: 345–357 [PMC free article] [PubMed] [Google Scholar]
- 35. Werdich XQ, Penn JS. Specific involvement of Src family kinase activation in the pathogenesis of retinal neovascularization. Invest Ophthalmol Vis Sci. 2006; 47: 5047–5056 [DOI] [PubMed] [Google Scholar]
- 36. Shen W, Fruttiger M, Zhu L, et al. Conditional Müller cell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J Neurosci. 2012; 32: 15715–15727 [DOI] [PMC free article] [PubMed] [Google Scholar]