

Abstract
We report the development of a structured illumination microscopy instrument specifically designed for the requirements for high-area-throughput, optically-sectioned imaging of large, fluorescently-stained tissue specimens. The system achieves optical sectioning frame-rates of up to 33 Hz (and pixel sampling rates of up to 138.4 MHz), by combining a fast, ferroelectric spatial light modulator for pattern generation with the latest large-format, high frame-rate scientific CMOS camera technology. Using a 10X 0.45 NA objective and a 7 mm/sec scan stage, we demonstrate 4.4 cm2/min area-throughput rates in bright tissue-simulating phantoms, and 2 cm2/min area-throughput rates in thick, highly-absorbing, fluorescently-stained muscle tissue, with 1.3 μm lateral resolution. We demonstrate high-contrast, high-resolution imaging of a fluorescently-stained 30.4 cm2 bovine muscle specimen in 15 minutes comprising 7.55 gigapixels, demonstrating the feasibility of the approach for gigapixel imaging of large tissues in short timeframes, such as would be needed for intraoperative imaging of tumor resection specimens.
OCIS codes: (170.2520) Fluorescence microscopy, (170.4730) Optical pathology, (170.1610) Clinical applications
1. Introduction
The current clinical reference standard for tissue diagnosis is permanent histopathology, in which excised tissue biopsies or surgical specimens are fixed, dehydrated, embedded in paraffin, sectioned into microscopically thin slices with a microtome, de-paraffinized, stained with histological stains, and mounted on microscope slides for observation by a pathologist. Although this process is the gold-standard by which histological diagnoses are made, the technology is laborious and time-consuming, and has remained little unchanged in the last century. A faster alternative for permanent histopathology replaces the chemical fixation and paraffin-embedding step with freezing of the tissue, after which the tissue may be physically sectioned on the microtome (or cryotome). However, the process still requires that the tissue be physically sectioned into thin slices, each of which must be stained and mounted on microscope slides for analysis.
A number of clinical situations could be benefited by a more rapid histological processing method, which reduces the number of steps between obtaining a fresh tissue specimen and rendering a diagnosis. For example, there is a need for rapid pathologic detection of residual cancer on the surface of tumor resection specimens (known as positive tumor margins) [1,2], such that the surgical procedure can be revised in time to prevent cancer from being left behind in the patient. Also, for diagnostic procedures such as prostate core needle biopsy, where false-negative rates can be as high as 50% [3,4], a rapid assessment of the biopsy diagnosis at the point-of-care could allow the clinician to continue to collect biopsies from the organ until the suspicious lesion is successfully sampled, preventing an unnecessary repeat biopsy procedure. In both of these cases, high quality microscopic images of the tissue must be obtained in rapid timeframes, in order for a pathologic assessment to be useful for guiding the intervention. With regard to tumor margin assessment, current pathology techniques are too laborious and slow to allow comprehensive analysis of a large surgical specimen, and it is furthermore not possible to preferentially shave microscopically-thin physical sections from the entire surface of the specimen. With regard to biopsy assessment, even with frozen section analysis, it is not practical to freeze, stain, and section a large number of core needle biopsies (between 6 and 20 for prostate biopsy) within the timeframe of a typical biopsy procedure in the clinic.
A number of groups have employed fluorescent staining of unfixed tissues followed by optical sectioning microscopy with laser-scanning microscopes (including confocal microscopy and multi-photon microscopy) as a means to obtain high-contrast pathologic images of tissues at the point-of-care [5–10]. The fluorescent stains highlight the microanatomy similarly to histological stains, and the rejection of out-of-focus background allows thin optical sections of the intact tissue to be obtained in lieu of thin physical sections. However, the need to perform beam scanning and sequential pixel acquisition limits the area-throughput of such methods, making coverage of large areas (i.e. 10’s to 100’s of cm2) challenging within point-of-care timeframes. In organs where positive surgical tumor margins are problematic, such as the breast and prostate, specimen areas routinely exceed 100 cm2 and can easily approach 300 cm2 [11].
We previously reported on the use of structured illumination microscopy (SIM) as a means to provide rapid optical sectioning microscopy of ex vivo tissue specimens [12]. Structured illumination microscopy is a wide-field method for optical sectioning first reported by Neil et al. [13]. The method involves modulating the in-focus plane of the tissue with a sinusoidal grid projected onto the specimen through the illumination system. By phase-stepping the grid pattern over the sample and collecting multiple images (typically 3), and employing a simple square-law detection algorithm, the modulated focal plane is retained in the final image while the un-modulated, out-of-focus plane is rejected. In our previous work, we determined that the concept of SIM for imaging of tumor resection specimens was feasible, using a transgenic mouse model of sarcoma as a pre-clinical test-bed. However, that specific implementation of the system used a standard CCD camera and a twisted nematic crystal spatial light modulator (SLM), which were limiting factors in terms of speed and resolution.
Here, we report on the development of a next-generation Rapid Optical Sectioning Specimen Scanner (ROS3), based on incoherent structured illumination microscopy, and developed specifically for the design requirements for rapid, high-area throughput fluorescence microscopy of intact surgical and biopsy specimens. We employ the latest high-speed digital SLM and sCMOS camera technology to achieve high resolution, optically-sectioned images of fluorescent samples at area-throughput rates of up to 4.4 cm2/min at current scan stage speeds, with 1.3 μm lateral resolution. In this manuscript, we report on the validation of the ROS3 system performance metrics on tissue simulating phantoms, and demonstrate the feasibility of the system for rapid optical sectioning microscopy of large tissue areas in clinically-relevant timeframes.
2. Materials and methods
2.1 ROS3 Instrumentation and SIM imaging
Figure 1 contains a schematic of the ROS3 system. The system is constructed around a modular automated epi-fluorescence microscope platform (RAMM, Applied Scientific Instrumentation), which incorporates a motorized XY stage and controller (7 mm/sec lateral translation speed) and piezoelectric Z objective positioner. Light from a blue LED (Thorlabs) is collected by an aspheric condenser lens, transmitted through a polarizing beam splitter (PBS) (Moxtek), and is imaged onto a ferroelectric liquid-crystal-on-silicon (LCoS) spatial light modulator (3DM, Forth Dimension Displays), which was placed in a plane conjugate to the sample plane. The LED die was imaged onto the SLM to increase pattern contrast and illumination uniformity, and to maximize étendue. Light reflected from the SLM was reflected by the PBS through a clean-up polarizer to improve contrast (Thorlabs), through the illumination tube lens, and through the bandpass excitation filter (Semrock). The filtered excitation light was reflected into the imaging objective (Nikon, Plan Apo 10X 0.45 NA) by a dichroic mirror, and formed an image of the SLM onto the sample. The approximate power at the sample was measured with a thermal power sensor (Thorlabs) placed above the objective on the microscope stage. Fluorescence from the sample was collected by the objective, transmitted through the dichroic mirror and the bandpass emission filter, and imaged by a Nikon tube lens onto a scientific CMOS camera (Orca Flash 4.0 v2, Hamamatsu). Synchronization and control of the LED, SLM, stage, objective, and camera was achieved via custom software written in LabVIEW 8.5 (National Instruments). Sinusoidal grid patterns were generated in MATLAB (Mathworks) and uploaded to the SLM on-board memory prior to imaging. The self-contained SIM module is mounted to the RAMM base, and the entire system fits comfortably on a custom 4′W x 3′D wheeled lab bench (OnePointe Solutions).
Fig. 1.
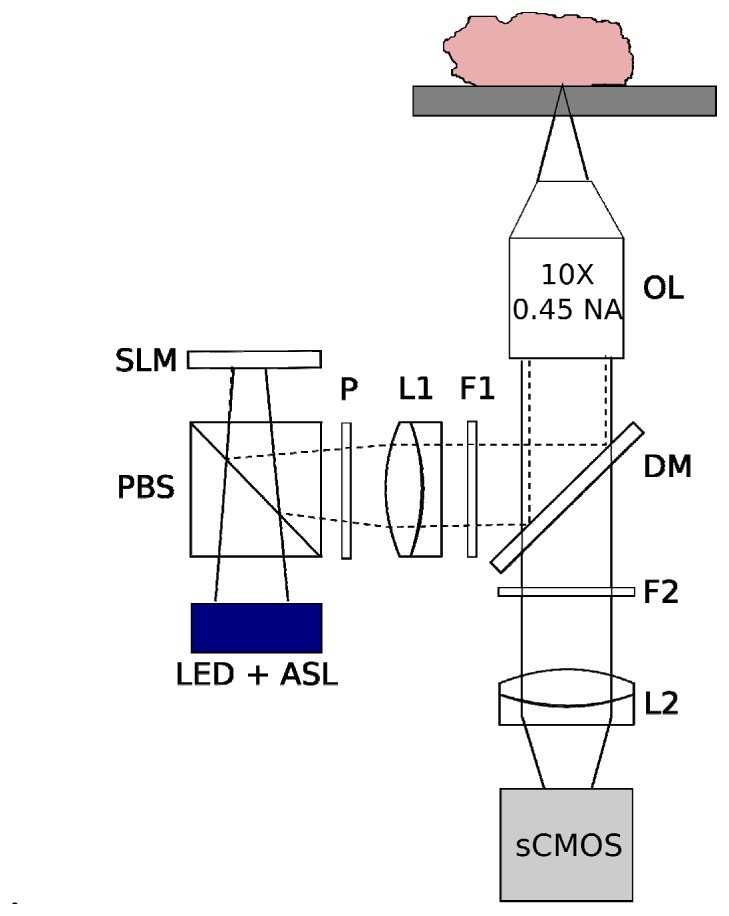
Schematic of the ROS3 instrumentation and implementation for tissue surface imaging. The objective lens (OL) is positioned underneath the tissue sample, and is translated in the z-direction via a piezoelectric transducer. The tissue rests above the objective lens on a standard X-Y motorized microscope stage. ASL, aspheric condenser lens. PBS, polarizing beam splitter. P, clean-up polarizer. L1, L2, achromat tube lenses. DM, dichroic mirror. F1, bandpass excitation filter. F2, bandpass emission filter.
Configured thusly, the ROS3 system has a single-frame field-of-view (FOV) of 1.3 mm x 1.3 mm, at 4.2 megapixel resolution (2048 x 2048 pixels). For the particular objective and camera used in this work, the limiting factor for resolution is the camera pixel size, which is 0.65 μm at the sample. The diffraction-limited optical resolution of the optical system is 0.745 μm, using the commonly accepted formula of R = 0.61λ/NA, where λ = 550 nm for the emission bandpass filter used in this work. Therefore, by the Nyquist criterion, the lateral resolution of the system is limited not by diffraction, but by the size of the camera pixels, and is limited to 1.3 μm. Different FOV-to-optical-resolution ratios can be achieved by changing the imaging objective to lower or higher magnification models. The SLM was operated in 8-bit bitplane weighting mode, and the pattern display was synchronized with the camera exposure. Ferroelectric liquid crystal displays must be operated in DC-balancing mode, in which each pixel is driven in positive and negative directions for equal durations. Therefore, to maintain contrast of the patterns, the LED was triggered to only illuminate the SLM during the positive portion of the display cycle. At the maximum full-resolution camera frame-rate of 100 frames-per-second (fps) in internal trigger mode, the corresponding full-resolution SIM frame-rate was 100/3 = 33 fps (since 3 sequential frames are needed for one SIM frame). This corresponded to a 138.4 MHz pixel sampling rate for obtaining optically-sectioned images. In practice, the camera frame-rate (given by the reciprocal of the integration time for integration times ≥ 10 ms) was adjusted to optimize the signal intensity from the sample. The duration of the 8-bit bitplane SLM sequence used in our work was 4.135 ms; we used integration times that were integer multiples of the sequence duration when using short integration times, to properly synchronize the pattern display with the camera exposure.
Images from FOVs larger than a single frame were achieved by translating the automated microscope stage in 1.3 mm increments in the X and Y directions (serpentine scanning), and creating a mosaic from the resulting images. Image mosaics were saved as 16-bit TIFF files for further analysis in ImageJ or MATLAB.
SIM was performed using the square-law detection algorithm described by Neil et al. [13]. Briefly, for each grid pattern frequency, 3 sequential images were taken with the pattern phase-shifted by one-third of the grid period between each image. Optically-sectioned images were computed using Eq. (1):
| (1) |
The computation of the SIM image and assembly of the images into mosaics was performed automatically during the acquisition procedure by the custom LabVIEW application.
2.2 Characterization on solid phantoms
Solid phantoms were used to characterize the performance metrics of the system and were prepared as previously described [12]. Briefly, to test the axial response of the system, a droplet of 1 μm fluorescent polystyrene beads (Fluoresbrite Yellow Green Microspheres, Polysciences, Inc.) was smeared onto the cover glass bottom of a culture dish (Mat-tek), and covered with a layer of optically-clear poly(dimethysiloxane) (PDMS) polymer (Phantom A). To simulate imaging of fluorescently-stained cell nuclei at the surface of tissue, 10 μm fluorescent beads were smeared onto the cover glass, and were covered with a layer of PDMS admixed with TiO2 powder, to simulate the scattering of tissue (reduced scattering coefficient μs′ ~20 cm−1) (Phantom B). In both phantoms, the fluorescent beads were located in a single layer at the cover glass surface, with either clear or turbid PDMS layers behind the beads.
The axial response of the system to defocus was determined as a function of grid pattern frequency using Phantom A. Absolute spatial frequency of the grid pattern at the sample, f, was converted to normalized spatial frequency, ν, via the relationship ν = fλ/NA, where NA is the numerical aperture of the objective lens. For each pattern frequency, the microscope objective was scanned through the sample in 1-3 μm increments (depending on the frequency), and a SIM image was acquired at each z-plane. The axial response of the system was determined by averaging the intensity of a region-of-interest (ROI) containing the smear of sub-diffraction polystyrene beads, and plotting the mean intensity versus z-position. Measured optical section thicknesses, determined as the full-width-half-maximum (FWHM) of the axial response, were compared to theoretical calculations for incoherent SIM according to Karadaglic and Wilson [14].
The imaging speed of the system and resulting signal-to-background ratio (SBR) was measured using Phantom B. A single region of the phantom was imaged in SIM mode using 12.4 ms, 25 ms, 50 ms, and 100 ms integration times (corresponding to 26.9, 13.3, 6.7, and 3.3 Hz SIM imaging rates, respectively). Corresponding non-SIM images were collected for comparison. Ten (10) sequential images were acquired for each integration time. The SBR for a single image was measured by averaging the intensity of a bead-containing image ROI, and dividing by the average intensity of an identically-sized ROI without any beads. The average and standard deviation of the SBR was calculated over the 10 sequential images using the same ROIs. The LED output was adjusted to obtain the highest available signal without saturating the sCMOS detector prior to imaging. In addition, mosaics of the phantom were obtained.
2.3 Tissue imaging
The ability of the system to practically image large (30.4 cm2) areas was tested on excised animal tissue. Slices of bovine muscle (~3 mm thick) were cut to dimensions of 10 x 5 cm area, stained in acridine orange solution (Sigma Aldrich) for 5 minutes, and placed on 10 x 5 cm cyclic olefin copolymer slides for imaging. Widefield (no optical sectioning) and SIM images of the tissue were obtained of a 3.8 x 7.9 cm region of the tissue using the ROS3 system (1,800 frames total). For this sample, the illumination power at the sample was set to 0.3 mW and the camera integration time was set to 100 ms (SIM frame-rate = 3.33 fps).
3. Results
Figure 2 contains a plot of the measured and theoretical FWHM of the axial response as a function of normalized spatial frequency (measured using Phantom A). As expected, the optical section thickness decreases as the grid pattern spatial frequency is increased, and there is fairly good agreement between the measured and theoretical axial responses. In all cases the measured axial FWHM is slightly larger than the computed value; this may be due to the fact that we used 1 μm beads instead of an infinitesimally thin fluorescent sheet. In our previous work with coherent SIM [12], we determined that there is little benefit to using normalized spatial frequencies larger than about 0.17, since the increase in optical sectioning power is small, and may be outweighed by decreasing SNR. Here, we show that for incoherent SIM, the optimum normalized spatial frequency is lower, at around 0.1, beyond which there is little decrease in optical section thickness. This is an advantage of incoherent SIM, in that the optical section thickness for a given normalized spatial frequency is smaller than that for coherent SIM [14], and allows lower pattern frequencies with higher SNR to be used to obtain the same optical sectioning effect.
Fig. 2.
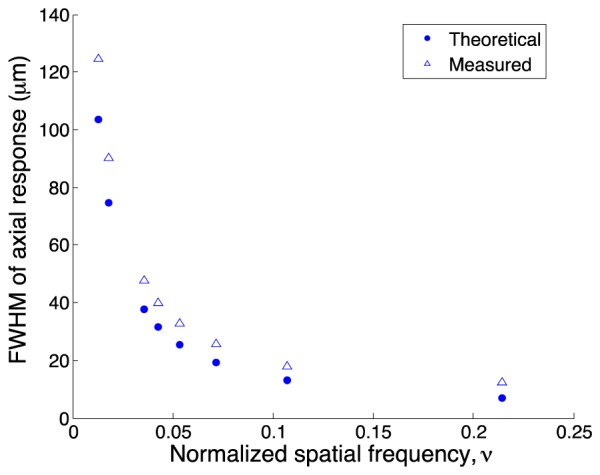
Full-width half-maximum (FWHM) of the measured and theoretical axial responses for incoherent SIM as a function of normalized spatial frequency.
Figure 3 contains mosaics of Phantom B demonstrating the improvement in SBR via the use of SIM, as well as a comparison of the image quality for 2 different SIM frame-rates. Figure 3(A) contains a mosaic of the phantom taken with uniform illumination (no optical sectioning). As expected, there is significant background from the scattering of the fluorescence by the TiO2 powder above the fluorescent microspheres (zoom shown in Fig. 3(D)). However, in Fig. 3(B), SIM was used to optically section the phantom surface using a normalized pattern frequency of 0.05 (33 μm section thickness) and an integration time of 12.4 ms (corresponding to a SIM frame-rate of 26.9 Hz). Although the system is capable of running at 10 ms integration time (for a 33 Hz SIM frame-rate), the time required to display one full pattern on the SLM is 4.135 ms. Unless integer multiples of this duration are used for the camera integration time, especially for short integration times, significant banding artifacts are present due to collection of a partial pattern image by the camera. Increasing the integration time slightly to 12.4 ms offered a closer synchronization between the SLM display bitplanes and the camera exposure time, and largely eliminated the banding (although some still remains, see white arrow in Fig. 3(B)). This 9.1 x 9.1 mm2 area was scanned in 11.62 seconds, only 1.82 seconds of which was image acquisition time; in this case, the stage speed was the limiting factor for the overall scan time. As seen in the zoom in Fig. 3(E), even at this high frame-rate, the image quality and contrast is significantly improved over the non-sectioned image in Fig. 3(D). The individual beads are difficult to distinguish in the non-sectioned image due to high background fluorescence in Fig. 3(D); however, in Fig. 3(E), the reduction in background fluorescence via SIM enables the beads to be clearly resolved. Figure 3(C) contains a mosaic of the phantom at a SIM frame-rate of 3.33 Hz. At this slower frame-rate, there are no banding artifacts, due to the longer integration time, which reduces the effect of imperfect synchronization between the SLM display switching and the camera exposure. In this case, the image acquisition time (14.7 seconds) becomes the limiting factor for the overall scan time of 24.5 seconds. As seen in the zoom in Fig. 3(F), again there is good image quality and contrast, although note that it is similar to that obtained in the much faster 26.9 Hz image. The dark areas to the right side of the phantom in the SIM mosaics correspond to an area where the spheres were out of focus and were sectioned out by the application of SIM.
Fig. 3.

Images of Phantom B taken with the ROS3 system. A-C are mosaics of the phantom, corresponding to an area of 9.1 x 9.1 mm2 (scale bar 1 mm), and D-F are zooms corresponding to the boxed regions in A-C (scale bar 50 μm). A) Non-sectioned image taken with uniform illumination. B) SIM image at 12.4 ms integration time (SIM frame-rate = 26.9 Hz). The total image acquisition time was 1.82 seconds, and the stage translation time was 9.8 seconds (11.62 seconds total scan time). White arrow is aligned with the striping artifact due to inexact timing between the SLM display sequence and end of the camera exposure. C) SIM image at 100 ms integration time (SIM frame-rate = 3.33 Hz). The total image acquisition time was 14.7 seconds, and the stage translation time was 9.8 seconds (24.5 seconds total scan time).
The signal-to-background ratio as a function of imaging speed was calculated from the series of phantom images shown in Fig. 4 . A normalized pattern spatial frequency of 0.05 (33 μm section thickness) was used for SIM in these images. Figure 4(A) is an image of the phantom with no optical sectioning from SIM (80.6 Hz frame-rate), whereas Fig. 4(B)-4(E) correspond to optically-sectioned SIM images at 26.9, 13.3, 6.7, and 3.3 Hz frame-rates, respectively. The horizontal banding artifacts noted previously can be clearly observed in Fig. 4(B) and 4(C). To quantify the effect of this banding artifact on image contrast, the SBR was calculated in the areas of the artifact as well as in the unaffected areas for each SIM frame-rate. Three (3) regions of interest (ROI’s) used to calculate the SBR are shown in Fig. 4(A), and are labeled S1, S2, and B, where S1 is the bead signal outside of the artifact region, S2 is the bead signal inside the artifact region, and B is the background in an artifact-free region. Each ROI was 75 x 75 pixels. Figure 4(F) contains a barchart demonstrating the effect of the artifact on SBR in the images as a function of frame-rate. The SBR in the artifact-free region is higher than that in the region of the artifact for 26.9 and 13.3 Hz frame-rates; however, at SIM frame-rates of 6.7 Hz or lower, there is no significant difference in SBR in these regions. In all cases, the SBR for the SIM images is several-fold higher than the SBR for the non-SIM image, demonstrating the improvement in contrast due to optical sectioning with SIM.
Fig. 4.

Single-frame (1.3 x 1.3 mm) images of Phantom B. A) Non-sectioned (standard wide-field) image at 80.6 Hz frame-rate. SIM-sectioned images at B) 26.9 Hz, C) 13.3 Hz, D) 6.7 Hz, and E) 3.3 Hz frame-rates. Scale bars = 100 μm. F) Average signal-to-background ratio (SBR) for each of the frame-rates calculated over 10 sequential images, computed using the ROI’s indicated in A. Error bars are ± one standard deviation. S1/B = SBR in artifact-free region, S2/B = SBR in region of horizontal banding artifact.
An image mosaic of bovine muscle stained with acridine orange is shown in Fig. 5 . This image corresponds to an image area of 30.4 cm2, and is composed of 1,800 individual frames. A normalized spatial frequency of 0.013, corresponding to an optical section thickness of 125 μm, was used for these images. To maximize the signal from the tissue, the camera integration time was set at 100 ms, which corresponded to a SIM frame-rate of 3.33 Hz. This resulted in a SIM imaging time of 300 ms for each frame, or 9 minutes total image acquisition time for the entire 30.4 cm2 area. Additionally, the translation stage time between successive frames was ~200 ms, which added a total stage translation time of 6 minutes. Altogether, the total scan time for this sample, including stage translation, was 15 minutes, for an area-throughput rate of 2 cm2/min.
Fig. 5.

ROS3 mosaic of a 7.8 x 3.9 cm piece of bovine muscle stained with acridine orange (30.4 cm2 total area). The image comprises 1,800 individual SIM frames, each of which are 1.3 x 1.3 mm at 2048 x 2048 pixel resolution. The entire mosaic comprises 7.55 gigapixels, and was acquired in 15 minutes (9 minutes image acquisition time + 6 minutes stage translation time). This corresponds to an area throughput rate of 2 cm2/min at 100 ms integration time (3.3 Hz SIM frame-rate). Areas labeled (NO SIM) correspond to standard wide-field imaging for comparison purposes, to demonstrate the increased contrast provided by SIM. Individual nuclei are clearly resolved in the zoomed-in SIM images of areas E, F, and G (white arrow indicates single nucleus in G). Note: The file size of the upper raw 16-bit mosaic was 15 GB; all images were converted to a lower resolution JPG for inclusion in this manuscript.
In the zoomed-in images of Fig. 5(E), 5(F), and 5(G), the detail of the tissue becomes more readily apparent. In Fig. 5(F) NO SIM, a non-sectioned image of the tissue is shown, whereas in Fig. 5(F) SIM, the corresponding optically-sectioned image is shown. The benefit of optical sectioning via SIM is clearly demonstrated when comparing the two images; the individual nuclei, seen as bright spots at the periphery of the muscle fibers, are apparent in the SIM image, but are obscured by background fluorescence in the non-sectioned image (5F NO SIM). Figure 5(E) contains yet another digital zoom, corresponding to the area enclosed by the box in Fig. 5(D). The nuclei of the adipose cells in this area of the tissue are clearly observed in Fig. 5(G), demonstrating the subcellular resolution of the system.
4. Discussion
A recent report demonstrated video-rate SIM for optical-sectioning imaging of large tissue areas, in which the maximum full-resolution frame-rate demonstrated was 16.6 Hz [15]. In this manuscript we improve upon those results, and demonstrate the feasibility of SIM for video-rate full-resolution (26.9 Hz at 2048 x 2048 pixel resolution) imaging of fluorescently-stained tissue surfaces. We used a high-speed sCMOS camera and a ferroelectric spatial light modulator with a 40 μs switching time, which allowed the grid pattern to be translated at speeds much faster than the camera frame-rate. We point out that in this type of mosaicking it is important to maximize the single frame FOV as much as possible while maintaining the desired resolution, since the number of scan steps required to cover a larger area is inversely proportional to the squared area of the single-frame FOV. This confers a speed advantage to the ROS3 system as compared to laser scanning confocal systems, since the small pixels and large image sensor area of the sCMOS camera enables high resolution imaging over large FOVs without any consideration of pixel dwell time, since all pixels are obtained in parallel.
One disadvantage of SIM compared to laser scanning confocal is that since the shot noise of the background is collected along with the in-focus signal, the SNR of the method is less than that of confocal when attempting to image deep into the tissue [16, 17]. The primary advantage of SIM for surface imaging of tissues is the reduction in out-of-focus background fluorescence, which serves to dramatically increase the contrast of features at the surface, as demonstrated in this work. As others have predicted, we observed qualitatively that the image quality of SIM degrades significantly when attempting to image into thick tissue at a depth of 2-3X the optical section thickness (which depends on the pattern frequency). However, since SIM is an overall light-efficient technique, for surface imaging applications it is expected to perform comparably or better than confocal in terms of SNR when the modulation depth is greater than 0.094 [17]. The achievable modulation depth depends on the pattern projection system and the sample under investigation, however in our previous work [12] we demonstrated modulation depths ranging from 0.17 - 0.24 in thick tumor tissues.
Tissue-simulating phantoms composed of a layer of bright fluorescent microspheres with a scattering background were used to test the limits of the system with respect to speed. We demonstrate that it is possible to perform SIM imaging at 26.9 Hz, although some artifact is present due to imperfect overlap between the SLM bitplane durations and the sCMOS exposure. The artifact resulted in a ~40% penalty in SBR in the area of the artifact, which improved to a 3% SBR penalty as the integration time was increased to 100 ms (3.33 Hz SIM frame-rate). The SBR for the SIM images compared to the non-SIM (i.e. widefield) images is improved by several-fold, even in the presence of artifact.
Figure 5 demonstrates the feasibility for imaging large area (30 cm2) areas in clinically relevant timeframes. For this particular sample, a 3.33 Hz SIM frame-rate was used, for an image acquisition time of 300 ms per frame or 9 min, and a total stage scanning time of 6 min (200 ms per step), for a real image scan time of 15 min. This particular sample was composed of red skeletal muscle with a high concentration of myoglobin, which significantly attenuated the excitation light at 470 nm. This contributed to decreased brightness of the sample, which necessitated the use of 100 ms integration time to maximize the sCMOS dynamic range. Less highly-absorbing samples would be expected to have a brighter signal, which would allow faster SIM frame-rates to be used. In any case, even at this relatively slow frame-rate, we achieved a real imaging area-throughput of 2 cm2/min at 1.3 μm resolution. A normalized pattern spatial frequency of 0.013 was used for this sample as it offered a good balance between recovered in-focus signal and background rejection. The longer depth-of-field also helped to keep the specimen in focus when scanning over large areas with varying topography. Thinner optical sections (albeit with lower recovered in-focus signal) can be achieved by simply displaying higher spatial frequencies on the SLM, and can be optimized for each individual sample to achieve the desired result.
In Fig. 3(B) and 3(E), as well as Fig. 4(B), we demonstrate in phantoms with bright fluorescent microspheres that it is possible to increase the SIM frame-rate to 26.9 Hz when the sample is sufficiently bright, giving an area throughput of 4.4 cm2/min at current scan-stage speeds. At this frame-rate, the limiting throughput factor for overall speed in our system was the microscope translation stage, which has a lateral translation speed of 7 mm/sec, and required ~200 ms for each 1.3 mm step between frames. Faster microscope stages (e.g., 28 mm/sec) are available, and could be used to improve the overall scanning speed. For example, if a 28 mm/s translation stage had been used for the phantoms shown in Fig. 4(B) (imaged at 26.9 Hz SIM frame-rate) then we would have achieved an area throughput of 11.7 cm2/min. We note that in the absence of stage scanning limitations, the inherent area throughput possible with our system is (0.13 x 0.13 cm2/frame) * (26.9 frames/s) * (60 s/min) = 27.3 cm2/min. Combined with an appropriate video mosaicking algorithm, area throughput could be maximized by continuously moving the sample over the objective. In any case, with our current stage (employing a “start-stop” scanning routine), and imaging at a SIM frame-rate of 3.33 Hz, we demonstrate that it is practical to image a 40 cm2 tissue area at 1.3 μm resolution in 20 minutes. This could increase to 234 cm2 in 20 minutes when the system is operated at the full 26.9 Hz imaging rate (assuming a bright sample) and a 28 mm/s stage is used. Twenty minutes is the commonly accepted time for an in-procedure frozen-section analysis, and is often used as the benchmark for how fast a point-of-care pathology imaging method must be to gain clinical acceptance [11].
The banding artifacts observed in the SIM images at short integration times are due to a software limitation with the camera external trigger-out. In our application, the camera is continuously operated in internal trigger (also called “free-running”) mode, in which the frame-rate is equal to the reciprocal of the integration time, for integration times greater than or equal to 10 ms. At the end of each frame readout, an external trigger pulse, slightly delayed from the end of the camera readout, is sent by the camera to the SLM to trigger the switch to the next pattern in the sequence. The consequence of this delay is that the next frame begins to read-out slightly before the SLM switches to the next pattern in the sequence, which is the source of the horizontal banding artifacts. These are most prominent when the integration times are short; longer integration times reduce the contribution of this asynchrony to the image as shown in Fig. 4. This artifact could be reduced or eliminated in the future by deriving a trigger signal from the camera at a user-programmed delay from the start of the readout (rather than the end of the readout). Such a signal is already able to be derived from the camera in the manufacturer-supplied software application, but is currently unavailable in the LabVIEW software development package provided for the camera. We anticipate that this will be rectified in future versions of the software development kit; if not, future work will investigate the use of software triggers implemented separately from the manufacturer-supplied software.
While the work here demonstrates that subcellular resolution imaging of very large tissue surfaces in short timeframes is feasible with topical fluorescent staining and video-rate SIM, a number of improvements could be made to increase the quality of the images. Most notably, the uneven illumination of the SLM display results in intensity fall-off at the edges of each individual image frame, which creates a distracting “lattice” artifact in the image mosaics. This is most noticeable when the image is viewed at low digital zoom as in the large mosaic of Fig. 5; when the images are zoomed in as in Fig. 5(A) and 5(C), the artifact becomes less distracting. However, the intended use for surgical margin assessment would require the pathologist to survey the zoomed-out mosaic to identify regions of interest, and then digitally zoom-in for a closer look. The lattice artifact makes this initial surveillance more difficult than necessary; therefore, future work will include efforts to eliminate the artifact by increasing the intensity and uniformity of the SLM illumination system, and/or flat-field correction in post-processing with the use of a uniform reference sample. In addition, even though relatively flat tissues were used for imaging, the varying topography of the tissue, air and/or liquid bubbles between the tissue and the slide, and microscopic drifts in focus when scanning over large areas, all presented a limitation that sometimes required imaging the tissue in intermediately small sections (~1 cm2) and pausing to refocus the objective manually. We are currently investigating the use of rapid autofocus algorithms, which could be used to correct the focus of each frame automatically, without user intervention.
5. Conclusions
The instrument described here was designed to meet the requirements for high contrast, high resolution images of large, intact surgical specimens within clinically-practical timeframes. Structured illumination microscopy is used to optically-section the surface of the tissue, which serves the purpose of rejecting out of focus light, thereby increasing contrast and effective resolution. By using a fast ferroelectric SLM for generating structured light, and a fast 4.2 megapixel sCMOS camera, we were able to achieve 138.4 MHz pixel sampling rates in the absence of stage-scanning limitations, which are to our knowledge the fastest pixel sampling rates demonstrated for optical sectioning using SIM to date. In practice, the duration of the SLM display bitplanes limited the maximum SIM frame-rate to 26.9 Hz; future work will address ways to circumvent this including using different bitplane sequences (for instance a 3.33 ms bitplane duration would allow 33 Hz SIM). Our initial results with a sub-optimum scan stage and a relatively slow SIM frame-rate (3.33 Hz) demonstrated imaging of a 30.4 cm2 fluorescently-stained tissue area in 15 minutes at 1.3 μm resolution (at a real pixel sampling rate of 0.5 gigapixels per minute). We further demonstrated on tissue-simulating phantoms containing intensely-fluorescent microspheres that the system can be operated at 26.9 Hz when the sample brightness is sufficiently high, enabling areas of 88 cm2 to be scanned in a 20 minute timeframe with the current 7 mm/sec scan stage (at a real pixel sampling rate of 1.1 gigapixels per minute). Since our system is limited by the scan stage speed and not the image acquisition rate, faster scan stages could increase this area-throughput rate to bring full microscopic analysis of entire surgical resection specimen surfaces within clinical reality. Future work will investigate the feasibility of the device for imaging of surgical tumor resection specimens intra-operatively.
Acknowledgments
This work was supported by a grant from the NIH National Cancer Institute R21 CA159936, and a grant from the Tulane University School of Science and Engineering. Katherine Elfer acknowledges support from the National Science Foundation Graduate Research Fellowship Program. The authors thank Henry Fu of Duke University for the kind gift of the solid tissue phantoms.
References and links
- 1.Yossepowitch O., Bjartell A., Eastham J. A., Graefen M., Guillonneau B. D., Karakiewicz P. I., Montironi R., Montorsi F., “Positive surgical margins in radical prostatectomy: outlining the problem and its long-term consequences,” Eur. Urol. 55(1), 87–99 (2009). 10.1016/j.eururo.2008.09.051 [DOI] [PubMed] [Google Scholar]
- 2.Jacobs L., “Positive margins: the challenge continues for breast surgeons,” Ann. Surg. Oncol. 15(5), 1271–1272 (2008). 10.1245/s10434-007-9766-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy D. A., Jones J. S., “Management of rising prostate-specific antigen after a negative biopsy,” Curr. Urol. Rep. 12(3), 197–202 (2011). 10.1007/s11934-011-0177-1 [DOI] [PubMed] [Google Scholar]
- 4.Ukimura O., Coleman J. A., de la Taille A., Emberton M., Epstein J. I., Freedland S. J., Giannarini G., Kibel A. S., Montironi R., Ploussard G., Roobol M. J., Scattoni V., Jones J. S., “Contemporary role of systematic prostate biopsies: indications, techniques, and implications for patient care,” Eur. Urol. 63(2), 214–230 (2013). 10.1016/j.eururo.2012.09.033 [DOI] [PubMed] [Google Scholar]
- 5.Liu J. T., Loewke N. O., Mandella M. J., Leigh S. Y., Levenson R. M., Crawford J. M., Contag C. H., “Real-time pathology through in vivo microscopy,” Stud. Health Technol. Inform. 185, 235–264 (2013). [PubMed] [Google Scholar]
- 6.Saldua M. A., Olsovsky C. A., Callaway E. S., Chapkin R. S., Maitland K. C., “Imaging inflammation in mouse colon using a rapid stage-scanning confocal fluorescence microscope,” J. Biomed. Opt. 17(1), 016006 (2012). 10.1117/1.JBO.17.1.016006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J., Mroz P., Hamblin M. R., Yaroslavsky A. N., “Dye-enhanced multimodal confocal microscopy for noninvasive detection of skin cancers in mouse models,” J. Biomed. Opt. 15(2), 026023 (2010). 10.1117/1.3394301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abeytunge S., Li Y., Larson B., Peterson G., Seltzer E., Toledo-Crow R., Rajadhyaksha M., “Confocal microscopy with strip mosaicing for rapid imaging over large areas of excised tissue,” J. Biomed. Opt. 18(6), 061227 (2013). 10.1117/1.JBO.18.6.061227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makhlouf H., Rouse A. R., Gmitro A. F., “Dual modality fluorescence confocal and spectral-domain optical coherence tomography microendoscope,” Biomed. Opt. Express 2(3), 634–644 (2011). 10.1364/BOE.2.000634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobbs J. L., Ding H., Benveniste A. P., Kuerer H. M., Krishnamurthy S., Yang W., Richards-Kortum R., “Feasibility of confocal fluorescence microscopy for real-time evaluation of neoplasia in fresh human breast tissue,” J. Biomed. Opt. 18(10), 106016 (2013). 10.1117/1.JBO.18.10.106016 [DOI] [PubMed] [Google Scholar]
- 11.Brown J. Q., Bydlon T. M., Richards L. M., Yu B., Kennedy S. A., Geradts J., Wilke L. G., Junker M. K., Gallagher J., Barry W. T., Ramanujam N., “Optical assessment of tumor resection margins in the breast,” IEEE J. Sel. Top. Quantum Electron. 16(3), 530–544 (2010). 10.1109/JSTQE.2009.2033257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu H. L., Mueller J. L., Javid M. P., Mito J. K., Kirsch D. G., Ramanujam N., Brown J. Q., “Optimization of a widefield structured illumination microscope for non-destructive assessment and quantification of nuclear features in tumor margins of a primary mouse model of sarcoma,” PLoS ONE 8(7), e68868 (2013). 10.1371/journal.pone.0068868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neil M. A. A., Juskaitis R., Wilson T., “Method of obtaining optical sectioning by using structured light in a conventional microscope,” Opt. Lett. 22(24), 1905–1907 (1997). 10.1364/OL.22.001905 [DOI] [PubMed] [Google Scholar]
- 14.Karadaglić D., Wilson T., “Image formation in structured illumination wide-field fluorescence microscopy,” Micron 39(7), 808–818 (2008). 10.1016/j.micron.2008.01.017 [DOI] [PubMed] [Google Scholar]
- 15.Xu D., Jiang T., Li A., Hu B., Feng Z., Gong H., Zeng S., Luo Q., “Fast optical sectioning obtained by structured illumination microscopy using a digital mirror device,” J. Biomed. Opt. 18(6), 060503 (2013). 10.1117/1.JBO.18.6.060503 [DOI] [PubMed] [Google Scholar]
- 16.Mertz J., “Optical sectioning microscopy with planar or structured illumination,” Nat. Methods 8(10), 811–819 (2011). 10.1038/nmeth.1709 [DOI] [PubMed] [Google Scholar]
- 17.Hagen N., Gao L., Tkaczyk T. S., “Quantitative sectioning and noise analysis for structured illumination microscopy,” Opt. Express 20(1), 403–413 (2012). 10.1364/OE.20.000403 [DOI] [PMC free article] [PubMed] [Google Scholar]