

Abstract
A novel series of amphiphilic macromolecules (AMs) composed of a sugar backbone, aliphatic chains, and branched, hydrophilic poly(oligoethylene glycol) methyl ether methacrylate (POEGMA)were developed for drug delivery applications. The branched, hydrophilic domains (POEGMA homopolymers with one hydroxyl group) were prepared via atom transfer radical polymerization (ATRP) of oligo(ethylene glycol) methyl ether methacrylate (OEGMA) monomers using 2-hydroxyethyl-2-bromoisobutyrate (HEBiB) as an initiator and copper bromide/bipyridine (CuBr/Bpy) as the catalyst system. To form the amphiphilic structures, the branched POEGMAs were coupled to hydrophobic domains that were formed via acylation of a sugar backbone. The impact of branching in the hydrophilic domain was investigated by comparing the AMs’ solution and thermal properties with those of the linear counterparts. Although these highly branched AMs showed similar critical micelle concentration (CMC) values as compared to linear analogues, they possessed quite low glass transition (Tg) temperatures. Consequently, these novel AMs with branched hydrophilic domain combine the desirable thermal properties of POEGMA with favorable solution properties of amphiphilic architectures, which make them suitable for injectable drug delivery systems.
Introduction
Amphiphilic molecules have been used as nanoscale drug delivery vehicles for hydrophobic drugs.1-16 Above a critical micelle concentration (CMC), these molecules aggregate spontaneously in aqueous solution to form micelles, with hydrophobic cores and hydrophilic shells. This aggregation is thermodynamically driven and the micelle stability is reflected in the CMC value.9 Within micelles, hydrophobic drug molecules can be solubilized in the hydrophobic cores and are protected by the hydrophilic barrier. We previously reported the successful development of amphiphilic macromolecules (AMs) comprised of sugar backbones, aliphatic chains, and poly(ethylene glycol) (PEG) for drug delivery applications.17-22 This design allows for the precise tuning of AM chemical structures and has generated a range of physicochemical properties, including different colloidal stability, aggregation sizes, and thermal properties. Our studies have shown that altering the hydrophobic region by increasing the length and number of aliphatic chains attached to the sugar backbone, greatly improves hydrophobic interactions, micelle formation, and hence micelle stability.18 Additionally, we reported that replacing the single linear PEG chain with two shorter PEG chains (pseudo-branched analogues)22 yielded AMs that formed micelles of smaller size and higher water solubility, thus higher micelle stability, as compared to analogues containing a single, longer PEG chain.22 This enhancement of micelle stability is highly favorable for effective drug delivery systems to achieve sustained release and targeted site accumulation.23
Thermo-sensitive hydrogels have also attracted attention as drug delivery systems. 24, 25 These liquid hydrogels can solidify upon exposure to physiological temperatures into a drug depot which provides controlled release of encapsulated drugs. Poly(oligo(ethylene glycol) methyl ether methacrylate (POEGMA) is a thermo-sensitive polymer; its low critical solution temperature (LCST) can be controlled by the oligo(ethylene glycol) (OEG) chain length.26, 27 POEGMA consists of biocompatible components: methacrylate and pendant OEG. This composition imparts water-solubility and hydrophilicity comparable to PEG, as it provides numerous sites for hydrogen bonding, and has increased chain flexibility. We envisioned that the introduction of a thermo-responsive polymer as the hydrophilic domain within the previously established AMs can maintain the AMs’ micelle stability, provide beneficial thermo-sensitivity, and make them suitable for formulation into hydrogels. Therefore we investigated the effect of structurally modifying the AMs’ hydrophilic domain on AMs’ solution and thermal properties.
Herein, we report the synthesis and physicochemical characterization of AMs with branched, hydrophilic domains shown schematically in Figure 1. Synthesis of the branched, PEG-containing chain was accomplished by atom transfer radical polymerization (ATRP). This method has emerged as one of the most powerful and widely used synthetic techniques in polymer science,28-30 as it enables polymer synthesis with predetermined molecular weights and narrow molecular weight distributions.28-30 In this study, linear palm-tree-like POEGMA homopolymers were successfully obtained (Figure 1) via ATRP of oligo(ethylene glycol) methyl ether methacrylate (OEGMA) monomers using 2-hydroxyethyl-2-bromoisobutyrate (HEBiB) as an initiator and copper bromide/bipyridine (CuBr/Bpy) as the catalyst system (Figure 2).
Fig 1.
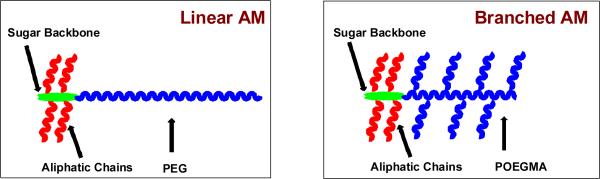
Schematic representation of linear and branched AMs
Fig 2.

Synthetic scheme for preparation of POEGMA homopolymers and AMs with branched hydrophilic domains
Previously published hydrophobic domains, consisting of a sugar (mucic acid or tartaric acid) acylated with lauroyl chloride to introduce four or two twelve carbon chains, respectively, were used.17 These hydrophobic domains were then coupled to hydrophilic POEGMA of various molecular weights to generate the branched AMs. The adapted nomenclature system for these molecules consisted of either M12P5 or T12P5 for the PEG-containing macromolecules: where M stands for mucic acid, T stands for tartaric acid, 12 refers to the number of carbon atoms of each aliphatic chain, and P5 indicates 5 KDa PEG. With POEGMA, however, the system consists of M12(Pn) or T12(Pn) where Pn refers to POEGMA with the specified number (n) of OEGMA repeats. The chemical structures of these novel polymers were confirmed by proton nuclear magnetic resonance (1H NMR) spectroscopy, infrared (IR) spectroscopy, and gel permeation chromatography (GPC). Additionally, aggregation size was quantitatively assessed via dynamic light scattering and micelle stability was determined by measuring CMCs using fluorescence spectroscopy. Lastly, thermal properties were determined using differential scanning calorimetry (DSC). These branched AMs were compared with linear AM analogues as shown in Figure 1, with the expectation that the branched, flexible hydrophilic domain would provide new AMs with similar solution stability but different thermal properties.
Results and Discussion
Amphiphiles can self-assemble to form micelles which show potential as colloidal drug delivery systems, especially for hydrophobic drug entities.1-16 Upon micellization, these molecules can solubilize hydrophobic drug molecules within their core11-16 and are capable of enhancing the bioavailability, increasing the blood circulation time, and minimizing the toxicity of the solubilized bioactives.1-16 A limitation for micelles’ in vivo applications, however, has been their solution stability, namely the ability to withstand drastic dilution.31 To overcome this limitation, we previously developed a series of AMs containing branched hydrophobic domains and linear PEG, a biocompatible and non-immunogenic hydrophilic domain. Our AMs possessed remarkably low CMC values (10−6 to 10−7 M) as compared to established surfactants (e.g., sodium dodecyl sulfate CMC = 8.6 × 10−3 M).32 This low CMC was attributed to the strong hydrophobic interactions between the hydrophobic domains, which facilitate micelle formation. The hydrophilic PEG domain then provides a hydrophilic barrier which prevents unwanted protein adsorption and cell adhesion.11 Therefore, we hypothesized that the introduction of branching in the hydrophilic domain within the previously established AMs would enhance the AMs’ in vivo stability. To investigate the relationship between the branched, hydrophilic domains and solution stability, we prepared several AMs with varying degrees of branching in the hydrophilic domain. Specifically, we investigated the importance of branching by synthesizing AM analogues with equivalent molecular weights yet with varying degrees of branching in the hydrophilic domain. POEGMA, a hydrophilic polymer consisting of repeat units of OEGMA was chosen as a branched equivalent of PEG. Inclusion of the thermo-sensitive POEGMA was also expected to decrease the melting point and provide AMs with potential for application as hydrogels.
Synthesis of POEGMA Homopolymers
POEGMA homopolymers with one hydroxyl end group were prepared via ATRP of OEGMA using HEBiB as an initiator with a CuBr/Bpy catalyst system as shown in Figure 2. The initial conditions and results of ATRP of OEGMA are listed in Table 1. These conditions allowed for the successful synthesis of polymers with predetermined molecular weights, high conversion (100%), and narrow polydispersities (PDI< 1.5).The new POEGMA polymers were characterized by both 1H NMR and GPC. The experimental degree of polymerization (Dp) of POEGMA was calculated according to 1H NMR by comparing signal at δ 3.3-3.4 ppm from OEGMA with signal at δ 4.0-4.2 ppm from both OEGMA and HEBiB, respectively. Good correlation was found between target Dp based on the [Monomer]0/[Initiaor]0 and experimental Dp, which indicated our good control over the polymerization under this reaction condition. Mn,GPC were determined from GPC using PMMA as standards while Mn,NMR were calculated from Dp determined by 1H NMR. These results are consistent with literature, considering that conventional GPC measurements are not necessarily suitable for branched polymers like POEGMA and NMR is a better measurement.33-35
Table 1.
Summary of the synthesized polymers POEGMA via ATRP
| Mn (g/mol) | |||||||
|---|---|---|---|---|---|---|---|
| Polymer | [OEGMA]0:[HEBiB]0:[CuBr]0:[Bpy]0 | Target Dpb | Exptl Dpc | M n,th d | M n,NMR e | M n,GPC f | PDIf |
| P25a | 25:1:1:2 | 25 | 21 | 7700 | 6500 | 15000 | 1.5 |
| P10a | 10:1:1:2 | 10 | 8 | 3200 | 2600 | 9000 | 1.3 |
Conditions: 30 °C, 24h, HEBiB as initiator, isopropanol as solvent.
Calculated from Dp=[M]o/[I]o.
Determined by 1H NMR analysis
Mn,th=[M]0/[I]0×% Conv.×Mw(monomer) + Mw(initiator). % Conversions were calculated from 1H NMR spectra and 100% conversion was obtained for all polymerizations at 24h.
Determined by 1H NMR analysis.
Determined by GPC using THF with 0.05 wt % butylated hydroxyl toluene (BHT) and 2 % triethylamine (TEA) as eluent with PMMA standard.
We also successfully practiced the ATRP of P50 and P100 (data not shown). It was observed that increasing the molecular weight resulted in qualitative increase in viscosity; therefore, properties can thus be tuned through careful choice of the OEG repeat number, which gives rise to the desired molecular weight. P10 and P25 were used to determine the impact of a branched, hydrophilic domain on AM physical properties (including thermal and solution properties).
Synthesis of Amphiphilic Macromolecules
POEGMA was conjugated onto the previously synthesized hydrophobic domains (M12 or T12) to form the final AM structures. The coupling of M12 or T12 onto the mono-hydroxyl substituted POEGMA was accomplished with 1,3-dicyclohexylcarbodiimide (DCC) as a coupling agent and DPTS as a catalyst to yield the AMs (Figure 2). 1H NMR data confirmed successful coupling of POEGMA to the acylated sugars. Although the spectra were dominated by the POEGMA peaks, the peaks corresponding to the terminal methyl and methine groups of the aliphatic chains were prominent. In IR spectra, disappearance of the hydroxyl group signal was the further evidence for successful reaction.
Thermal Properties
Thermal characterization of polymers was performed using DSC methods to evaluate the thermal stability, which may correlate to their in vivo stability. POEGMA homopolymers were viscous liquids which exhibited low glass transition temperatures between −56 and −59 °C. The low glass transition temperatures of POEGMA homopolymers is attributed to a plasticizer effect exerted by the OEG side chains.26, 27 Similarly, the branched AMs were thick liquids exhibiting glass transition temperatures at slightly higher temperatures of −50 and −53 °C (Table 2). AMs with linear PEG chains; however, were solids which possessed a melting temperature of 56 and 58 °C and did not exhibit glass transition temperatures. This result implies that the glass transition temperature is mainly determined by the more flexible OEG side chains of POEGMA. Overall, the chemical structure of the hydrophilic OEG domain dictates thermal properties of the new molecules.
Table 2.
Thermal and solution properties of POEGMA homoploymers and AMs
| Thermal Properties | Solution Properties | ||||
|---|---|---|---|---|---|
| Polymer | Tg (°C) | Tm (°C) | CMC (M) | HLB Valuea | Dhb (nm) |
| M12(P25) | −52 | - | 7.57 × 10−7 | 62.5 | 138 |
| T12(P25) | −50 | - | 4.35 × 10−6 | 69.1 | 49 |
| M12P5 | - | 56 | 1.20 × 10−7 | 37.4 | 20 |
| T12P5 | - | 58 | 6.46 × 10−5 | 44.0 | 7 |
| M12(P10) | −53 | - | 1.48 × 10−6 | 24.3 | 135 |
| T12(P10) | −51 | - | 4.36 × 10−6 | 30.9 | 47 |
| M12P2 | - | 45 | 1.27 × 10−6 | 13.6 | 16 |
| T12P2 | - | 53 | 6.35 ×10−5 | 20.2 | 7 |
These values were calculated using Davies’formula35 to assign hydrophilic balance (HLB) group numbers to chemical groups; HLB = 7 + Σ (hydrophilic group numbers) + Σ (lipophilic group numbers).
Hydrodynamic size determined by DLS.
Hydrodynamic Size
Formation of nanoscale aggregates in aqueous solutions is highly desirable for drug carriers. The aggregation sizes of the macromolecules were determined by using dynamic light scattering. All hydrodynamic sizes of the POEGMA homopolymers were in the low nanoscale range (5-18 nm) (Table 2). The size of PEG-containing AMs ranged from 7 to 20 nm, whereas the POEGMA-containing AMs were relatively larger (45-140 nm). This size difference can be attributed to efficiency of packing of these molecules into aggregates. By comparing the sizes of linear PEG-based AMs with branched POEGMA-based AMs which have same hydrophobic domain and molecular weight of hydrophilic component (e.g., M12P5 and M12(P25)), it's interesting to find the sizes of branch POEGMA-based AMs were about seven times of their linear counterparts. By comparing the sizes of mucic acid-based AMs with tartaric acid-based AMs, we note that the increase in branching in the hydrophobic domain increases the overall hydrodynamic size - regardless of the hydrophilic domain. This effect is more pronounced in the POEGMA-containing AMs. These results indicate that aggregation sizes are dependent on the interactions of both hydrophobic and hydrophilic domains within each macromolecule. Despite this difference most aggregation sizes were within the nanoscale range36 required for drug delivery purposes.
Critical Micelle Concentration (CMC) values
CMC values provide relevant information regarding solution stability of the micelles in aqueous media; lower CMC values indicate higher stability upon drastic dilutions, as is experienced under physiological conditions.37 Therefore, preventing such micelle dissociation increases the stability of micelle-encapsulated hydrophobic drugs.23 Assessment of the CMC values was performed by fluorescence spectroscopy using a pyrene probe.23, 38 Figure.3 shows the change of intensity ratio (I334.5nm/332nm) of pyrene as a function of M12(P10) concentration. As indicated in Table 2, all AMs had very low CMC values (ranging from 10−5 to 10−7) as compared to commercially available surfactants.39 These values indicate their ability to form stable micelles and withstand dilution that would occur upon injection, for example. Comparing AMs with mucic acid versus those with tartaric acid reveals that mucic acid derivatives display lower CMC values. However, similar CMC values were obtained for branched and linear polymers, indicating that the hydrophobic domain is the key factor influencing the overall CMC. These observations directly correlate with the calculated hydrophiliclipophilic balance (HLB) values, which reflect the degree of hydrophilicity.40 As indicated in Table 2, although higher HLB values were obtained for branched hydrophilic chains, owing to their more hydrophilic nature, their CMC values were similar to those of their linear counterparts. This confirms that CMC is dictated by the stronger hydrophobic interactions of the hydrophobic domain alkyl chains. The low micromolar values indicate the ability of these novel AMs to withstand dilution and show their potential for drug delivery applications.
Fig 3.

Fluorescence intensity ratios of pyrene excitation bands (I334.5nm/I332nm) as a function of the concentration of M12(P10) aqueous solutions
Experimental Section
Materials
Oligo(ethylene glycol) methyl ether methacrylate (OEGMA) with an average molecular weight of 300 g/mol was obtained from Sigma-Aldrich and purified by passing through a neutral alumina column prior to use. HEBiB, CuBr and Bpy, mucic acid, tartaric acid, lauroyl chloride, zinc chloride, dimethyl amino pyridine (DMAP), p-toluene sulphonic acid, and DCC were purchased from Aldrich and used as received. Dimethyl amino pyridine ptoluene sulphonate (DPTS), M12 (acylated mucic acid derivative with four twelve carbon aliphatic chains (Figure 2)), and T12 (acylated tartaric acid derivative with two twelve carbon aliphatic chains (Figure 2)) were prepared as previously published.17
Measurements
1H NMR spectra were recorded on Varian VNMRS 400 MHz spectrometer. Samples were dissolved in CDCl3 with tetramethylsilane (TMS) as an internal reference. IR spectra were recorded on a Thermo Nicolet/Avatar 360 spectrophotometer by solvent-casting onto a NaCl plate. Each spectrum was an average of 32 scans. Molecular weights were determined by gel permeation chromatography using a PerkinElmer series 200 LC system equipped with a PL-Gel column. Tetrahydrofuran (THF) was used as eluent for analysis and solvent for sample preparation. The sample (10 mg/mL) was dissolved into THF and filtered using a 0.45 μm PTFE syringe filter (Whatman, Clifton, NJ) before injection into the column at a flow rate of 0.5 mL/min. The average molecular weight of the sample was calibrated against narrow molecular weight poly(methyl methacrylate) (PMMA) (Poly[Anayltik]n, Ontario, CA). Thermal properties (melting temperature, Tm and glass transition temperature, Tg) were determined by a TA instrument Q200. Data was analyzed by TA Instruments Universal Analysis 2000 software version 4.5 A. Samples (4-8 mg) were heated under dry nitrogen gas from −70 °C to 100 °C, and data were collected at heating and cooling rates of 10 °C/min with a three-cycle minimum. CMC measurements were carried out by fluorescence studies on a Spex fluoro Max spectrofluorometer at 25 °C. A stock solution of 5 × 10−7 M pyrene in water was prepared and used as the probe molecule. Samples were dissolved in water, diluted to specific concentrations, then added to the stock pyrene solution,. Excitation was performed from 300 to 360 nm, with 390 nm as the emission wavelength. Upon micelle formation, the pyrene maximum absorption shifted from 332 to 334.5 nm. The ratio of absorption of pyrene with polymer (334.5 nm) to pyrene only (332 nm) was plotted as the logarithm of polymer concentrations and the inflection point was taken as the CMC value. Hydrodynamic diameter and zeta potential analyses were performed using a NanoZS90 instrument (Malvern Instruments, UK). Samples (10 mg/mL were prepared using HPLC grade water and filtered through 0.45 µm PTFE syringe filters before measurement. Each sample was run at room temperature three separate times with 20 measurements per run.
Synthesis of POEGMA
ATRP of OEGMA was carried out using HEBiB as an initiator and CuBr/Bpy as the catalyst system in isopropanol at 30 °C. The general procedures of the polymerization were as follows: OEGMA, CuBr, BPy and solvent (isopropanol) were transferred into a dry round-bottomed flask equipped with a magnetic stirring bar, placed in an ice-bath. The flask was sealed and the solution degassed by purging with argon for 15 min. The initiator (HEBiB) was mixed separately with a small amount of solvent and the resulting solution degassed with argon for 15 min. To start the polymerization, the initiator solution was added to the monomer solution, and placed in an oil-bath at 30 °C for 24 h. Aliquots were withdrawn from the flask at timed intervals to monitor the polymerization progress. Conversion of the OEGMA monomer was determined by comparison of the intensity of the 1H NMR signals corresponding to vinylic protons (5.6 and 6.1 ppm) with aliphatic protons of CH3O (~3.4 ppm) or -OCOCH2CH2O- (~4.1 ppm). Using this method, 100% conversion was reached by 24 hr.
The catalyst was removed by passing the polymer solution through a silica column. Subsequently, the polymers were precipitated from THF into cold n-hexane, filtered and lyophilized. 1H NMR spectrum of POEGMA homopolymer reveals the characteristic signals of POEGMA at (δ): 4.1 (br s, 2H, CO2CH2CH2O-), 3.65 (br s, 14H,OCH2CH2-), 3.55 (s, 2H, CH2OCH3), 3.38 (s, 3H, OCH3), 1.74-2.1(m, 2H, -CH2C(CH3)-) and 0.8-1.02 (m, 3H, -CH2C(CH3)-). IR spectroscopy of POEGMA homopolymers with one hydroxyl end group confirmed the successful ATRP of OEGMA. IR (NaCl, cm−1): 3513 (OH), 2874 (C-H), 1728 (C=O) and 1109 (C-O-C). The molecular weight results are listed in Table 1.
Synthesis of branched amphiphilic macromolecules
The preparation of M12(P25) is presented as an example. (P25) refers to POEGMA homopolymers with 25 OEG repeat units, (P10) refers to POEGMA homopolymers with 10 OEG repeat units POEGMA (Mn = 7711) (1.1 g, 0.14 mmol) was dehydrated by azeotropic distillation from toluene (50 mL) under vacuum. M12 (0.40 g, 0.42 mmol) and DPTS (36 mg, 0.12 mmol) were dissolved in THF (3.0 mL) and methylene chloride (10 mL), and the solution then added at room temperature to POEGMA. After 10 min under nitrogen, 0.50 mL of DCC solution (1.0 M in methylene chloride) was added dropwise over 15 min. After 24 h, the DCC side product (dicyclohexylurea) was removed by vacuum filtration. The filtrate was washed with 0.1 N HCl, then twice with brine. The organic layer was dried over anhydrous magnesium sulfate, and evaporated to dryness. The crude product was purified by precipitation into diethyl ether (100 mL). Product was obtained as colorless oil (360 mg, 30% yield).The ethylene oxide of POEGMA comprises a significant component of branched AMs and dominates the 1H NMR and IR spectra. Thermal and properties are listed in Table 2.
M12(P25): Product was obtained as colorless oil (360 mg, 30% yield). 1H NMR (CDCl3) (δ): 4.08 (br s, 52H, CH), 3.65 (br s, 350H, CH2), 3.55 (s, 52H, CH2), 3.38 (s, 75H, CH3), 1.89 (m, 50H, CH2), 1.25 (m, 64H, CH2), 1.02 (m, 81H, CH3), 0.88 (t, 12H, CH3). IR (NaCl, cm−1): 2921 (C-H), 1728 (C=O) and 1109 (C-O-C).
T12(P25): Product was obtained as brown oil (460 mg, 40% yield).1H NMR (CDCl3) (δ): 4.08 (br s, 52H, CH), 3.65 (br s, 350H, CH2), 3.57 (s, 52H, CH2), 3.38 (s, 75H, CH3), 1.89 (m, 50H, CH2), 1.25 (m, 32H, CH2), 1.02 (m, 81H, CH3), 0.88 (t, 12H, CH3). IR (NaCl, cm−1): 2874 (C-H), 1728 (C=O) and 1111 (C-O-C).
M12(P10): Product was obtained as colorless oil (170 mg, 30% yield). 1H NMR (CDCl3) (δ): 4.08 (br s, 22H, CH), 3.65 (br s, 140H, CH2), 3.56 (s, 22H, CH2), 3.38 (s, 30H, CH3), 1.88 (m, 20H, CH2), 1.26 (m, 64H, CH2), 1.02 (m, 36H, CH3), 0.88 (m, 12H, CH3). IR (NaCl, cm−1): 2874 (C-H), 1728 (C=O) and 1107 (C-O-C).
T12(P10): Product was obtained as brown oil (210 mg, 40% yield).1H NMR (CDCl3) (δ): 4.08 (br s, 22H, CH), 3.65 (br s, 140H, CH2), 3.55 (s, 22H, CH2), 3.38 (s, 30H, CH3), 1.89 (m, 20H, CH2), 1.25 (m, 32H, CH2), 1.02 (m, 36H, CH3), 0.88 (t, 12H, CH3). IR (NaCl, cm−1): 2873 (C-H), 1728 (C=O) and 1108 (C-O-C).
Conclusions
A series of new AMs were synthesized to study the impact of branched versus linear hydrophilic domains on solution and thermal properties. Solution properties of branched and linear AMs, including hydrodynamic size, and CMC, were similar. This result indicated that solution properties are dictated by the interactions of the hydrophobic domain. Adversely, a significant difference in thermal properties, on the other hand was observed. These studies show that the hydrophilic domain contributes significantly to thermal properties and affects the AMs’ overall physical characteristics. Together the low CMC and glass transition temperature make these new AMs envisioned to be appropriate for drug delivery of hydrophobic drug entities in the form of hydrogels or injectables. Further studies are underway to determine their behavior under simulated physiological conditions.
Acknowledgements
This work was supported by the National Institutes of Health (5 R01 HL107913) to Kathryn E. Uhrich. Dr. Bryan Langowski (Rutgers, Department of Chemistry & Chemical Biology) is thanked for intellectual discussions. Hülya Arslan acknowledges support by Presidency of The Council of Higher Education of Turkey.
Notes and references
- 1.Kwon GS, Kataoka K. Adv Drug Deliver Rev. 1995;16:295–309. [Google Scholar]
- 2.La SB, Okano T, Kataoka K. J Pharm Sci. 1996;85:85–90. doi: 10.1021/js950204r. [DOI] [PubMed] [Google Scholar]
- 3.Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T. J Control Release. 1997;48:157–164. [Google Scholar]
- 4.Jeong B, Bae YH, Lee DS, Kim SW. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 5.Allen C, Yu YS, Maysinger D, Eisenberg A. Bioconjugate Chem. 1998;9:564–572. doi: 10.1021/bc9702157. [DOI] [PubMed] [Google Scholar]
- 6.Inoue T, Chen GH, Nakamae K, Hoffman AS. J Control Release. 1998;51:221–229. doi: 10.1016/s0168-3659(97)00172-7. [DOI] [PubMed] [Google Scholar]
- 7.Kim SY, Shin ILG, Lee YM, Cho CS, Sung YK. J Control Release. 1998;51:13–22. doi: 10.1016/s0168-3659(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 8.Bae YH, Huh KM, Kim Y, Park KH. J Control Release. 2000;64:3–13. doi: 10.1016/s0168-3659(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 9.Kataoka K, Harada A, Nagasaki Y. Adv Drug Deliver Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Li CX, Li XC, Yuan Z, An YL, He BL. J Appl Polym Sci. 2001;80:1976–1982. [Google Scholar]
- 11.Otsuka H, Nagasaki Y, Kataoka K. Curr Opin Colloid In. 2001;6:3–10. [Google Scholar]
- 12.Riley T, Stolnik S, Heald CR, Xiong CD, Garnett MC, Illum L, Davis SS, Purkiss SC, Barlow RJ, Gellert PR. Langmuir. 2001;17:3168–3174. [Google Scholar]
- 13.Rosler A, Vandermeulen GWM, Klok HA. Adv Drug Deliver Rev. 2001;53:95–108. doi: 10.1016/s0169-409x(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 14.Gillies ER, Frechet JMJ. J Am Chem Soc. 2002;124:14137–14146. doi: 10.1021/ja028100n. [DOI] [PubMed] [Google Scholar]
- 15.Liggins RT, Burt HM. Adv Drug Deliver Rev. 2002;54:191–202. doi: 10.1016/s0169-409x(02)00016-9. [DOI] [PubMed] [Google Scholar]
- 16.Yu JJ, Jeong YI, Shim YH, Lim GT. J Appl Polym Sci. 2002;85:2625–2634. [Google Scholar]
- 17.Tian L, Yam L, Zhou N, Tat H, Uhrich KE. Macromolecules. 2004;37:538–543. [Google Scholar]
- 18.Tao L, Uhrich KE. J Colloid Interf Sci. 2006;298:102–110. doi: 10.1016/j.jcis.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 19.Djordjevic J, Del Rosario LS, Wang JZ, Uhrich KE. J Bioact Compat Pol. 2008;23:532–551. [Google Scholar]
- 20.Harmon AM, Uhrich KE. J Bioact Compat Pol. 2009;24:185–197. [Google Scholar]
- 21.del Rosario LS, Demirdirek B, Harmon A, Orban D, Uhrich KE. Macromol Biosci. 2010;10:415–423. doi: 10.1002/mabi.200900335. [DOI] [PubMed] [Google Scholar]
- 22.Wang JZ, del Rosario LS, Demirdirek B, Bae A, Uhrich KE. Acta Biomater. 2009;5:883–892. doi: 10.1016/j.actbio.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 23.Wilhelm M, Zhao CL, Wang YC, Xu RL, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991;24:1033–1040. [Google Scholar]
- 24.Gong C, Qi T, Wei X, Qu Y, Wu Q, Luo F, Qian Z. Curr Med Chem. 2013;20:79–94. [PubMed] [Google Scholar]
- 25.Wolinsky JB, Colson YL, Grinstaff MW. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iizawa T, Yamamoto D, Gotoh T, Sakohara S. Polymer. 2012;53:3417–3420. [Google Scholar]
- 27.Roth PJ, Jochum FD, Theato P. Soft Matter. 2011;7:2484–2492. [Google Scholar]
- 28.Wang JS, Matyjaszewski K. J Am Chem Soc. 1995;117:5614–5615. [Google Scholar]
- 29.Kato M, Kamigaito M, Sawamoto M, Higashimura T. Macromolecules. 1995;28:1721–1723. [Google Scholar]
- 30.Percec V, Barboiu B. Macromolecules. 1995;28:7970–7972. [Google Scholar]
- 31.Torchilin VP. J Control Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 32.Lavasanifar A, Samuel J, Kwon GS. Colloids and surfaces. B, Biointerfaces. 2001;22:115–126. doi: 10.1016/s0927-7765(01)00147-3. [DOI] [PubMed] [Google Scholar]
- 33.Yau WW, Kirkland JJ, Bly DD. Modern size-exclusion liquid chromatography : practice of gel permeation and gel filtration chromatography. Wiley; New York: 1979. [Google Scholar]
- 34.O'Brien N, McKee A, Sherrington DC, Slark AT, Titterton A. Polymer. 2000;41:6027–6031. [Google Scholar]
- 35.Robinson KL, de P-BMV, Wang XS, Armes SP. Macromolecules. 2001;34:5799–5805. [Google Scholar]
- 36.Kabanov AV, Batrakova EV, Meliknubarov NS, Fedoseev NA, Dorodnich TY, Alakhov VY, Chekhonin VP, Nazarova IR, Kabanov VA. J Control Release. 1992;22:141–157. [Google Scholar]
- 37.Allen C, Maysinger D, Eisenberg A. Colloid Surface B. 1999;16:3–27. [Google Scholar]
- 38.Astafieva I, Zhong XF, Eisenberg A. Macromolecules. 1993;26:7339–7352. [Google Scholar]
- 39.Rosen HB, Chang J, Wnek GE, Linhardt RJ, Langer R. Biomaterials (Guildford, Engl.) 1983;4:131–133. doi: 10.1016/0142-9612(83)90054-6. [DOI] [PubMed] [Google Scholar]
- 40.Jönsson B. Surfactants and polymers in aqueous solution. John Wiley & Sons; Chichester ; New York: 1998. [Google Scholar]