

Abstract
The serine protease granzyme B (GrB) induces apoptosis through both caspase-dependent and caspase-independent multiple-cascade mechanisms. Vascular endothelial growth factor 121 (VEGF121) binds to both VEGFR-1 and VEGFR-2 receptors. We engineered a unique GrB/VEGF121 fusion protein and characterized its properties in vitro and in vivo. Endothelial and tumor cells lines demonstrated varying levels of sensitivity to GrB/VEGF121 that correlated closely to total VEGFR-2 expression. GrB/VEGF121 localized efficiently into VEGFR-2 expressing cells while the internalization into VEGFR-1 expressing cells was significantly reduced. Treatment of VEGFR-2+ cells caused mitochondrial depolarization in 48% of cells by 48 h. Exposure to GrB/VEGF121 induced apoptosis in VEGFR-2+, but not in VEGFR-1+, cells and rapid caspase activation was observed that could not be inhibited by treatment with a pan-caspase inhibitor. In vivo, GrB/VEGF121 localized in perivascular tumor areas adjacent to microvessels and in other areas in the tumor less well vascularized, while free GrB did not specifically localize to tumor tissue. Administration (i.v.) of GrB/VEGF121 to mice at doses up to 40 mg/kg showed no toxicity. Treatment of mice bearing established PC-3 tumor xenografts with GrB/VEGF121 showed significant antitumor effect vs. treatment with GrB or saline. Treatment with GrB/VEGF121 at 27 mg/kg resulted in the regression of 4 of 5 tumors in this group. Tumors showed a two-fold lower Ki-67 labeling index compared to controls. Our results demonstrate that targeted delivery of granzyme B to tumor vascular endothelial cells or to tumor cells activates apoptotic cascades and this completely human construct may have significant therapeutic potential.
Keywords: Growth factor receptors and other cell surface molecules as targets for therapy, effectors of apoptosis, VEGF, Granzyme B, vascular targeting
INTRODUCTION
Angiogenesis is a critical process in numerous diseases, and intervention in neovascularization has therapeutic value in several disease settings including ocular diseases (1), arthritis (2) and in tumor progression and metastatic spread (3). Numerous groups have focused drug development strategies on targeting tumor neovascularization by inhibitors of various growth factor receptor tyrosine kinases (4), blocking antibodies that interfere with receptor signal transduction (5) and strategies that trap growth factor ligands (6). These approaches have all been used with varying degrees of success in preclinical and clinical settings.
While the control of tumor neovascularization has been found to be a highly complex process involving many driving critical events, vascular endothelial growth factor-A (VEGF-A) and its receptors have been found to play a central role. VEGF receptors VEGFR-1 (Flt-1/FLT-1) and VEGFR-2 (Flk-1/ KDR) are rarely expressed on tumor cells, exist at low levels on the endothelium of resting blood vessels, but are upregulated in the tumor neovasculature (7, 8). As a result, numerous laboratories have focused blocking tumor growth by interfering with the VEGF-VEGFR interaction. An alternative approach is to leverage this interaction for drug delivery, and numerous research groups have developed recombinant growth factor fusion constructs of VEGF-A and various toxins (9–11) to target cells bearing receptors of VEGF-A. Our laboratory has developed fusion proteins that exploit the VEGFR-targeting ability of VEGF121, the smallest VEGF-A isoform, that binds only VEGFR-1 and VEGFR-2. Studies of VEGF121 fused with gelonin, a potent plant toxin that results in irreversible inhibition of protein synthesis (“VEGF121/rGel”) have shown excellent efficacy in various subcutaneous, xenograft, orthotopic, and experimental metastasis models by reducing the overall tumor burden of non-VEGFR-expressing tumor cells via targeting of tumor neovasculature and normal bone morrow derived cells that are recruited during tumor development (12–15). However, concerns relating to the development of immunogenicity to gelonin have led us to also develop completely human cytotoxic proteins for use as payloads.
Granzyme B (GrB) is a member of the granzyme family of serine proteases that play a critical role in the body’s defense against viral infection and tumor development. Cytotoxic T lymphocyctes (CTL) and natural killer (NK) cells directly deliver granzymes to target cells. Once endocytosed, granzyme B remains trapped in endocytic vesicles until released by perforin into the cytosol, where it induces intense cellular apoptosis by causing the release of cytochrome c from mitochondria and initiating apoptosis by activating procaspases -3, -7 and -9. Apoptosis can also be initiated via caspase-independent methods such as direct cleavage of the pro-apoptotic molecule Bid. Thus, granzyme B is capable of apoptotic activity via both caspase-dependent and caspase-independent mechanisms (16, 17).
Our laboratory has previously described GrB-based fusion proteins that efficiently delivered GrB to the cytoplasm of target cells in vitro and induced pro-apoptotic responses including caspase activation (18–20). Limited anti-tumor efficacy studies have suggested that the targeted delivery of human pro-apoptotic GrB to tumor cells has a significant potential for cancer treatment. Unfortunately, the low yields of GrB-based proteins in prokaryotic expression systems are a major bottleneck in characterizing these proteins for therapeutic use.
In this report, we describe the expression of a fusion construct of granzyme B and VEGF121 (GrB/VEGF121) in a mammalian expression system and characterize the in vitro properties of this molecule against VEGFR-expressing endothelial and non-endothelial cells. We also examine, for the first time, anti-tumor efficacy of a granzyme B-based construct targeting the tumor vasculature. GrB/VEGF121 efficiently internalized and targeted cells expressing both VEGFR-1 and VEGFR-2, initiating apoptosis via both caspase-dependent and –independent mechanisms without the use of endo-osmolytic agents. In addition, we demonstrate robust in vivo anti-tumor efficacy of GrB/VEGF121 with significant inhibition of tumor growth and efficient localization into the tumor neovasculature and the tumor core.
MATERIALS AND METHODS
Construction of the GrB/VEGF121 expression vector
The cDNA encoding human granzyme B and VEGF121 has been previously described (19). The recombinant protein was fused into the mammalian cell expression vector pSecTag (Life Technologies, Carlsbad, CA) by using the splice overlap extension PCR method with VEGF121 and granzyme B cDNA as templates. A gene sequence encoding the hexa-histidine tag and an enterokinase cleavage site was cloned behind the secretion leader at the N-terminus with the SfiI restriction site. An internal (Gly4Ser) linker was inserted between the GrB and VEGF121 molecule, and an amber stop-codon with the restriction site XhoI was subcloned at the C-terminus of the fusion molecule (Figure 1A). Primers used were:
Figure 1.

Construction, expression and purification of GrB/VEGF121. (A) The Granzyme B/VEGF121 cassette encoding the hexa-histidine tag and an enterokinase cleavage site (H6D4K) was cloned in frame 3′ to the secretion leader peptide in the 5.2 kb pSecTag plasmid. LS, Igκ leader sequence and Myc tag with the following amino acid sequence: METDTLLLWVLLLWVPGSTGDAAQPAEQKLISEEDLNSAVDGSGSGHM. (B) Coomassie Blue staining showing purification of pro-Granzyme B/VEGF121 from transiently transfected HEK-293T cells using Cobalt-IMAC. The activated GrB/VEGF121 was purified as an 80 kDa homodimer. (C) Granzyme B enzymatic activity assay indicating that the granzyme B portion of the fusion construct was comparable to commercially prepared granzyme B.
Primer GrB-1: 5′ – CATCATCATTCTTCTGGTGGTACCGACGACGACGACAAGATCATCGGGGGA-3′
Primer GrB-2: 5′ – CTGAATAGCGCCGTCGACGGTTCTGGTTCTGGCCATATGCACCATCATCATCATCAT TCTTCTGGT-3′
Primer GrB-3: 5′ – GGTTCCACTGGTGACGCGGCCCAGCCGGCCGAACAAAAACTCATCTCAGAAGAGGA TCTGAATAGCGCCGTC-3′
Primer GB Link: 5′ - ACCATGAAACGCTACGGTGGCGGTGGCTCC - 3′
Primer Link VEGF: 5′ - GGTGGCGGTGGCTCCGCACCCATGGCAGAAGGA - 3′
Primer VEGF Bac: 5′ - GGAGCCACCCTCGAGTTATCACCGCCTCGGCTTGTCACA - 3′
Expression and purification of GrB/VEGF121
Proteins were expressed in HEK-293T cells. Transient transfection of the plasmid containing the pSecTag-GrB/VEGF121 expression vector with PEI was performed overnight at a 1:3 ratio, followed by incubation of cells in serum-free DMEM media for 72 h. Following dialysis of conditioned media against 20 mM Tris, 150 mM NaCl, pH 7.6, recombinant protein was purified using cobalt-immobilized metal affinity chromatography (Co-IMAC). Pro-Granzyme B/VEGF121 was activated by cleaving the N-terminus with recombinant enterokinase (10 Units/mg overnight). The fusion toxin was stored in sterile PBS at −20°C.
Granzyme B activity assay
The specific activity of GrB/VEGF121 was compared to that of free human granzyme B (Alexis Biochemicals) by assessing the cleavage of Boc-Ala-Ala-Asp-SBzl (BAADT) (MP Biomedical), a granzyme B substrate, using a protocol described previously (18). The reaction was monitored at 2 min intervals for up to one hour at 405 nm.
In vitro cytotoxicity
SK-N-SH neuroblastoma cells, previously shown to express VEGFR-2 (21), were a kind gift from Dr. V. Gopalakrishnan (M.D. Anderson Cancer Center). Cytotoxicity of GrB/VEGF121 and GrB against log phase cells was performed over 72 hours as previously described (14). Porcine aortic endothelial (PAE) cells transfected with the human VEGFR-2 receptor (PAE/VEGFR-2) or the human VEGFR-1 receptor (PAE/VEGFR-1) were a generous gift from Dr. Johannes Waltenberger (University Hospital, Maastricht, The Netherlands) and were propagated as previously described (22). The numbers of VEGFR-2 and VEGFR-1 receptor sites on these cells have been determined to be 150,000 and 50,000 per cell, respectively (23). Cytotoxicity against confluent PAE/VEGFR-2 and PAE/VEGFR-1 cells was assessed by 72 hour treatment of cells following confluence. Competitive inhibition of cytotoxicity was assessed on PAE/VEGFR-2 cells by co-treatment of cells with VEGF121 in the presence of GrB/VEGF121 or GrB. To assess if the activity of GrB/VEGF121 was affected by the exposure time to endothelial cells, log-phase PAE/VEGFR-2 cells were treated with GrB/VEGF121 and media containing the cytotoxic agent was removed at varying time-points and replaced with fresh media. Cytotoxicity was assessed by crystal violet staining as previously described (14).
In vitro immunofluorescence
Cells were treated with 1.6 μg/ml (20 nM) GrB/VEGF121 for 24 h, then washed with Glycine buffer (500 mM NaCl, 0.1 M glycine, pH 2.5) to remove cell surface-bound GrB/VEGF121. Cells were incubated with a mouse anti-GrB monoclonal antibody (1:100) (sc-8022; Santa Cruz Biotechnology) followed by a goat anti-mouse AF488 antibody (1:500) (A-11017; Invitrogen). Nuclei were counterstained with propidium iodide (1μg/ml) in PBS. The slides were mounted with 1,4-Diazabicyclo (2,2,2)-octane (DABCO) reagent and visualized under fluorescence (Nikon Eclipse TS1000) and confocal (Zeiss LSM 510) microscopes.
Activation of Caspase-3 and -9
Log-phase cells grown in 10-cm dishes were scraped and lysed in cold 1× cell culture lysis reagent (Promega, Madison, WI). In vitro activation of caspases by GrB or GrB/VEGF121 was assessed by mixing 2 μl cell lysate with the Caspase 3 (Ac-DEVD-pNA) or Caspase 9 (Ac-LEHD-pNA) chromogenic subtrates (AnaSpec Inc, Fremont CA) in the Chromogenic Assay buffer (0.1 M HEPES pH7.5, 0.5 mM EDTA, 20% glycerol, 5 mM dithiothreitol). Substrate specificity was assessed incubation with the pan-caspase inhibitor z-VAD-FMK (Enzo Life Sciences). Assay development was monitored at 405 nm.
Measurement of apoptosis with Annexin V/propidium iodide
Cells were treated with GrB/VEGF121 (50 nM), granzyme B (50 nM), or staurosporine (2 ug/ml) for up to 48 hours. Cells were harvested by trypsinization, washed with PBS, then incubated with Annexin V Alexa Fluor 488 conjugate (AF3201; Invitrogen) and propidium iodide (1 ug/ml) and analyzed by flow cytometry on a BCI XL Analyzer.
Western blot analysis
Whole cell extracts of PAE/VEGFR-2 and PAE/VEGFR-1 cells untreated or treated with 20 nM GrB/VEGF121 in the absence or presence of 20 μM z-VAD-FMK were prepared as described previously (13). Western blotting was conducted using antibodies for actin (loading control), PARP-1 and VEGFR-2.
Mitochondrial depolarization
PAE/VEGFR-1 and PAE/VEGFR-2 cells in 6 well plates (5 × 105 cells) were untreated or treated with 20 nM GrB/VEGF121 or GrB for 4, 24 or 48 h, or with 5 μM Staurosporine (positive control) for 4 h. Cells were treated with JC-1 reagent from the JC-1 mitochondrial membrane potential assay kit (Cayman Chemical Co., Ann Arbor, MI) according to the manufacturer’s protocol. Apoptotic or unhealthy cells with low mitochondrial transmembrane potential were quantified by flow cytometry via green fluorescence of the JC-1 cationic dye in its monomeric form.
Determination of maximal tolerated dose and lethal dose for GrB/VEGF121 in mice
All mice were maintained under specific pathogen–free conditions according to the American Association for Accreditation of Laboratory Animal Care (AAALAC) standards. Female BALB/c mice (4–6 weeks old, 2 mice per group) were treated with GrB/VEGF121 (i.v., every other day × 5) at 40 mg/kg or treated with Saline. Weight change and survival of mice was monitored throughout treatment and for three weeks after the final dose.
In vivo efficacy in a tumor xenograft model
Nu/nu male mice (5–6 weeks old, 5 mice per group) were injected subcutaneously (right flank) with 5 × 106 log-phase PC-3 prostate cancer cells. This cell line has previously been shown to be insensitive to VEGF121-mediated targeting suggesting that it expresses insufficient receptor levels to mediate specific VEGF121 driven cytotoxicity (14). However, PC-3 tumor xenografts are highly vascularized and VEGF121-mediated targeting has a significant anti-tumor therapeutic effect (14). Tumors were allowed to establish for three days. Once tumors were measurable, mice received intravenous injections of either saline, GrB (15 mg/kg) or GrB/VEGF121 (11 or 27 mg/kg) every other day for 11 days, for a total of six treatments. Tumor volume was monitored twice weekly and calculated according to the formula: Volume = length (L) × width (W) × height (H). At the end of the experiment, tumor tissues were harvested in ornithine carbamyl transferase (OCT) on dry ice.
In vivo localization of GrB/VEGF121
Mice with injected with 14 × 106 PC-3 cells in the right flank. After eight days, mice were injected (i.v.) with either GrB (30 μg) or GrB/VEGF121 (100 μg). Normal and tumor tissues were harvested after four hours in OCT on dry ice.
Tissue processing and histology
Histopathologic analysis included H&E as well as immunofluorescence staining for human granzyme B and mouse vasculature with anti-granzyme B (ab53097, Abcam) and anti-CD31 MEC 13.3 (553370, BD Pharmingen), respectively. Tissue sections were thawed and immediately fixed with 4% formaldehyde. Sections were incubated overnight at 4°C in a humidified container with the appropriate primary antibody (1:100). Fluorochrome-conjugated secondary antibody (1:60) was added for 1 hour at room temperature in a humidified light-tight container. Sections were counterstained with Hoechst (1:2000) for 30 min, followed by mounting and analysis. All images were taken under identical conditions. Computerized quantification of immunostained vascular structures was performed with MetaMorph 7.7 imaging software (Molecular Devices, Downingtown, PA). CD-31+ pixels were selectively detected using the threshold function. Regions of counter-staining and background were adjusted from the threshold until all the CD-31+ pixels were selectively thresholded. Identical threshold settings were defined for all images quantified. At least five fields per image were quantified and averaged. The percent threshold area (which corresponds to the endothelial area) was measured. Representative images are shown in Supplementary Figure 1.
Immunohistochemical analysis of proliferation of PC-3 tumor cells
Analysis was performed on fresh frozen tumor sections. To determine the number of cycling cells, sections were stained with the Ki-67 antibody followed by antimouse IgG HRP conjugate. Sections were analyzed at a magnification of ×40. The number of cells positive for Ki-67 was assessed per viewing field. The mean ± SEM per GrB/VEGF121 and control group is presented. The average numbers derived from analysis of each slide were combined per either GrB/VEGF121 or saline control group and analyzed for statistical differences.
Cell line authentication
All human cell lines used in this study were authenticated by STR DNA fingerprinting analysis by the Characterized Cell Line Core Facility at MD Anderson Cancer Center. KS1767 cells could not be authenticated as the STR profile for these cells was not available. Non-human cell lines were analyzed by G banding and confirmed to be of the stated origin.
Statistical analysis
All statistical analyses were done with Microsoft Excel software (Microsoft, Redmond, WA). Data are presented as means ± SEM. P values were obtained using the 2-tailed t test with 95% confidence intervals to evaluate statistical significance; P < 0.05 was considered statistically significant.
RESULTS
GrB/VEGF121 protein expression and purification
Pro-GrB/VEGF121 was secreted out of HEK-293T cells by a leader sequence (Figure 1A) and purified from conditioned media using Co-IMAC. The protein was activated by cleavage of the poly-histidine tag with recombinant enterokinase (10 U/mg, overnight). One liter of conditioned medium yielded approximately 5 mg purified protein. The protein was expressed as an 80 kDa homodimer, as indicated by SDS-PAGE analysis under non-reducing conditions (Figure 1B and Supplementary Figure 2).
Granzyme B activity assay
The specific activity of GrB/VEGF121, as determined by the BAADT assay, was determined to be 880 units/nmol GrB. This activity was slightly more than that found for the commercially prepared GrB (330 units/nmol GrB) (Figure 1C).
Cytotoxicity of GrB/VEGF121 in vitro
The cytotoxicity of HEK-239T-expressed GrB/VEGF121 was assessed against various cell types. GrB/VEGF121 was preferentially toxic to log-phase hVEGFR-2-expressing porcine aortic endothelial cells in vitro (IC50 ~10 nM), compared to PAE/VEGFR-1 cells (IC50 ~ 2000 nM) (Table 1). These results match our previous observations with aglycosylated GrB/VEGF121 obtained via bacterial expression (19). The neuroblastoma cell line SK-N-SH was also highly sensitive to GrB/VEGF121 treatment (IC50 of 27 nM), while U-87 MG cells were moderately sensitive (IC50 of 204 nM), as were mVEGFR-2-expressing b.END3 cells and mVEGFR-1-expressing RAW264.7 cells (IC50 of 60 and 156 nM, respectively). The log-phase PAE/VEGFR-2 cells were more sensitive to GrB/VEGF121 and GrB itself than were the confluent cells, suggesting that the quiescence of confluent cells impacts their sensitivity to both targeted and non-targeted GrB. The Ewing’s sarcoma cell line TC-71 indicated low specific targeting of GrB/VEF121 compared to the non-targeting control. All other cells lines tested were at least 50-fold resistant to GrB/VEGF121 compared to PAE/VEGFR-2 cells. In order to understand the impact of receptor expression on cytotoxicity, we performed Western blot and flow cytometry analysis to compare VEGFR-2 expression. With the exception of b.END3 cells, the relative cytotoxicity of GrB/VEGF121 correlated closely to total VEGFR-2 expression (Supplementary Figure 3).
Table 1.
Cytotoxicity of GrB/VEGF121 on various cell lines
| Category | Cell line | Cell type | VEGFR-2 Receptor sites per cell | IC50 (nM) GrB/VEGF121 | IC50 (nM) Granzyme B | Targeting Index* |
|---|---|---|---|---|---|---|
| Endothelial | PAE/hVEGFR-2 (log-phase) | Endothelial | +++++ | 10 | 500 | 50 |
| PAE/hVEGFR-2 (confluent) | Endothelial | + | 140 | 4800 | 34 | |
| PAE/hVEGFR-1 | Endothelial | − | 2000 | 6900 | 3.5 | |
| PAE/hVEGFR-1 (confluent) | Endothelial | − | >2000 | >6900 | N/A | |
| b.End3 | Endothelial | +++ | 156 | 3076 | 20 | |
| KS1767 | Kaposi’s sarcoma | + | 660 | > 4700 | >7 | |
| Non-endothelial | RAW264.7 | Monocyte | + | 60 | 1200 | 20 |
| SK-N-SH | Neuroblastoma | ++ | 27 | 1809 | 67 | |
| TC-71 | Ewing’s sarcoma | + | 190 | 1300 | 6.8 | |
| U-87 MG | Glioblastoma | + | 204 | 2900 | 14 | |
| MDA-MB-231/luc | Breast adenocarcinoma | − | 500 | 2300 | 4.6 |
Targeting index defined as (IC50 GrB)/(IC50 GrB/VEGF121)
VEGF121 blocks GrB/VEGF121-mediated cytotoxicity
To determine whether cytotoxicity of GrB/VEGF121 is mediated by its binding to VEGF121-receptors, we pre-incubated PAE/VEGFR-2 cells with 1 μM VEGF121 for one hour, followed by addition of various amounts of GrB/VEGF121 and GrB. As shown in Table 2, preincubation with VEGF121 strongly reduced GrB/VEGF121-mediated cytotoxicity, confirming that binding of the VEGF121 moiety of the fusion protein to VEGFR-2 is required to initiate cytotoxicity.
Table 2.
Cytotoxicity of GrB/VEGF121 and GrB in the presence and absence of 1 μM VEGF121.
| Treatment | IC50 (nM) |
|---|---|
| GrB/VEGF121 | 40 |
| GrB/VEGF121 + VEGF121 | 290 |
| GrB | 730 |
| GrB + VEGF121 | 770 |
GrB/VEGF121 cytotoxicity on PAE/VEGFR-2 cells correlates with exposure time
We next studied the cytotoxic effect of GrB/VEGF121 as a function of exposure time of this agent on PAE/VEGFR-2 cells. Cells were treated with GrB/VEGF121/rGel from 4–72 hours and the cytotoxic effect was assessed at the end of the 72-hour period. As summarized in Table 3, the lowest IC50 doses of GrB/VEGF121 (≤22 nM) were observed after 24 h of exposure although longer exposure times did result in lower IC50’s. GrB/VEGF121 demonstrated relatively low targeted toxicity after short (4–8 hour) exposure, with IC50’s over 100 nM.
Table 3.
Minimal contact time for GrB/VEGF121 on PAE/VEGFR-2 cells
| Time of Exposure (h) | IC50 (nM) |
|---|---|
|
| |
| 4 | N. D. (> 150) |
| 8 | 139.8 ± 50.1 |
| 24 | 22.2 ± 14 |
| 48 | 5.8 ± 2.3 |
| 72 | 18.2 ± 2.2 |
VEGF121/rGel is internalized into PAE/VEGFR-2 cells but not into PAE/VEGFR-1 cells
GrB/VEGF121 internalization into PAE/VEGFR-2 cells was assessed by immunofluorescence microscopy following stripping of the plasma membrane. Efficient localization of the construct into the cytoplasm was observed at the 24 h timepoint (Figure 2). Localization of GrB/VEGF121 into PAE/VEGFR-1 cells was much less efficient than into PAE/VEGFR-2 (Supplementary Figure 4), which appeared to correlate to the cytotoxicity profiles of these two cell lines.
Figure 2.
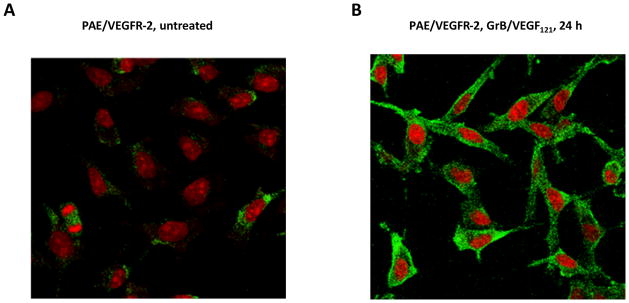
GrB/VEGF121 internalization into PAE/VEGFR-2 was driven by VEGF121. Cells were (A) untreated or (B) treated with 20 nM GrB/VEGF121 for 24 h, followed by detection of GrB. Nuclei were stained with propidium iodide. Only nuclei were visible in untreated PAE/VEGFR-2 cells whereas fluorescent GrB staining was observed in the cytoplasm of GrB/VEGF121-treated cells.
GrB/VEGF121 treatment results in mitochondrial depolarization and triggers apoptosis in target cells
Mitochondrial depolarization is a common event in numerous forms of cell death (24). Exposure of PAE/VEGFR-1 cells to GrB/VEGF121 over 48 h resulted in a slight increase in the number of cells with low mitochondrial transmembrane potential, from 10% to 16.2%, compared to GrB (12.5%) (Figure 3A). In contrast, 48% of PAE/VEGFR-2 cells underwent mitochondrial depolarization, compared to 13% of untreated cells and 14.4% of cells treated with GrB. The impact of GrB/VEGF121 on PAE/VEGFR-2 cells appeared to be dependent on exposure time, but a significant degree of mitochondrial depolarization occurred within the first four hours of treatment (33.8%) compared to later timepoints (39.9% at 24 h) (Figure 3B). The GrB/VEGF121-mediated mode of cell death was investigated by flow cytometry twenty four hours after treatment of cells with GrB/VEGF121, and subsequent incubation with Annexin V and propidium iodide. As expected, GrB/VEGF121 had a minimal effect on Annexin V and PI uptake in PAE/VEGFR-1 cells, with 9.3% of cells mobilized into early apoptosis, compared to 6.8% of control cells (Figure 3C). Non-targeted MDA-MB-435 cells were similarly inert (5.8% of treated cells in early apoptosis vs 5.4% control cells). In contrast, 34.5% of GrB/VEGF121-treated PAE/VEGFR-2 cells were found to have mobilized into early apoptosis, compared to 4.3% of control cells. Plasma membrane integrity, as assessed by propidium iodide exclusion, was not lost over the 24 hour period, indicating that necrosis is not a major mechanism of cell death over the observed period of time. Treatment of PAE/VEGFR-2 cells with 20 nM GrB/VEGF121 over time resulted in a time-dependent increase in the number of Annexin V+ cells, which did not change in cells treated with GrB (Figure 3D).
Figure 3.

GrB/VEGF121 triggers apoptosis in target cells via mitochondrial depolarization. (A–B). Mitochondrial depolarization of PAE/VEGFR-2 and PAE/VEGFR-1 cells following treatment with GrB or GrB/VEGF121. Granzyme B treatment (20 nM) did not trigger mitochondrial depolarization in either cell. GrB/VEGF121 treatment resulted in significant levels of mitochondrial depolarization in PAE/VEGFR-2 cells within 4 h of treatment. (C–D) Detection of apoptosis in VEGFR-2+ cells. Cells were treated with GrB or GrB/VEGF121 for various times. Apoptosis was detected by flow cytometry following incubation with Annexin V and propidium iodide. N.D., not done. NT, no treatment.
GrB/VEGF121 cytotoxicity is caspase-dependent and -independent
GrB/VEGF121, as well as GrB, activated both Caspase 3 (Figure 4A) and Caspase 9 (Figure 4B), as assessed by an in vitro chromogenic assay for caspase activation. Because apoptosis can occur via caspase-dependent and –independent mechanisms, we then assessed the ability of z-VAD-fmk, a pan-caspase inhibitor, to mitigate GrB/VEGF121 cytotoxicity. Pre-treatment of cells in vitro with z-VAD-fmk prior to incubation with GrB/VEGF121 significantly inhibited the cleavage of the caspase 3 (Figure 4C) and caspase 9 (data not shown) chromogenic substrates, indicating inactivation of the respective proteins. However, this had no effect on GrB/VEGF121-mediated cytotoxicity on PAE/VEGFR-2 cells over 72 hours (Figure 4D) while, as expected, no effect was observed on PAE/VEGFR-1 cells (Figure 4E). GrB/VEGF121-mediated caspase-3 cleavage was observed within one hour of treatment of PAE/VEGFR-2 cells, in the presence of z-VAD-fmk concentrations ranging from 0–100 μM (Figure 4F), suggesting that caspase-3 cleavage occurred independently of z-VAD-fmk mediated inhibition of its enzymatic activity. To assess the impact of blocking caspase activation on GrB/VEGF121 on downstream mediators of apoptosis, PAE/VEGFR-1 and PAE/VEGFR-2 cells were untreated or pre-treated with 20 μM z-VAD-fmk for one hour prior to treatment with GrB/VEGF121 for various timepoints up to 48 h. Cells were then harvested and assessed for PARP-1 cleavage by Western blotting. As expected, little to no PARP-1 cleavage was observed in PAE/VEGFR-1 cells regardless of treatment conditions (Figure 4G). In contrast, cleaved PARP was observed in PAE/VEGFR-2 lysates when treated with GrB/VEGF121 (Figure 4H). Interestingly, PAE/VEGFR-2 cells pretreated with z-VAD-fmk also had cleaved PARP-1. Together, these results suggest that GrB/VEGF121 activates PARP-1-mediated apoptosis independently of caspase activation.
Figure 4. PARP-1 cleavage by GrB/VEGF121 is caspase-dependent and –independent.

(A–B) GrB/VEGF121 activates caspase-3 (4A) and -9 (4B) in whole cell lysates. Chromogenic substrates for caspase-3 and caspase-9 were incubated in PAE/KDR whole cell lysates with or without GrB or GrB/VEGF121. Activation of the caspases was indicated by an increase in absorbance of the cleaved substrate. (C) Pre-incubation of PAE/VEGFR-2 cells with z-VAD-fmk (20 or 100 μM) for 1 h prior to GrB/VEGF121 treatment for 2 h significantly blocked caspase-3 activation. (D–E) GrB/VEGF121-mediated cytotoxicity on PAE/VEGFR-2 and PAE/VEGFR-1 cells over 72 h is unchanged despite co-incubation with various concentrations of z-VAD-fmk. (F) Granzyme B internalization results in caspase-3 cleavage in vitro. PAE/VEGFR-2 cells were pre-treated with up to 100 μM z-VAD-fmk prior to addition of 20 nM GrB/VEGF121 for one hour. GrB/VEGF121-mediated cleavage of caspase-3 cleavage occurred in the presence of up to 100 μM z-VAD-fmk. (G–H) Caspase-independent PARP cleavage. PAE/VEGFR-2 and PAE/VEGFR-1 cells were treated with up to 48 with 20 nM GrB/VEGF121 in the presence or absence of 20 μM z-VAD-fmk. PARP cleavage was observed in PAE/VEGFR-2, but not PAE/VEGFR-1, cells.
In vivo localization of GrB/VEGF121 in PC-3 tumor-bearing mice
Mice bearing PC-3 subcutaneous tumors were injected intravenously with equimolar doses of GrB/VEGF121 or GrB. Tumor and other organs were harvested four hours later, preserved frozen, and then analyzed for localization of GrB or GrB/VEGF121 by immunofluorescence. As shown in Figure 5A–D, GrB/VEGF121 was detected in the tumors of GrB/VEGF121-injected mice. GrB/VEGF121 diffused into the perivascular tumor areas adjacent to microvessels over the four hour period, although some GrB/VEGF121 was detected as localized within CD31+ tumor vessels. Free GrB did not localize to tumor tissue (Figure 5E–F). Thus, GrB/VEGF121 appears to localize specifically to tumor tissue after i.v. injection. Normal organs were unstained by GrB/VEGF121 or free GrB (data not shown). These results indicate that GrB/VEGF121 localized specifically to tumor tissue after i.v. injection.
Figure 5. Specific localization of GrB/VEGF121 to PC-3 tumors.

Nu/nu mice bearing human prostate PC-3 tumors were injected i.v. with GrB/VEGF121 or GrB at molar equivalent doses. Four hours after administration, tissues were removed and snap frozen. Sections were stained with immunofluorescent reagents to detect murine blood vessels (CD31, red), nuclei (Hoechst, blue) and granzyme B (green). Co-localization of GrB into CD31+ tumor vessels appear yellow (representative areas indicated with white arrows). (A–D) GrB/VEGF121 localized to the tumor tissue, whereas (E–F) GrB did not. Robust GrB staining was observed in the GrB/VEGF121-treated tumor core (B–C) as well as tumor vessels (D). In contrast, no GrB was detected in GrB-treated tumors. Vessels in all normal organs were unstained by GrB/VEGF121 or GrB.
Determination of Maximal Tolerated Dose and Lethal Dose for GrB/VEGF12 in BALB/c Mice
All mice treated with saline (control) and 40 mg/kg GrB/VEGF121 survived when treated with the described regimen. No impact on weight was observed in GrB/VEGF121-treated mice compared to control mice. Accordingly, both the LD50 and MTD of GrB/VEGF121 at this schedule in mice are above 40 mg/kg (120 mg/m2).
Inhibition of tumor growth in vivo by GrB/VEGF121
We examined the efficacy of GrB/VEGF121 against subcutaneously injected PC-3 cells in male nude mice. In all cases differences in tumor volume were not seen until about day 28, well after cessation of treatment (Days 3–13) (Figure 6A). Saline-treated mice were euthanized on Day 53 (cachexia) and Day 59 (tumor volume > 1500 mm3). Thus, on average, tumors from saline-treated mice grew about 15-fold over 30 days. Over the same time span, granzyme-B treated tumors grew at a similar rate. In contrast, tumors from mice treated with the 11 mg/kg dose of GrB/VEGF121 only grew about 3-fold by Day 60, whereas tumors treated with the 27 mg/kg dose of GrB/VEGF121 did not grow during this period and remained the same size as at the onset of treatment. Tumors from 11 mg/kg GrB/VEGF121-treated mice did not reach the maximum tumor volume of 1500 mm3 until Day 80. Only one of the tumors from the 27 mg/kg GrB/VEGF121-treated group eventually grew, reaching 1500 mm3 at Day 100, terminating the study. Overall, GrB/VEGF121 treatment appeared to be well tolerated by mice and resulted in significant anti-tumor efficacy at doses below the MTD. Histopathological analysis of tumors was performed at the end of the study. The vascular area from the tumor sample from mice treated with GrB/VEGF121 was found to be significantly lower compared to that from saline-treated mice (mean area 0.3% vs 4.2%; P < 0.0001, t test, two-tail) (Figure 6B). Thus, GrB/VEGF121 treatment appears to directly impact tumor vasculature.
Figure 6. Anti-tumor efficacy of GrB/VEGF121.

(A) Mice with PC-3 tumor xenografts were intravenously injected with saline, GrB or GrB/VEGF121 at the indicated times (arrows). Mean tumor volume was calculated as W × H × L as measured with digital calipers. (B) Representative immunofluorescent CD31 staining of saline- and GrB/VEGF121-treated tumors. Quantification of percent CD31+ area revealed that GrB/VEGF121 significantly reduced the overall vascular area compared to the saline treatment. (C) Quantification of the growth rate of PC-3 tumor cells by Ki-67. A minimum of 5 fields were assessed per slide. GrB/VEGF121 treatment reduced the number of cycling cells by 50% compared to tumors from Saline-treated mice.
Effect of GrB/VEGF121 treatment on the number of cycling cells in PC-3 tumors
The growth rate of PC-3 cells was determined by staining cells with Ki-67 antibody. The number of cycling tumor cells in lesions from the GrB/VEGF121 group was reduced by 50% compared to controls (mean number of cells per field reduced from 59.6 ± 4.7 to 29.3 ± 4.1; P < 0.0002, t test, two-tail) (Figure 6C).
DISCUSSION
Interruption of signaling through VEGF is a clinically-validated strategy in a number of malignancies including breast and colon cancer (25). Signaling through VEGF and c-Kit have also been found to contribute to the pathology of AML (26). In addition to Avastin which reduces free VEGF levels, there are a number of small molecule approaches to suppress VEGFR related signaling events generally based on receptor tyrosine-kinase inhibition (27, 28). Some pharmacokinetic limitations have been observed, including limited half-lives and absorption (29). The development of targeted protein therapeutics focusing on interrupting the VEGF pathway has been more limited. Lu and colleagues and Witte and colleagues have identified antibodies specific for the external domain of the KDR receptor for VEGF (30, 31) to block signaling by interfering with ligand-binding.
Utilizing a ligand-based approach to target VEGFR expressing cells, there have also been a number of studies employing VEGF-A isoforms fused to shiga toxin (9), diphtheria toxin (10, 32) and abrin (33). Our laboratory has described the capabilities of a fusion construct of VEGF121 fused to rGel toxin (12–14). This construct was found to be highly active against a number of endothelial cells in culture and against a number of solid tumor xenograft models. In addition, this agent demonstrated impressive activity against skeletal tumor metastases, which is a frequent cause or mortality and morbidity in late-stage breast and prostate tumors, via a multi-targeted approach (34).
Concerns relating to immunogenicity of the toxin components during long-term treatment have led to the development of powerful, cytotoxic but non-immunogenic proteins for targeted therapeutic applications. A number of groups have developed non-immunogenic variants of toxins such as pseudomonas exotoxin (35) and deBouganin (36) by mutating the B-cell recognition epitopes on the original molecule. Other groups have developed targeted therapeutics with highly cytotoxic human proteins such as RNAse (37) and constitutively active DAPK2 (38). Several groups, including ours, have developed GrB as a novel payload which is completely human in origin. This well-studied 25 kDa serine protease is capable of inducing intense cellular apoptosis through both caspase-dependent and caspase-independent multiple-cascade mechanisms. In addition to the GrB payload, we have developed a number of human pro-apoptotic molecules such as BAX345 and IκB for use as unique payloads fused or conjugated to cell-targeting molecules. When delivered to cells, these payloads were shown to generate unique and impressive cytotoxic effects (39, 40).
We previously described the generation of GrB/VEGF121 and the in vitro effects of this molecule against a limited number of cells in culture (19). The current study expands and confirms our original observations regarding the activity of this agent. Our cytotoxicity findings against a panel of endothelial, tumor, and bone marrow-derived cells indicate that GrB/VEGF121 effectively targets endothelial cells over-expressing VEGFR-2. In addition, this agent was found to be effective against some non-endothelial cells which over-express the VEGFR-1(FLT-1) receptor. We also found that the SK-N-SH neuroblastoma cell line, previously shown to express VEGFR-2 but not VEGFR-1 (21), was particularly sensitive to the direct effects of GrB/VEGF121.
Entrapment of targeted therapeutics into cell protective vesicles such as endosomes can significantly impact the amount of payload delivered to the cell. Dälken et al (41) noted the entrapment of their GrB-based, anti-Her2 fusion proteins in the endosomal compartment, requiring treatment of cells with the endosomolytic agent chloroquine to achieve cytotoxicity in the nanomolar range. Other constructs expressed in insect, yeast, and mammalian expression systems employing GrB at the C- or N-terminus have also been tested, showing varying requirements for endosomal release (42–46). We did not observe a significant impact of addition of chloroquine to cells treated with GrB/VEGF121 (data not shown), suggesting that endosomal entrapment may not be a feature of this particular construct. This suggests that endosomal entrapment observed with other agents may be a unique feature of the target antigen or the target cells themselves.
Although GrB/VEGF121 appeared to be less cytotoxic in vitro against the same cell lines compared to VEGF121/rGel (12, 14), both molecules were found to localize exclusively to tumor tissue over normal tissue, as determined by immunohistochemistry. While VEGF121/rGel overwhelmingly localized to the tumor vasculature, we found that significant amounts of GrB/VEGF121 was able to penetrate into the perivascular tumor areas adjacent to microvessels and in other areas in the tumor less-well vascularized. This may be the result of a more rapid cytotoxic onset of the targeted GrB construct resulting in a more rapid loss of tumor vascular integrity. The GrB/VEGF121 localization to the tumor appears to be VEGFR-mediated since immunohistochemical analysis of tissues from mice treated with GrB alone showed no evidence of tumor localization.
Both GrB/VEGF121 and VEGF121/rGel (14) exhibited similar levels of anti-tumor efficacy against subcutaneous PC-3 prostate xenograft models. However, the MTD for VEGF121/rGel was determined to be 18 mg/kg (47) while the MTD of GrB/VEGF121 was not achieved and appeared to be greater than 40 mg/kg since no toxicity was observed in mice treated at this dose. Treatment of mice with GrB/VEGF121 at 27 mg/kg resulted in the regression of 4 of 5 tumors in this group and we observed a dramatic reduction in overall tumor vessel density in the remaining GrB/VEGF121-treated tumor compared to the control. The lack of toxicity suggests that improved anti-tumor efficacy with higher doses may be possible.
Several investigators have shown that the recruitment of bone marrow derived VEGFR-1+ cells of various lineages plays a role in the establishment and growth of metastases (48–50). The IC50 (60 nM) of GrB/VEGF121 against RAW264.7 cells, which primarily express VEGFR-1, is similar to the IC50 (40 nM) previously observed for VEGF121/rGel (12), suggesting that the progression of metastatic disease by targeting vascular as well as stromal components may be possible. The unique mechanism of action of GrB-based fusion-targeted therapeutic agents may also provide for novel interactions with other types of therapeutic agents. Investigation of GrB/VEGF121 efficacy in other tumor models, particularly bone metastasis and in combination with clinically-relevant therapeutics, is underway.
In summary, we produced recombinant GrB/VEGF121 in a mammalian expression system and found that this molecule (a) demonstrated varying levels of cytotoxicity to VEGFR-positive cells, (b) internalized efficiently into VEGFR-2+ cells, (c) triggered apoptosis via multi-modal mechanisms (d) localized in both vascularized and less vascularized areas in the tumor, and (e) significantly delayed tumor growth. Delivery of granzyme B to tumor vascular endothelial cells or to tumor cells has significant therapeutic potential and our studies suggest that further studies with GrB/VEGF121 as an antitumor agent for treating cancer patients are warranted.
Supplementary Material
Acknowledgments
Research supported, in part, by the Clayton Foundation for Research (M. G. Rosenblum) and by the MD Anderson Cancer Center Support Grant CA016672.
Footnotes
Conflicts of interest: The authors have no potential conflicts of interest
Reference List
- 1.Caputo M, Zirpoli H, Di BR, De NK, Tecce MF. Perspectives of choroidal neovascularization therapy. Curr Drug Targets. 2011;12:234–42. doi: 10.2174/138945011794182791. [DOI] [PubMed] [Google Scholar]
- 2.Paleolog EM. Angiogenesis in rheumatoid arthritis. Arthritis Res. 2002;4 (Suppl 3):S81–S90. doi: 10.1186/ar575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–95. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruce D, Tan PH. Blocking the interaction of vascular endothelial growth factor receptors with their ligands and their effector signaling as a novel therapeutic target for cancer: time for a new look? Expert Opin Investig Drugs. 2011;20:1413–34. doi: 10.1517/13543784.2011.611801. [DOI] [PubMed] [Google Scholar]
- 6.Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–8. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de VC, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–91. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 8.Shinkaruk S, Bayle M, Lain G, Deleris G. Vascular endothelial cell growth factor (VEGF), an emerging target for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2003;3:95–117. doi: 10.2174/1568011033353452. [DOI] [PubMed] [Google Scholar]
- 9.Hotz B, Backer MV, Backer JM, Buhr HJ, Hotz HG. Specific targeting of tumor endothelial cells by a shiga-like toxin-vascular endothelial growth factor fusion protein as a novel treatment strategy for pancreatic cancer. Neoplasia. 2010;12:797–806. doi: 10.1593/neo.10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hotz HG, Gill PS, Masood R, Hotz B, Buhr HJ, Foitzik T, et al. Specific targeting of tumor vasculature by diphtheria toxin-vascular endothelial growth factor fusion protein reduces angiogenesis and growth of pancreatic cancer. J Gastrointest Surg. 2002;6:159–66. doi: 10.1016/s1091-255x(01)00040-3. [DOI] [PubMed] [Google Scholar]
- 11.Ramakrishnan S, Wild R, Nojima D. Targeting tumor vasculature using VEGF-toxin conjugates. Methods Mol Biol. 2001;166:219–34. doi: 10.1385/1-59259-114-0:219. [DOI] [PubMed] [Google Scholar]
- 12.Mohamedali KA, Poblenz AT, Sikes CR, Navone NM, Thorpe PE, Darnay BG, et al. Inhibition of prostate tumor growth and bone remodeling by the vascular targeting agent VEGF121/rGel. Cancer Res. 2006;66:10919–28. doi: 10.1158/0008-5472.CAN-06-0459. [DOI] [PubMed] [Google Scholar]
- 13.Ran S, Mohamedali KA, Luster TA, Thorpe PE, Rosenblum MG. The vascular-ablative agent VEGF(121)/rGel inhibits pulmonary metastases of MDA-MB-231 breast tumors. Neoplasia. 2005;7:486–96. doi: 10.1593/neo.04631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veenendaal LM, Jin H, Ran S, Cheung L, Navone N, Marks JW, et al. In vitro and in vivo studies of a VEGF121/rGelonin chimeric fusion toxin targeting the neovasculature of solid tumors. Proc Natl Acad Sci U S A. 2002;99:7866–71. doi: 10.1073/pnas.122157899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang M, Gao H, Sun X, Yan Y, Quan Q, Zhang W, et al. Multiplexed PET probes for imaging breast cancer early response to VEGF(1)(2)(1)/rGel treatment. Mol Pharm. 2011;8:621–8. doi: 10.1021/mp100446t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17:616–23. doi: 10.1038/cdd.2009.206. [DOI] [PubMed] [Google Scholar]
- 17.Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC. Granzyme B: a natural born killer. Immunol Rev. 2003;193:31–8. doi: 10.1034/j.1600-065x.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Cheung LH, Hittelman WN, Rosenblum MG. Targeted delivery of human pro-apoptotic enzymes to tumor cells: In vitro studies describing a novel class of recombinant highly cytotoxic agents. Mol Cancer Ther. 2003;2:1341–50. [PubMed] [Google Scholar]
- 19.Liu Y, Cheung LH, Thorpe P, Rosenblum MG. Mechanistic studies of a novel human fusion toxin composed of vascular endothelial growth factor (VEGF)121 and the serine protease granzyme B: directed apoptotic events in vascular endothelial cells. Mol Cancer Ther. 2003;2:949–59. [PubMed] [Google Scholar]
- 20.Liu Y, Zhang W, Niu T, Cheung LH, Munshi A, Meyn RE, Jr, et al. Targeted apoptosis activation with GrB/scFvMEL modulates melanoma growth, metastatic spread, chemosensitivity, and radiosensitivity. Neoplasia. 2006;8:125–35. doi: 10.1593/neo.05556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meister B, Grunebach F, Bautz F, Brugger W, Fink FM, Kanz L, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors in human neuroblastoma. Eur J Cancer. 1999;35:445–9. doi: 10.1016/s0959-8049(98)00387-6. [DOI] [PubMed] [Google Scholar]
- 22.Kroll J, Waltenberger J. A novel function of VEGF receptor-2 (KDR): rapid release of nitric oxide in response to VEGF-A stimulation in endothelial cells. Biochem Biophys Res Commun. 1999;265:636–9. doi: 10.1006/bbrc.1999.1729. [DOI] [PubMed] [Google Scholar]
- 23.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–95. [PubMed] [Google Scholar]
- 24.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–30. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 25.Satoh T, Yamaguchi K, Boku N, Okamoto W, Shimamura T, Yamazaki K, et al. Phase I results from a two-part Phase I/II study of cediranib in combination with mFOLFOX6 in Japanese patients with metastatic colorectal cancer. Invest New Drugs. 2012;30:1511–8. doi: 10.1007/s10637-011-9693-6. [DOI] [PubMed] [Google Scholar]
- 26.Fiedler W, Mesters R, Heuser M, Ehninger G, Berdel WE, Zirrgiebel U, et al. An open-label, Phase I study of cediranib (RECENTIN) in patients with acute myeloid leukemia. Leuk Res. 2010;34:196–202. doi: 10.1016/j.leukres.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 27.Normanno N, Morabito A, De LA, Piccirillo MC, Gallo M, Maiello MR, et al. Target-based therapies in breast cancer: current status and future perspectives. Endocr Relat Cancer. 2009;16:675–702. doi: 10.1677/ERC-08-0208. [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Tan C, Ding H, Xin T, Jiang Y. Selective VEGFR inhibitors for anticancer therapeutics in clinical use and clinical trials. Curr Pharm Des. 2012;18:2921–35. doi: 10.2174/138161212800672732. [DOI] [PubMed] [Google Scholar]
- 29.Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med. 2012;2:a006577. doi: 10.1101/cshperspect.a006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int J Cancer. 2002;97:393–9. doi: 10.1002/ijc.1634. [DOI] [PubMed] [Google Scholar]
- 31.Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, et al. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev. 1998;17:155–61. doi: 10.1023/a:1006094117427. [DOI] [PubMed] [Google Scholar]
- 32.Koshikawa N, Takenaga K. Hypoxia-regulated expression of attenuated diphtheria toxin A fused with hypoxia-inducible factor-1alpha oxygen-dependent degradation domain preferentially induces apoptosis of hypoxic cells in solid tumor. Cancer Res. 2005;65:11622–30. doi: 10.1158/0008-5472.CAN-05-0111. [DOI] [PubMed] [Google Scholar]
- 33.Smagur A, Boyko MM, Biront NV, Cichon T, Szala S. Chimeric protein ABRaA-VEGF121 is cytotoxic towards VEGFR-2-expressing PAE cells and inhibits B16-F10 melanoma growth. Acta Biochim Pol. 2009;56:115–24. [PubMed] [Google Scholar]
- 34.Mohamedali KA, Li ZG, Starbuck MW, Wan X, Yang J, Kim S, et al. Inhibition of prostate cancer osteoblastic progression with VEGF121/rGel, a single agent targeting osteoblasts, osteoclasts, and tumor neovasculature. Clin Cancer Res. 2011;17:2328–38. doi: 10.1158/1078-0432.CCR-10-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci U S A. 2011;108:5742–7. doi: 10.1073/pnas.1102746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Entwistle J, Brown JG, Chooniedass S, Cizeau J, MacDonald GC. Preclinical evaluation of VB6-845: an anti-EpCAM immunotoxin with reduced immunogenic potential. Cancer Biother Radiopharm. 2012;27:582–92. doi: 10.1089/cbr.2012.1200.271. [DOI] [PubMed] [Google Scholar]
- 37.Rybak SM, Arndt MA, Schirrmann T, Dubel S, Krauss J. Ribonucleases and immunoRNases as anticancer drugs. Curr Pharm Des. 2009;15:2665–75. doi: 10.2174/138161209788923921. [DOI] [PubMed] [Google Scholar]
- 38.Tur MK, Neef I, Jost E, Galm O, Jager G, Stocker M, et al. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J Immunother. 2009;32:431–41. doi: 10.1097/CJI.0b013e31819f1cb6. [DOI] [PubMed] [Google Scholar]
- 39.Lyu MA, Cheung LH, Hittelman WN, Liu Y, Marks JW, Cho MJ, et al. Bax345/BLyS: a novel, completely human fusion protein targeting malignant B cells and delivering a unique mitochondrial toxin. Cancer Lett. 2012;322:159–68. doi: 10.1016/j.canlet.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 40.Zhou H, Liu Y, Cheung LH, Kim S, Zhang W, Mohamedali KA, et al. Characterization and mechanistic studies of a novel melanoma-targeting construct containing IkappaBa for specific inhibition of nuclear factor-kappaB activity. Neoplasia. 2010;12:766–77. doi: 10.1593/neo.10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalken B, Giesubel U, Knauer SK, Wels WS. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006;13:576–85. doi: 10.1038/sj.cdd.4401773. [DOI] [PubMed] [Google Scholar]
- 42.Jabulowsky RA, Oberoi P, Bahr-Mahmud H, Dalken B, Wels WS. Surface charge-modification prevents sequestration and enhances tumor-cell specificity of a recombinant granzyme B-TGFalpha fusion protein. Bioconjug Chem. 2012;23:1567–76. doi: 10.1021/bc3000657. [DOI] [PubMed] [Google Scholar]
- 43.Kanatani I, Lin X, Yuan X, Manorek G, Shang X, Cheung LH, et al. Targeting granzyme B to tumor cells using a yoked human chorionic gonadotropin. Cancer Chemother Pharmacol. 2011;68:979–90. doi: 10.1007/s00280-011-1573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurschus FC, Kleinschmidt M, Fellows E, Dornmair K, Rudolph R, Lilie H, et al. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004;562:87–92. doi: 10.1016/S0014-5793(04)00187-5. [DOI] [PubMed] [Google Scholar]
- 45.Stahnke B, Thepen T, Stocker M, Rosinke R, Jost E, Fischer R, et al. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol Cancer Ther. 2008;7:2924–32. doi: 10.1158/1535-7163.MCT-08-0554. [DOI] [PubMed] [Google Scholar]
- 46.Zhao J, Zhang LH, Jia LT, Zhang L, Xu YM, Wang Z, et al. Secreted antibody/granzyme B fusion protein stimulates selective killing of HER2-overexpressing tumor cells. J Biol Chem. 2004;279:21343–8. doi: 10.1074/jbc.M312648200. [DOI] [PubMed] [Google Scholar]
- 47.Mohamedali KA, Niu G, Luster TA, Thorpe PE, Gao H, Chen X, et al. Pharmacodynamics, tissue distribution, toxicity studies and antitumor efficacy of the vascular targeting fusion toxin VEGF121/rGel. Biochem Pharmacol. 2012;84:1534–40. doi: 10.1016/j.bcp.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.