

Abstract
The P23H mutation in rhodopsin (RhoP23H) is a prevalent cause of autosomal dominant retinitis pigmentosa. We examined the role of the ER stress proteins, Chop and Ask1, in regulating the death of rod photoreceptors in a mouse line harboring the RhoP23H rhodopsin transgene (GHL+). We used knockout mice models to determine whether Chop and Ask1 regulate rod survival or retinal degeneration. Electrophysiological recordings showed similar retinal responses and sensitivities for GHL+, GHL+/Chop−/− and GHL+/Ask1−/− animals between 4–28 weeks, by which time all three mouse lines exhibited severe loss of retinal function. Histologically, ablation of Chop and Ask1 did not rescue photoreceptor loss in young animals. However, in older mice, a regional protective effect was observed in the central retina of GHL+/Chop−/− and GHL+/Ask1−/−, a region that was severely degenerated in GHL+ mice. Our results show that in the presence of the RhoP23H transgene, the rate of decline in retinal sensitivity is similar in Chop or Ask1 ablated and wild-type retinas, suggesting that these proteins do not play a major role during the acute phase of photoreceptor loss in GHL+ mice. Instead they may be involved in regulating secondary pathological responses such as inflammation that are upregulated during later stages of disease progression.
Introduction
Retinitis pigmentosa (RP) is a group of inherited retinal diseases that are characterized by the degeneration of the retina. RP has an incidence rate of ∼1 in 4000 [1] and can be classified based on the mode of inheritance [2], [3]. To date, mutations in over 50 genes have been associated with RP [4]. Mutations in rhodopsin (Rho), the visual pigment of rod photoreceptors, accounts for ∼10% of all cases of RP [1]. These mutations have been grouped on the associated functional defects and cellular distribution of the protein [5]. In North America, the class II missense mutation P23H (RhoP23H), is the most common and accounts for >25% of all autosomal dominant forms of RP [4]. This mutation leads to a misfolded protein that is characterized by abnormal glycosylation pattern, abnormal disulfide bond formation and oligomerization [6]. In RP patients with the RhoP23H allele, retinal degeneration is characterized by the initial disorganization and shortening of rod outer segments, followed by the loss of rod photoreceptors and the ensuing death of cone cells [7], [8]. Similar findings are observed in various RhoP23H animal models [9]–[12]. However, the cellular mechanisms that lead to cell death are not completely understood.
Since RhoP23H is a misfolded protein, recent studies have examined the up-regulation of endoplasmic reticulum stress (ER-stress)/unfolded protein response (UPR) pathways with retinal degeneration [13]–[15]. The UPR is an intracellular response to perturbations of ER homeostasis. It is regulated by the Hsp70 family protein chaperone, BiP [16]. Under normal conditions, BiP binds to three ER transmembrane proteins, ATF6, IRE1, and PERK. In a stressed state, BiP dissociates, and ATF6, IRE1 and PERK pathways are activated, resulting in the upregulation of protective events to decrease ER stress (reviewed in [17]). However, failure to restore ER homeostasis results in the upregulation of cell death pathways [18]–[21]. Two genes, C/EBP homologous protein-10 (Chop) and apoptosis signal-regulating kinase 1 (Ask1), have been identified as important regulators of UPR induced cell death/apoptosis [22]–[26].
Chop is a transcription factor [27] that is regulated mainly by the PERK branch of the UPR response and has a functional role in the other pro-apoptotic pathways of the UPR [25], [28], [29]. Ask1 is a kinase activated by the IRE1 division of the UPR pathway [30]–[32]. It activates downstream kinases that induce apoptosis, and post-translationally phosphorylates Chop to positively modify its activity [33]. Several studies have shown that altering the expression of UPR genes, including Chop and Ask1, can change the pathology of different diseases [22]–[24], [26], [34]–[36]. The role of the UPR in RP is not completely understood. Although CHOP was elevated in cells transfected with RhoP23H [14], it is not clear whether that also occurs in rod photoreceptors. In transgenic rats expressing RhoP23H, expression of Chop and BiP were increased in degenerated retinas [13]–[15]. Moreover, overexpression of BiP can restore some visual function in this animal [13]. Upregulation of Ask1 has also been linked to retinal degenerative diseases [37]. More recently, the Ask1 pathway has been implicated as a regulator of photoreceptor death in a transgenic RP mouse model expressing a RhoT17M mutation [38]. Taken together, these findings suggest that UPR genes may regulate photoreceptor death in RP, and regulating the ER stress response might be beneficial to the treatment of RP. However, direct evidence is lacking that establishes the upregulation of UPR proteins as a necessary response for photoreceptor death.
To determine whether Chop or Ask1 regulate photoreceptor cell death, we utilized knockout mice and a well characterized RhoP23H transgenic mouse model (GHL+) [9]. Similar to human patients, RP in GHL+ mice is characterized by the progressive decline in visual function, and morphological changes in the retina that include shortening of rod outer segments and thinning of the outer nuclei layer due to death of photoreceptor cells [9], [39]. The disease pathology of GHL+ mice is also comparable to recently generated RhoP23H/+ heterozygote knock-in mice [11], which also show a slow progressive degeneration of the retina characterized by shortening of rods followed by death of rod photoreceptors. We assessed the role of Chop and Ask1 proteins as possible modifiers of GHL+ induced pathology by generating GHL+/Chop−/− and GHL+/Ask1−/− mice and characterized the progression of retinal degeneration electrophysiologically and histologically over a 7-month period. We observed that ablation of either of these genes did not affect the rate of photoreceptor death in early stages of the disease, however a regional protective effect was observed in the central retina at later stages of the disease. In GHL+ mice, the central retina degenerates faster than more peripheral regions of the retina. Therefore, our findings suggest that deleting Chop and Ask1 may have an indirect role in protecting the retina by modifying late stage responses to ongoing photoreceptor death, resulting in a slight delay of photoreceptor death.
Materials and Methods
Animal models
All animal handling and experiments were in agreement with the animal care and use guidelines of the Association for Research in Vision and Ophthalmology (ARVO). This study was done under the approval of the SUNY Upstate Medical University Committee on the Human Use of Animals (CHUA no. 182 and 323). C57BL/6 and Chop−/− animals were purchased from Taconic and the Jackson Laboratory respectively. Ask1−/− mice were obtained from H. Ichijo (University of Tokyo, Japan). GHL + transgenic mice [9] were obtained from C. Cepko (Harvard Medical School, Boston, MA). The GHL+ mice express a transgene encoding a mutated mouse opsin gene containing three amino acid substitutions at the N-terminus: V20G, P23H, and P27L (GHL+ mice). Mice were housed in a 14 h light–10 h dark cycle.
Genotyping
Genomic DNA from tail snips were isolated either by boiling in NaOH or using the Phire® Animal Tissue Direct PCR Kit (Fisher Scientific Pittsburgh, PA) according to manufacturer's instructions. The genotype of each gene was confirmed by PCR: Chop, [40], GHL+ [9], Ask1−/− [32], SOX21 (Phire® Animal Tissue Direct PCR Kit) and β-actin (Promega, Madison, WI) were used as loading controls.
Electroretinogram (ERG) recordings
ERG recordings were performed as described by [41]–[43] with some modifications. Animals were dark adapted overnight in a light-proof cage and prepared for ERG recordings under dim red light and infrared (IR) illuminators. Animals were anesthetized with an i.p. injection of 90 mg/kg Nembutal. The pupil of the stimulated eye (right eye) was dilated with 1% Tropicamide, and corneal moisture of the non-stimulated eye (left eye) was maintained with 0.3%glycerin/1% propylene glycerol eye drops or Gonak™ Hypromellose Solution, 2.5%. The mice were placed on a heating pad in a light-tight Faraday cage. Reference and ground electrodes were placed in the mouth and subcutaneously in the tail, respectively. A Burian-Allen electrode (Hansen Ophthalmic Development Laboratory, Coralville, IA) was placed on the corneal surface of the right eye. Following electrode placement under IR illumination, animals were dark adapted for 7–10 minutes prior to presentation of the first light stimulus. The response signal was amplified, band-pass filtered (0.3 Hz and 100 Hz) and digitized at 1 kHz. The light stimulus consisted of a series of 1 ms LED flashes (λ = 520 nm) presented in increments from −3.25 log cds/m2 to +3.17 log cds/m2. +3.17 log cds/m2 = 5.76 log photoisomerizations/rod (log Rh*/rod). To reduce the effects of noise, records for −3.25, −2.25, and −1.25 log cds/m2 are the average of 9 stimulus presentations presented at 30 second intervals, stimulus for −0.25, +0.74, +1.74, and +3.17 logcds/m2 were presented 4 times at 2 minute intervals. The a-wave was measured from the baseline to the trough of the initial negative wave, while the b-wave was measured from the trough of the a-wave to the peak of the initial positive wave. All responses were recorded with Clampex and analyzed using Clampfit 10.0 software (Molecular Devices LLC, Sunnyvale, CA). Averaged data was analyzed for graphical presentation with SigmaPlot 11.0 (Systat Software Inc., San Jose, CA). ERG recordings were repeated every 28–31 days over a 7-month period. The numbers of animals used for each experiment are presented in the figure legends.
Data analysis
Statistical analysis was performed using Student's t-test with SigmaPlot 11.0 software. All values are expressed as ± standard deviation (SD). The amplitudes of a- and b-wave intensity response function plots were fitted with Hill function (n = 0.8). Threshold intensities (It) required to evoke criteria a- and b-wave amplitudes of 50 µV and 75 µV respectively were extrapolated from Hill function fits of a- and b-wave intensity response function plots.
Histology
Eyes were enucleated and placed in a fixative mixture of 2% glutaraldehyde/4% paraformaldehyde in PB followed by embedding in JB-4 embedding media according to manufacturer's protocol (JB-4 embedding kit, Polysciences, Inc. Warrington, PA), sectioned at 2 µm thickness using the Sorvall MT-5000 Ultra Microtome, and stained with 1% (w/v) toluidine blue stain (Ricca Chemical Company, Arlington, TX). Changes in retinal morphology were determined by counting the number of nuclei per column in the outer nuclear layer (ONL). To account for the non-uniform degenerative changes in the retina, the total number of nuclei per column represents averaged nuclei counts from multiple regions, superior, central, and inferior zones, over a distance of 100 µm. The superior and central zones were in the dorsal portion of the retina and were measured ∼600 µm from the ciliary marginal zone and optic nerve respectively. The inferior zone was measured 1.1 mm from the CMZ in the ventral portion of the retina.
Quantitative Real time PCR (qRT-PCR)
Retinas were separated from the RPE and surrounding eye tissues, quickly frozen on dry ice and stored at −80°C until use. Total RNA was prepared using the RNeasy kit (Qiagen). RNA integrity was assessed using the Agilent 2100 bioanalyzer. mRNA was reverse transcribed using the Quantitect Reverse Transcription kit (Qiagen). PCR reaction was prepared using the LightCycler ® 480 SYBR Green I Master (Roche). Primers used were obtained from the PrimerBank database (http://pga.mgh.harvard.edu/primerbank/). PCR thermal cycles were as follows: heat activation: 95°C for 10 min, amplification (42 cycles): 95°C for 15 s, 60°C for 15 s, 72°C for 15 s, 80°C for 5 s, and melt curve: 95°C for 10 s, 45°C for 1 min with a heating rate of 0.1°C. The threshold cycle crossing point (Cp) of the gene of interest (GOI) in each sample was normalized to the Cp of β-actin of the same sample [44]. The derived normalized Cp value for each animal was then averaged to get the mean normalized Cp for each genotype and was used to define the relative expression of each gene.
Results
Effects of Chop on photoreceptor sensitivity
Since Chop has been shown to regulate the expression of rhodopsin through a microRNA, miR-708, in 293T cells [45], deleting Chop could alter rhodopsin levels in photoreceptors and potentially influence retinal degeneration. Therefore, we analyzed mRNA levels of rhodopsin, arrestin and transducin using RT-PCR analysis on RNA from 4 week old Chop −/− and C57BL/6 mice (Fig. 1A). There was no significant difference in the normalized mean expression levels of rhodopsin between Chop−/− and C57BL/6 mice (p = 0.962, n = 4) and there was less than a 5% difference in arrestin (p = 0.033, n = 4) and transducin (p = 0.021, n = 4) mRNA levels. Therefore in mouse photoreceptors, Chop does not appear to regulate the expression of rhodopsin and minimally affects the expression of transducin and arrestin.
Figure 1. Effect of Chop on the expression of phototransduction genes and visual function.

(A). Quantitative RT-PCR analysis of 4 week old C57BL/6 and Chop−/− mice (n = 4). Samples were normalized to β-actin levels. (B). Average scotopic ERG a- and b-wave waveforms from 4 week old C57BL/6 (blue trace) and Chop−/− (orange trace) mice in response to 1 ms flashes (n = 7–9). (C) Intensity-response functions of a- and b-waves recorded from the same C57BL/6 and Chop−/− mice. Horizontal lines and vertical intercepts on the abscissa (a-wave It: 50 µV, b-wave It: 75 µV) show the method used to determine threshold light intensities (It). (D) Age related changes in a- and b-wave threshold light intensities in C57BL/6 and Chop−/− mice. Error bars are ± SD.
To characterize the retinal sensitivity in Chop−/− mice, we used electrophysiology to record scotopic ERG waveforms as a function of light intensities. Representative ERG traces recorded in response to a saturating flash (Fig. 1B) and to a series of flashes covering the range of intensities used in this study (Fig. 2A) are shown. The cornea-negative leading edge of the ERG waveform, the a-wave, originates from photoreceptor cells [46], and the maximal positive potential gives the b-wave amplitude and represents responses by second order bipolar cells [47]–[49]. The intensity-response curves for both a-wave (Fig. 1C) and b-wave (Fig. 1C) from Chop−/− and C57BL/6 animals were indistinguishable at 4 weeks.
Figure 2. ERG responses in 4 week old GHL+ and GHL+/Chop−/− mice.

(A) Average scotopic ERG a- and b-wave waveforms from C57BL/6 (blue traces), Chop−/− (orange traces) GHL+ (black traces) and GHL+/Chop−/− (red traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 7–9). (B) Intensity-response functions of a- and b-waves. Error bars: ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent the onset of light stimulus.
We characterized changes in visual sensitivity by determining the light intensity, defined as the threshold intensity (It), required to evoke neural responses at criteria a- and b-wave amplitudes of 50 µV and 75 µV respectively over a 7-month period (Fig. 1D), which falls in the linear range of the response of the retinal cells to the presented stimulus. At 4 weeks, the mean a-wave was comparable between C57BL/6 and Chop−/− mice (p = 0.820, Table 1). Over the 7-month period, the a-wave I t was similar in C57BL/6 and Chop−/− mice (p>0.05) (Table 1). Similarly, over the 7-month period there was no significant difference between the b-wave I t in C57BL/6 and Chop−/− mice (p = 0.461, Table 2). Since we did not observe differences in a-wave I t between C57BL/6 and Chop−/− mice, the results show that photoreceptor sensitivity is not altered by ablation of Chop.
Table 1. a-wave threshold intensity (It) values.
| Age (weeks) | ||||||
| 4 | 8 | 12 | 16 | 24 | 28 | |
| a-wave It (cds/m2) | ||||||
| C57 | 2.1±0.7 | 2.4±1.1 | 2.6±1.0 | 2.3±0.8 | 2.8±1.2 | 2.9±1.0 |
| Chop−/− | 2.0±0.4 | 2.9±1.1 | 2.5±1.1 | 3.1±1.2 | 3.0±1.6 | 3.9±1.1 |
| Ask1−/− | 2.3±1.3 | 3.4±1.0 | 3.6±2.7 | 2.7±0.9 | 2.8±0.7 | 5.1±2.4 |
| GHL+ | 26.7±25.6 | 25.6±14.6 | 38.3±34.5 | 61.6±46.3 | 162±109 | ND |
| GHL+ / Chop−/− | 18.0±13.5 | 37.5±31.5 | 59.1±66.3 | 163±137 | 68.1±50.9 | ND |
| GHL+/Ask1−/− | 12.2±5.0 | 17.0±11.0 | 44.2±28.4 | 95.7±80.9 | 68.8±44.7 | ND |
Threshold amplitude: 50 µV, It: Threshold intensity, ND: not detectable.
Table 2. b-wave threshold intensity (It) values.
| Age (weeks) | ||||||
| 4 | 8 | 12 | 16 | 24 | 28 | |
| b-wave It (mcds/m2) | ||||||
| C57 | 21.3±10.8 | 16.8±12.6 | 16.8±10.1 | 15.8±5.5 | 10.3±5.0 | 9.7±2.5 |
| Chop−/− | 17.8±7.7 | 15.3±7.1 | 11.4±4.3 | 13.4±5.6 | 16.1±7.4 | 17.2±5.9 |
| Ask1−/− | 35.2±52.1 | 12.6±6.3 | 18.0±15.0 | 19.7±18.2 | 19.6±11.6 | 32.2±16.1 |
| GHL+ | 28.5±12.4 | 39.2±25.1 | 33.7±14.6 | 128±142 | 482±1190 | 252±313 |
| GHL+/Chop−/− | 35.5±30.8 | 31.1±16.5 | 33.4±17.6 | 80.7±55.3 | 142±179 | 281±232 |
| GHL+/Ask1−/− | 18.8±7.3 | 16.8±7.8 | 26.2±9.5 | 72.9±79.8 | 90.5±55.3 | 146±167 |
Threshold amplitude: 75 µV, It: Threshold intensity.
Effect of Chop on GHL+ retinal degeneration at 4 weeks
GHL+ transgenic mice exhibit a progressive loss in a-wave and b-wave amplitudes over time compared to non-transgenic animals [9]. We compared the retinal sensitivity of GHL+ and GHL+/Chop−/− transgenic mice by measuring scotopic ERG responses (Fig. 2A) over a range of light intensities. Representative ERG waveforms show that the a- and b-wave responses increased progressively with increasing light intensity (Fig. 2A). We used responses at maximal amplitudes (saturating light intensity) and criteria a- and b-wave amplitudes (threshold intensity = It, see above) to characterize physiological changes in the retina. We defined differences in retinal function between the genotypes as biologically relevant if statistical significance was observed for maximal amplitude and threshold intensity measurements.
At the saturating light intensity, there was a ∼62% reduction in the a-wave amplitude of 4 week old GHL+ (p<0.001) compared to C57BL/6 and Chop−/− mice (Fig. 2B). Similarly, threshold intensities decreased by ∼13 fold in 4 week old GHL+ compared to age matched control mice (Table 1). The b-wave amplitude of GHL+ mice at saturating light intensity decreased by ∼15% (p<0.01) compared to control mice, the threshold intensity (It) was higher than control mice, however the difference was not statistically significant when compared to in C57BL/6 mice (p = 0.242).
In 4 week old GHL+/Chop−/− mice, there was a ∼53% reduction in the a-wave amplitude at the saturating light intensity compared to C57BL/6 and Chop−/− mice (p<0.001). This was 24% higher than GHL+ mice at the same light intensity (Fig. 2B, see above), but the difference was not statistically significant (p = 0.126, n = 7–9). Also, a-wave I t was significantly different from C57BL/6 and Chop−/− mice (p = 0.009), but not significantly different compared to GHL+ mice (p = 0.424).
4 week old GHL+/Chop−/− mice had an ∼11% reduction in b-wave amplitude of compared to C57BL/6 (p = 0.153) and Chop−/− mice (p = 0.033). The maximum b-wave amplitudes of GHL+/Chop−/− and GHL+ mice were not statistically different (p = 0.515) (Fig. 2B). Similarly, the b-wave I t was not significantly different from GHL+ mice (p = 0.541) (Table 2).
We determined the number of photoreceptor nuclei across the retina at 4 weeks. In C57BL/6 and Chop−/− mice, the ONL had 8.6±1.5 and 8.6±1.2 nuclei per column respectively (Fig. 3). In GHL+ mice, the overall architecture of the retina was similar to control mice, however there was a decrease in the number of photoreceptor nuclei in the ONL. To account for the possibility of variation in the extent of photoreceptor loss across the retina, we divided the whole retina into three different zones, the superior zone, the central zone and the inferior zone (Fig. 3). In 4 week old GHL+ mice (n = 3), the number of photoreceptor nuclei per column in superior zone was 6.7±1.2. There were 5.7±1.7 in the central zone and 7.2±1.1 nuclei per column in the inferior zone, representing an overall average of 6.5±1.5 nuclei per column. On average this represents a ∼24% reduction in the number of photoreceptors compared to control (p<0.001) animals. The reduction in the number of photoreceptor nuclei in the ONL and shortening of the OS [9], [51] are consistent with observed reductions in the a-wave amplitude of GHL+ mice.
Figure 3. Histological analysis of 4 week old GHL+ and GHL+/Chop−/− retinas.

(A) Representative sections of retinas from 4 week old C57BL/6, Chop−/−, GHL+ and GHL+/Chop−/− mice (n = 2–3). (B) Mean number of photoreceptor nuclei per column counted in the ONL (left panel), and the variance in the number of photoreceptor nuclei per column counted in different zones of the retina in GHL+ and GHL+/Chop−/− mice, (right panel). Superior zone: 630 µm from the CMZ, central zone: 630 µm from optic nerve, and inferior zone: midpoint of inferior hemisphere. Error bars: ± SD OPL: outer plexiform layer, ONL: outer nuclear layer, OS: outer segment. Scale bar, 50 µm.
In 4 week old GHL+/Chop−/− mice (n = 3), the number of photoreceptor nuclei in superior zone was 5.9±0.7 per column. There were 5.6±1.4 in the central zone and 6.6±1.3 in the inferior zone, representing an overall average of 6.0±1.3. Accordingly, there was no significant difference between the mean number of photoreceptors in GHL+/Chop−/− and GHL+ mice (p = 0.086) (Fig. 3). Therefore, our results demonstrate that the reduction in a-wave and b-wave maximal amplitudes, threshold intensities, and the number of remaining photoreceptors in 4 week old GHL+- expressing mice appears to be independent of expression of Chop.
Effect of Chop on GHL+ retinal degeneration at 16 weeks
At 16 weeks, the approximate midpoint in retinal degeneration in GHL+ mice [51], the maximum a-wave amplitude at saturating light intensity decreased by 52% compared to 4 week old GHL+ mice (Fig. 4). The a-wave amplitude was 22% of C57BL/6 and Chop−/− mice. In some animals the a-wave could not be detected at intensities <+1.74 log cds/m2. The a-wave I t decreased by ∼20 fold compared to control mice (p<0.01). At the saturating light intensity, there was a ∼44% decline in the b-wave amplitude of GHL+ mice compared to age matched control animals (p<0.001). The b-wave I t was significantly different compared to C57BL/6 mice (p = 0.041) (Table 2).
Figure 4. ERG responses in 16 week old GHL+ and GHL+/Chop−/− mice.

(A) Average scotopic ERG a- and b-wave waveforms from C57BL/6 (blue traces), Chop−/− (orange traces) GHL+ (black traces) and GHL+/Chop−/− (red traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 8–13). (B) Intensity-response functions of a- and b-waves. Error bars ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent the onset of light stimulus.
In 16 week old GHL+/Chop−/− mice, the maximum a-wave amplitude at saturating light intensity decreased by 61% compared to 4 week old mice. The a-wave amplitude was 22% (p<0.001) that of 16 week old C57BL/6 and Chop−/− mice. There was no significant difference between maximum a-wave amplitudes in GHL+/Chop−/− and GHL+ mice (p = 0.994) (Fig. 4). In 16 week old GHL+/Chop−/− mice, a-wave I t was significantly different compared to C57BL/6 and Chop−/− mice (p<0.01), but not significantly different from GHL+ mice (p = 0.089) (Table 1).
At the saturating light intensity, there was a ∼30% reduction in the b-wave amplitude of 16 week old GHL+/Chop−/− mice compared to age matched control mice. The maximum b-wave in GHL+/Chop−/− mice was ∼25% higher compared to GHL+ mice (p = 0.018) (Fig. 4B). In 16 week old GHL+/Chop−/− mice, the b-wave I t was not significantly different compared to GHL+ mice (p = 0.279).
We determined the number of photoreceptor nuclei across the retina at 16 weeks. In C57BL/6 and Chop−/− mice, the ONL had 8.5±1.7 and 8.4±1.5 nuclei per column, respectively (Fig. 5A). By this time, retinas of GHL+ mice were severely degenerated [9], with shortened OS and a disorganized ONL with far fewer nuclei per column (Figure 5B). In 16 week old GHL+ mice (n = 5), the number of photoreceptor nuclei in superior zone was 4.0±1.2. There were 3.1±1.0 in the central zone and 4.7±1.0 in the inferior zone (Fig. 5A, B). On average the total number of photoreceptor nuclei per column in the ONL was 3.9±1.3.
Figure 5. Histological analysis of 16 and 28 week old GHL+ and GHL+/Chop−/− mice.

(A) Representative sections of retinas from 16 week old C57BL/6, Chop−/−, GHL+ and GHL+/Chop−/− mice. GHL+ and GHL+/Chop−/− retinas had shortened OS (arrowheads). (B, right panel) Mean number of photoreceptor nuclei per column counted in the ONL at 16 weeks. The variance in the number of photoreceptor nuclei per column counted in different zones of the retina in (B, left panel) in 16 week. (C) Representative sections of retinas from 28 week old GHL+ and GHL+/Chop−/− mice. (D) The variance in the number of photoreceptor nuclei per column counted in different zones of the retina in 28 week old GHL+ and GHL+/Chop−/− mice. Superior zone: 630 µm from the CMZ, central zone: 630 µm from optic nerve, and inferior zone: midpoint of inferior hemisphere. Error bars: ± SD OPL: outer plexiform layer, ONL: outer nuclear layer, OS: outer segment. Scale bar 50 µm.
In 16 week old GHL+/Chop−/− mice (n = 3), the number of photoreceptor nuclei in superior zone was 4.1±0.6 per column. There were 4.9±0.6 nuclei per column in the central zone and 4.6±0.6 in the inferior zone, representing an overall average of 4.5±0.7 nuclei per column (Fig. 5A, B). Unlike GHL+ mice, the pattern of degeneration in GHL+/Chop−/− mice was more uniform across the retina. When we compared the nuclei count in the different zones between GHL+/Chop−/− and GHL+ mice, the degree of degeneration in the central zone was significantly higher in GHL+ animals than in GHL+/Chop−/− mice (p<0.001), while the differences in the superior and inferior zones were more comparable with p values of 0.937 and 0.788 respectively (Fig. 5B). This, results suggest Chop may influence late stage responses that occur as the disease progresses.
Age related changes in retinal degeneration of GHL+/Chop−/− mice
We determined the effect of Chop on the rate of degeneration during the major phase of rod loss, between 4–28 weeks. In control C57BL6 and Chop−/− mice there were ∼26% (p = 0.001) and ∼23% (p = 0.008) losses in maximum a-wave amplitudes, respectively, which is similar to what has been reported previously [50] (Fig. 6A). Linear regression for a-wave amplitudes between 4 and 28 weeks showed a downward trend with age in C57BL/6 mice (−4.15±0.51 µV/week) and Chop−/− mice (−3.8±0.73 µV/week) (Fig. 6A). Likewise, there were 24% (p = 0.004) and ∼26% (p<0.001) losses in maximum b-wave amplitudes respectively, in C57BL6 and Chop−/− mice (Fig. 6B, Fig. S1). Linear regression of b-wave amplitudes trended downwards in C57BL/6 mice (−12.7±2.3 µV/week) and Chop−/− mice (−13.3±2.5 µV/week) (Fig. 6B). Therefore the rate of loss of retinal responses was similar in C57BL/6 and Chop−/− mice.
Figure 6. ERG changes as a function of age in GHL+ and GHL+/Chop−/− mice.
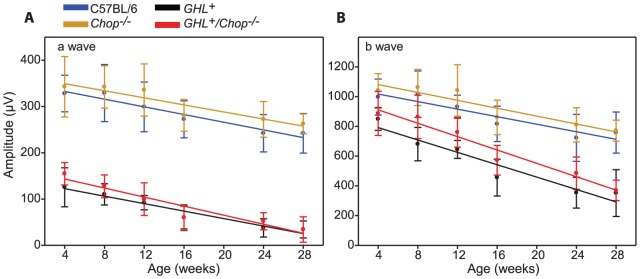
Maximal a- and b-wave amplitudes as a function of age in C57BL/6 (blue trace), Chop−/− (orange trace) GHL+ (black trace) and GHL+/Chop−/− (red trace) mice. Colored lines are polynomial fits.
In GHL+ mice, both maximal a-wave and b-wave amplitudes were severely reduced at all time points compared to control animals. The maximal a-wave amplitude, while significantly lower than control animals (see above) at 4 weeks, continued to decline so that by 28 weeks it was only 27% (p<0.001) of the initial 4 week value (and ∼13% that of 28 week old C57BL/6 and Chop−/− mice) (Fig. 6A). In a few GHL+ animals, the a-wave was no longer detectable at 28 weeks. Regression of a-wave amplitudes with age was −4.04±0.43 µV/week in GHL+ mice (Fig. 6A). The maximal b-wave amplitude declined by ∼60% (p<0.001) between 4 and 28 weeks in GHL+ mice, and was ∼45% (p<0.001) that of C57BL/6 and Chop−/− mice (Fig. 6B). Regression of b-wave amplitudes with age was −20.8±3.04 µV/week in GHL+ mice (Fig. 6B).
In GHL+/Chop−/− mice, both maximal a-wave and b-wave amplitudes were also severely reduced at all time points compared to control animals. The maximal a-wave amplitude declined to 22% (p<0.001) of the 4 week value (and ∼13% that of 28 week old C57BL/6 and Chop−/− mice). Regression of a-wave amplitudes with age was −4.9±0.72 µV/week in GHL+/Chop−/− mice (Fig. 6A). The rate of decline in a-wave responses was similar in C57BL/6, Chop−/−, GHL+ and GHL+/Chop−/− mice, however, there were significant reductions in maximal a-wave amplitudes and higher threshold intensities (It) in GHL+ and GHL+/Chop−/− mice compared to control mice (see above). The maximal a-wave amplitudes and threshold intensities were not significantly different between GHL+ and GHL+/Chop−/− mice, suggesting similar ongoing changes in the loss of photoreceptor cells and decrease in rhodopsin concentrations in the GHL+ and GHL+/Chop−/− mice retinas.
The maximal b-wave amplitude declined by 58% (p<0.001) of the 4 week value in GHL+/Chop−/− mice (Fig. 6B). Between 8–24 weeks, the maximum b-wave amplitudes were 15–27% higher (p<0.05) in GHL+/Chop−/− compared to GHL+ mice. However, at 4 and 28 weeks there was no statistical difference in between GHL+/Chop−/− and GHL+ b-wave maximal amplitudes. Regression of b-wave amplitudes with age was −22.5±2.23 µV/week in GHL+/Chop−/− mice (Fig. 6B). The rate of decline in b-wave responses was similar in GHL+ and GHL+/Chop−/−, and faster than C57BL/6, Chop−/− control mice. Thus, the overall loss of visual sensitivity in GHL+/Chop−/− and GHL+ mice declined at similar rates.
Since the b-wave was higher in GHL+/Chop−/− compared to GHL+ mice, we assessed the signal transmission from photoreceptor cells to second order cells by determining the b/a wave ratio [52]. We determined the b/a ratio by normalizing maximal b-wave amplitudes to maximal a-wave amplitudes and plotted the values as a function of age (Fig. 7A). The b/a ratio is constant in healthy retinas, therefore, deviations from normal b/a ratio represent pathological/degenerative changes in the retina [52]. The b/a ratio was similar between C57BL/6 and Chop−/− mice (Fig. 7A). In GHL+ mice, the b/a ratio was abnormally high at all ages compared to control mice, which is consistent with ongoing degeneration. Similarly, in GHL+/Chop−/− mice, the b/a ratio was higher than C57BL/6 and Chop−/− animals. At all ages, b/a ratio was comparable between GHL+/Chop−/− and GHL+ mice (p>0.05), suggesting that there are no significant differences in signal transmission from photoreceptors to second order cells in GHL+/Chop−/− and GHL+ mice.
Figure 7. Age related changes in retinal function and sensitivity of GHL+ and GHL+/Chop−/− mice.
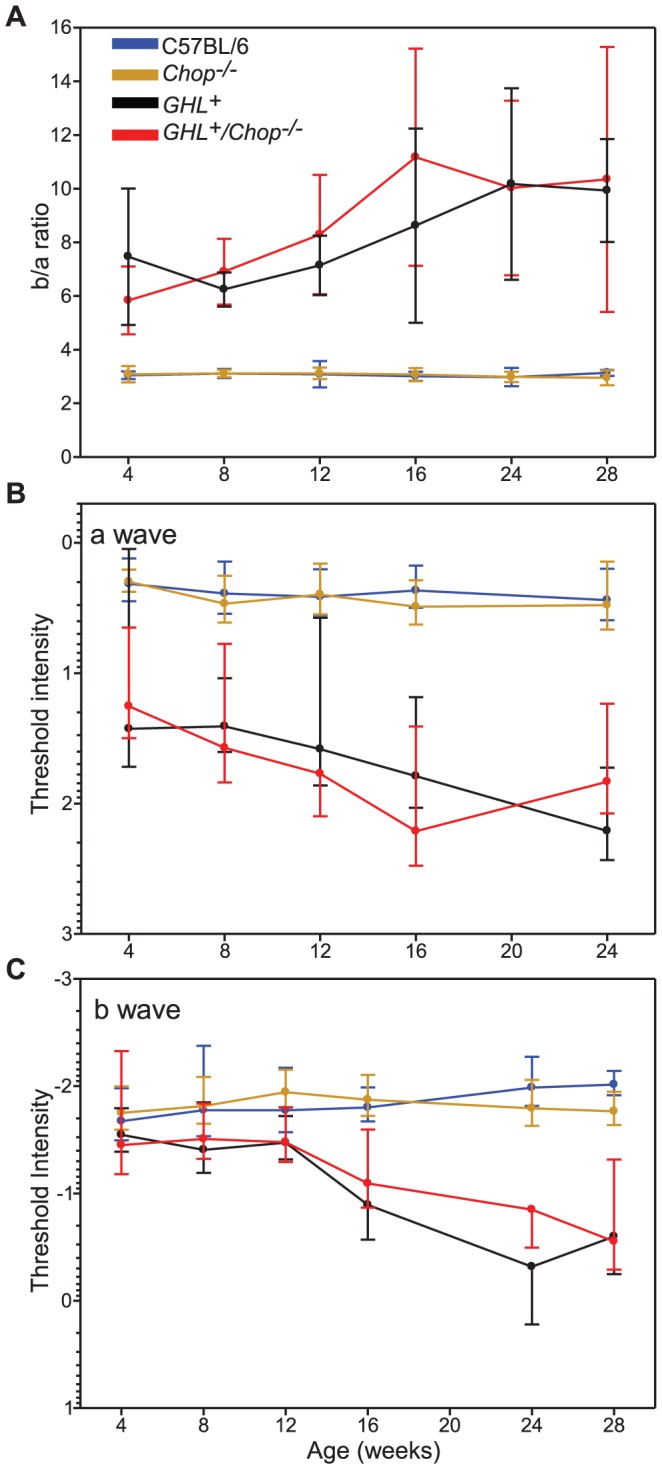
(A) b/a wave ratio in C57BL/6, Chop−/−, GHL+, and GHL+/Chop−/− mice. (B and C) Age related changes in a- and b-wave threshold intensities in C57BL/6, Chop−/−, GHL+, and GHL+/Chop−/− mice (a- wave It = 50 µV, b- wave It = 75 µV). Error bars: ± SD.
In addition, we determined the number of photoreceptor nuclei across the retina at 28 weeks. Since no significant differences were observed in the pattern of survival of photoreceptor cells between C57BL/6 and Chop−/− mice, at 28 weeks we focused on the degenerative changes between GHL+ and GHL+/Chop−/− mice. By 28 weeks, retinas of GHL+ mice (n = 5) were almost completely degenerated [9], with absent OS and the number of nuclei per column ranged from zero to four (Fig. 5C). The number of photoreceptor nuclei in superior zone was 2.1±1.5. There were 1.6±1.1 in the central zone and 3.1±1.3 in the inferior zone. Similar to the pattern observed at 16 weeks, the central zone was more degenerated than the more peripheral superior and inferior zones (Fig. 5C, D).
In 28 week old GHL+/Chop−/− mice (n = 3), the number of photoreceptor nuclei per column in superior zone was 3.5±0.7, there were 3.1±0.9 nuclei per column in the central zone, and 3.6±0.5 in the inferior zone (Fig. 5C, D). Again, the retina continued to uniformly degenerate in different zones of the retina, which was consistent with the pattern we observed at 16 weeks. When we compared the nuclei count in the different zones between GHL+/Chop−/− and GHL+ mice, the degree of degeneration in the central zone was significantly higher in GHL+ animals compared to GHL+/Chop−/− mice (p<0.001). In addition, the degree of degeneration in the superior zone was also significantly higher in GHL+ animals than GHL+/Chop−/− mice (p = 0.003) (Fig. 5D). However, inferior zones were more comparable with a p value of 0.171 (Fig. 5D). These results suggest that although Chop does not directly prevent photoreceptor death, it may influence late stage responses of surrounding cells and/or other ongoing cellular responses in the retina to dying photoreceptors and as such can prolong cell survival in older animals.
Effect of Ask1 on GHL+ retinal degeneration at 4 weeks
At the saturating light intensity, there was a ∼55% decreased in the a-wave amplitude of 4 week old GHL+/Ask1−/− mice compared to C57BL/6 and Ask1−/− mice (p<0.001). The maximum a-wave amplitude was 17% higher than the maximum a-wave of GHL+ mice (Fig. 8A), however the difference was not statistically significant (p = 0.294, n = 7–9). Similarly, the a-wave I t (Table 1) was significantly different from C57BL/6 and Ask1−/− mice (p<0.001), but not significantly different from GHL+ mice (p = 0.163).
Figure 8. ERG responses in GHL+ and GHL+/Ask1−/− mice.

Intensity-response functions of a- and b-waves in (A) 4, (B) 16, and (C) 28 week old C57BL/6 (blue), Ask1−/− (green) GHL+ (black) and GHL+/Ask1−/− (purple) mice (n = 7–14). Error bars: ± SD.
At the saturating light intensity, the b-wave decreased by 3% and 12% compared to C57BL/6 (p = 0.663) and Ask1−/− mice (p = 0.162), respectively. The maximum b-wave amplitudes of GHL+/Ask1−/− and GHL+ mice were statistically different (p = 0.027) (Fig. 8A). The b-wave I t was not statistically different from GHL+ mice (p = 0.089) (Table 2).
Effect of Ask1 on GHL+ retinal degeneration at 16 weeks
In 16 week old GHL+/Ask1−/− mice, the maximum a-wave amplitude at the saturating light intensity decreased by 53% compared to 4 week old animals. The a-wave amplitude was 25% that of C57BL/6 and Ask1−/− mice (p<0.001). There was no significant difference between maximum a-wave amplitudes in GHL+/Ask1−/− and GHL+ mice (p = 0.360) (Fig. 8B). The a-wave I t was significantly different from C57BL/6 and Ask1−/− mice (p<0.01), but not significantly different from GHL+ mice (p = 0.345) (Table 1).
At the saturating light intensity, there was a ∼25% decrease in b-wave amplitudes of 16 week old GHL+/Ask1−/− mice compared to C57BL/6 and Ask1−/− mice. The maximum b-wave in GHL+/Ask1−/− mice was ∼42% higher compared to GHL+ mice (p = 0.002) (Fig. 8B). The b-wave I t was not statistically different from GHL+ mice (p = 0.338) (Table 2).
Age related changes in retinal degeneration of GHL+/Ask1−/− mice
We determined the effect of Ask1 on the rate of degeneration during the major phase of rod loss, between 4–28 weeks. In control Ask1−/−, mice there was a ∼30% (p = 0.003) age related loss in maximum a-wave amplitudes, (Fig. 9A, Fig. S2). Linear regression for a-wave amplitudes between 4 and 28 weeks showed a downward trend with age in Ask1−/− mice (−4.2±0.87 µV/week) (Fig. 9A). Likewise, there was a 30% (p<0.001) age related loss in maximum b-wave amplitude. Linear regression of b-wave amplitudes trended downwards in Ask1−/− mice (−16.1±3.13 µV/week) (Fig. 9B).
Figure 9. Rate of retinal degeneration in GHL+ and GHL+/Ask1−/− mice.
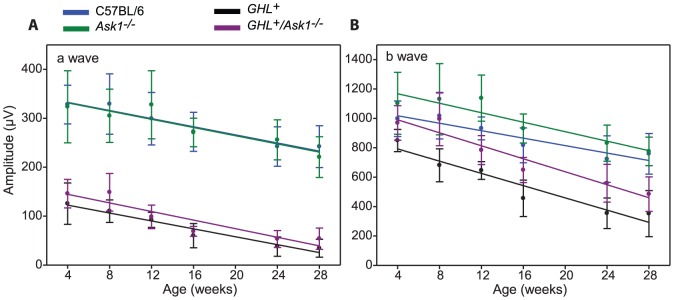
Maximal a- and b-wave amplitudes as a function of age in C57BL/6 (blue), Ask1−/− (green) GHL+ (black) and GHL+/Ask1−/− (purple) mice. Error bars: ± SD. Colored lines are polynomial fits.
In GHL+/Ask1−/− mice, both maximal a-wave and b-wave amplitudes were severely reduced at all time points compared to control animals. At 28 weeks, the maximal a-wave amplitude was 54±22 µV, which declined to 37% (p<0.001) of the 4 week value (and ∼22% that of C57BL/6 and Ask1−/− mice at 28 weeks, p<0.001). Regression of a-wave amplitudes with age was −4.4±0.89 µV/week in GHL+/Ask1−/− mice (Fig. 9A). The rate of decline in a-wave responses was similar in C57BL/6 (see above), Ask1−/−, GHL+ and GHL+/Ask1−/− mice, however, there were significant reductions in maximal a-wave amplitudes compared to control mice. The maximal a-wave amplitudes were not significantly different between GHL+ and GHL+/Ask1−/− mice, suggesting similar ongoing changes with respect to loss of photoreceptor cells and decrease in rhodopsin concentrations.
At 28 weeks, the maximal b-wave amplitude (484±117 µV) declined by 50% (p<0.001) of the 4 weeks value in GHL+/Ask1−/− mice (Fig. 9B). Between 4–28 weeks, the maximum b-wave amplitudes were 14–57% higher (p<0.01) in GHL+/Ask1−/− compared to GHL+ mice. Regression of b-wave amplitudes with age was −22.1±3.11 µV/week in GHL+/Ask1−/− mice (Fig. 9B). The rate of decline in b-wave responses was similar in GHL+ and GHL+/Ask1−/− but faster than C57BL/6, Ask1−/− control mice. However, there were significant differences in maximal b-wave amplitudes in GHL+ and GHL+/Ask1−/− mice compared to control mice. To assess signal transmission through the retina we determined the b/a ratio of GHL+/Ask1−/− mice (Fig. 10A). The b/a ratio was similar between C57BL/6 and Ask1−/− mice (Fig. 10A). In GHL+/Ask1−/− mice, the b/a ratio was higher than control C57BL/6 and Ask1−/− animals, but was comparable to GHL+ mice (p>0.05) (Fig. 10A).
Figure 10. Age related changes in retinal function and morphology of GHL+ and GHL+/Ask1−/− mice.
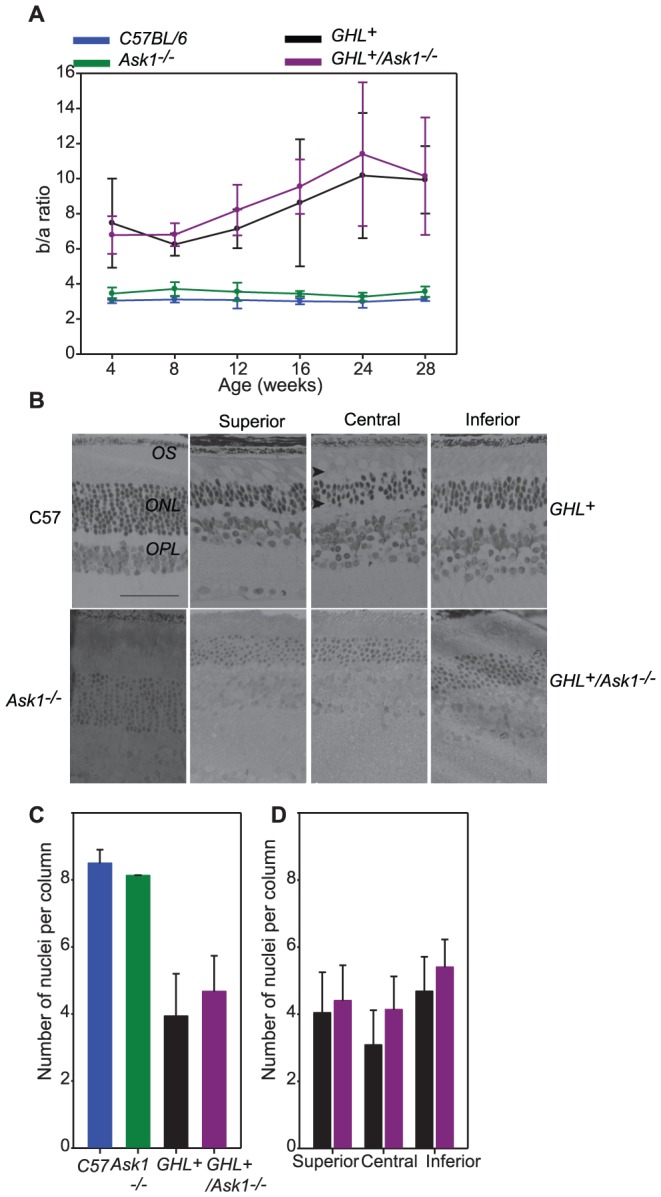
(A) b/a wave ratio in C57BL/6, Ask1−/−, GHL+, and GHL+/Ask1−/− mice. (B) Representative sections of retinas from 16 week old C57BL/6, Ask1−/−, GHL+ and GHL+/Ask1−/− mice. GHL+ and GHL+/Ask1−/− retinas had shortened OS (arrowheads). (C) Mean number of photoreceptor nuclei per column counted in the ONL. Error bars: ± SD. OPL: outer plexiform layer, ONL: outer nuclear layer, OS: outer segment. Scale bar 50 µm. (D) The variance in the number of photoreceptor nuclei per column counted in different zones of the retina in GHL+ and GHL+/Ask1−/− mice, (right panel). Superior zone: 630 µm from the CMZ, central zone: 630 µm from optic nerve, and inferior zone: midpoint of inferior hemisphere.
We determined the number of photoreceptor nuclei across the retina at 16 weeks. In Ask−/− mice, the ONL had 8.1±1.7 nuclei per column (Fig. 10B). In 16 week old GHL+/Ask1−/− mice (n = 3), the number of photoreceptor nuclei in superior zone was 4.4±1.1 per column, there were 4.1±1.0 nuclei per column in the central zone, and 5.4±0.8 nuclei per column in the inferior zone, representing an overall average of 4.7±1.1 (Fig. 10B, C). Unlike GHL+ mice, this pattern of degeneration was more uniform across the retina. When we compared the nuclei count in the different zones between GHL+/Ask1−/− and GHL+ mice, the degree of degeneration in the central zone was significantly higher in GHL+ animals than GHL+/Ask1−/− mice (p = 0.003), while the differences in the superior and inferior zones were more comparable with p values of 0.345 and 0.027 respectively (Fig. 10B, C). This, results suggest that similar to Chop, Ask1 may also influence late stage responses of surrounding cells and/or other cellular responses in the retina to dying photoreceptors and as such can prolong cell survival in older animals.
Discussion
The purpose of this study was to determine whether Chop or Ask1 regulate photoreceptor death in the RhoP23H transgenic mouse model of RP. If Chop or Ask1 are positive regulators of photoreceptor death, deficiency of these genes should preserve a- and b-wave response amplitudes, threshold intensities, and retinal morphology. On the other hand if Chop or Ask1 are negative regulators of cell death, ablation of these genes should enhance the loss of retinal function and structure. However, ablation of Chop or Ask1 genes did not alter the rate of loss of maximal a-wave amplitudes in GHL+-expressing mice. Electrophysiological recordings showed that deleting Chop or Ask1 genes did not preserve a-wave amplitudes, threshold intensities (It), and the rate of loss of photoreceptor responses in GHL+/Chop−/− and GHL+/Ask1−/− mice from 4 to 28 weeks. At all light intensities, the a-wave amplitude and It values were not significantly different between GHL+ and GHL+/Chop−/− or GHL+/Ask1−/− mice. These results imply that Chop and Ask1 do not preserve overall photoreceptor function and sensitivity in GHL+-expressing mice.
Histological analysis of 4,16, 28 week old GHL+ mice retinas revealed a regional difference in the rate of photoreceptor loss. Central zones of the retina degenerated faster than the more peripheral zones, consistent with other reports using these mice [9]. The superior and central zones are in the dorsal portion of the retina, while the inferior zone is in the ventral portion. One possible explanation for the differences in the rate of loss of photoreceptor cells in different zones of the retina may be attributable to a variance in the expression of the transgene in the dorsal and ventral retina. Since there is no way to differentiate between protein levels of the GHL transgene and endogenous opsin, we cannot confirm this regional effect in transgene expression.
At 4 weeks, the magnitude of decline in the a-wave amplitude was higher compared to the b-wave amplitude between control animals and mice expressing the GHL transgene. A possible explanation is that at this age, the photoreceptor responses can still elicit saturating responses in bipolar cells, which is represented by the smaller decline in b-wave amplitudes in GHL+ expressing animals. While in 16 to 28 week old mice, more photoreceptors have been lost resulting in a larger decline in the generated a-wave amplitudes driving the b-wave responses, which is represented by the larger decline in b-wave amplitudes in GHL+ expressing animals.
The b-wave amplitude was higher in GHL+/Chop−/− and GHL+/Ask1−/− compared to GHL+ mice. At most time points, the increase was significant at the higher light intensities compared to low/medium light intensities. b-wave amplitudes are driven by photoreceptor responses (a-wave) and synaptic connections with bipolar cells. The transfer of signals from photoreceptors to second order cells is non-linear [53]. Therefore changes in synaptic connections, cellular integrity and inter-cellular connections between rods, cones and bipolar cells may alter b-wave amplitudes. One possible explanation for the differences in b-wave amplitudes at the higher intensities is that Chop and Ask1 may have pleiotropic effects on the retina, and the loss of these genes may have influence other processes in inner retinal cells. To determine the physiological effects of deleting Chop and Ask1 genes, we assessed signal transmission from photoreceptors to second order cells by characterizing the b/a ratios [52]. At all ages (4–28 weeks), the b/a ratios were not significantly different between GHL+ and GHL+/Chop−/− or GHL+/Ask1−/− mice. Also, there was no significant difference in the rate of loss of b-wave amplitudes between GHL+ and GHL+/Chop−/− or GHL+/Ask1−/− mice. Therefore, physiological analysis suggests that signal transmission through the retina and number of inner retinal cells was similar in GHL+ and GHL+/Chop−/−, GHL+/Ask1−/− mice at all ages.
When we compared the pattern of degeneration and number of nuclei per column in the ONL between GHL+ and GHL+/Chop−/−, GHL+/Ask1−/− mice, we observed an age dependent difference between the pattern of degeneration and number of surviving cells in the different zones of the retina. There were no significant differences in number of photoreceptor nuclei per column between 4 week old GHL+ and GHL+/Chop−/−, GHL+/Ask1−/− mice. By contrast, in older mice, the central zone was particularly less degenerated in GHL+/Chop−/− and GHL+/Ask1−/− mice compared to GHL+ mice. This was an interesting finding because ERG a-wave amplitudes at 16 and 28 weeks showed no differences in retinal sensitivity between GHL+ and GHL+/Chop−/− or GHL+/Ask1−/− mice. One possible explanation for this is that since ERG recordings represent a summed response of all photoreceptor cells, and differences ∼1–2 row variance in nuclei count between GHL+ and GHL+/Chop−/−, GHL+/Ask1−/− mice, cannot be readily measured by this technique. Therefore, although ablation of Chop and Ask1 do not preserve overall functional response of the retina, the benefits of targeting the UPR in treating RP may be more apparent during later stages of the disease when the difference in the number of photoreceptors remaining is between no cells to one or two rows of cells.
In a recent study by Nashine et al. [54], ablation of Chop did not protect against retinal degeneration over a 3 month period in the RhoT17M transgenic mice RP model. That finding is similar to our results on GHL+. However, the RhoT17M transgenic mice may have exhibited a cytoprotective effect with Chop in the early stages of retinal degeneration since one month old RhoT17M Chop−/− mice had lower a-wave amplitudes and thinner ONL compared to age matched RhoT17M mice. The authors suggested that Chop deficiency causes a decrease in the rhodopsin expression via transcriptional mechanisms. These findings differ from our results on GHL+/Chop−/− mice, which exhibited some protection from cell death at later stages. Although photoreceptor death is the common fate in both RhoT17M and GHL+ transgenic mice, each model may lead to cell death via different intracellular pathways [55]. Besides the different rhodopsin mutations used, there are also differences in the rhodopsin protein sequence (RhoT17M transgene encodes the human protein while GHL+ transgene encodes the mouse protein). Previous studies using mammalian cell culture showed that there is a species difference in the expression and biochemical properties of RhoT17M [6], [56]. In addition, there are also possible differences in transgene expression or mosaicism between the two models. In fact, overexpression of wild-type opsin transgenes in mouse retinas has been shown to induce retinal degeneration [10]. Thus it is not clear what accounts for the differing effects of Chop on cell death in RhoT17M and GHL+ transgenic mice. To understand whether various rhodopsin mutants cause cell death by similar pathways it will be necessary to compare knock-in lines.
The ablation of Ask1 has a protective effect in different neurodegenerative disease models [24], [57]. In addition, an increase in the expression of Ask1 in the tubby mouse model of retinal degeneration suggests a role for Ask1 in other retinal degenerative diseases [37]. Ask1 was recently linked to photoreceptor death in the RhoT17M mice model [38]. This occurs by the Ask1 regulation of the c-Jun N-terminal kinase (c-JNK) pathway via the tumor necrosis factor (TNF)-dependent pathway ultimately regulating caspase-7 [32]. Together, these results suggest that Ask1 may have an indirect role in regulating cell death in RhoT17M. Our results are consistent with these findings, however the effects are small and only apparent at later stages. The delayed and partial rescue of photoreceptor cells suggests that Ask1 may influence chronic degenerative changes in the retina by modifying late onset pathological responses like inflammatory pathways.
The involvement of UPR pro-apoptotic genes in many diseases, such as diabetes, Huntington's disease and Parkinson's disease, is well established [22], [24], [26], [34], [35], and Chop and Ask1 have been shown to prolong cell survival in the aforementioned diseases. However, based on our results these branches of the UPR do not regulate the acute loss of photoreceptor cells in transgenic GHL+ mice. In fact, it appears that the influence on photoreceptor death may be through indirect mechanisms of modulating later pathological changes in the retina during disease progression. A possible explanation for the regional effect of deleting Chop and Ask1 is that since older photoreceptors reside in the central retina and are known to die faster than younger peripheral photoreceptors in different RP models [9], [58], deleterious effects associated with the expression of Chop and Ask1 occur earlier in these cells than cells in the retinal periphery. Therefore, accounting for the regional protection of deleting Chop and Ask1 in retinas of older animals.
Morphological changes in the outer segment are one of the earliest findings associated with disease pathology in RP patients. It has been established that the proper formation of the OS is required for the survival of rod photoreceptors [59]–[61]. Thus, it is possible that the observed early onset decrease in a-wave amplitudes in GHL+, GHL+/Chop−/− and GHL+/Ask1−/− mice is associated with the observed changes in the rod OS structure. The consequences of the presence of RhoP23H in the OS are unknown. Based on recent findings, we showed that RhoP23H forms aggregates that lead to the disruption of discs in the OS [62]. In another study [63], we showed via biophysical modeling of OS stability, that changes in protein composition in OS discs may lead to breakage of the OS. If the OS is the site of toxicity, enhancing the folding and transport of RhoP23H to the OS may be detrimental to the cell. Since haploinsufficiency does not cause severe degeneration [60], [61] and RhoP23H is preferentially degraded over wild type Rho [11], investigating the effects of enhancing the degradation of RhoP23H may determine if decreasing the transport of RhoP23H to the OS prevents the disorganization/displacement of OS discs and protects against photoreceptor death. Therefore, the role of the OS and protein degradation mechanisms in RP need to be investigated in the future.
Supporting Information
Representative ERG waveforms from GHL+ and GHL+/Chop−/− mice. Average scotopic ERG a- and b-wave waveforms from (A) 4 and 28 week old C57BL/6 (blue traces), Chop−/− (orange traces) (B) 4–28 week old GHL+ (black traces) and GHL+/Chop−/− (red traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 6–14). Error bars ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent onset of light stimulus.
(EPS)
Representative ERG waveforms from GHL+ and GHL+/Ask1−/− mice. Average scotopic ERG a- and b-wave waveforms from (A) 4 and 28 week old C57BL/6 (blue traces), Ask1−/− (green traces) (B) 4, 16, and 28 week old GHL+ (black traces) and GHL+/Ask1−/− (purple traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 7–14). Error bars ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent onset of light stimulus.
(EPS)
Acknowledgments
We thank Y. Umino for assistance in electrophysiological recordings and histology, S. Reks for helpful comments and suggestions for the manuscript, A. Truex for help with the genotyping the mice, J. Qi for assistance with microscopy.
Funding Statement
This work was supported in part by the National Institutes of Health grants EY-11256, EY-12975 (B.E.K.), Research to Prevent Blindness (Unrestricted Grant to SUNY UMU Department of Ophthalmology) and the Lions of CNY. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hartong DT, Berson EL, Dryja TP (2006) Retinitis pigmentosa. Lancet 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 2. Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH (1984) Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 97: 357–365. [DOI] [PubMed] [Google Scholar]
- 3. Grondahl J (1987) Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin Genet 31: 255–264. [DOI] [PubMed] [Google Scholar]
- 4. Papermaster DS, Dreyer WJ (1974) Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry 13: 2438–2444. [DOI] [PubMed] [Google Scholar]
- 5. Mendes HF, van der Spuy J, Chapple JP, Cheetham ME (2005) Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med 11: 177–185. [DOI] [PubMed] [Google Scholar]
- 6. Kaushal S, Khorana HG (1994) Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 33: 6121–6128. [DOI] [PubMed] [Google Scholar]
- 7. Fariss RN, Li ZY, Milam AH (2000) Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol 129: 215–223. [DOI] [PubMed] [Google Scholar]
- 8. Milam AH, Li ZY, Fariss RN (1998) Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res 17: 175–205. [DOI] [PubMed] [Google Scholar]
- 9. Naash MI, Hollyfield JG, al-Ubaidi MR, Baehr W (1993) Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci U S A 90: 5499–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, et al. (1992) Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron 9: 815–830. [DOI] [PubMed] [Google Scholar]
- 11. Sakami S, Maeda T, Bereta G, Okano K, Golczak M, et al. (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 286: 10551–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tso MO, Li WW, Zhang C, Lam TT, Hao Y, et al. (1997) A pathologic study of degeneration of the rod and cone populations of the rhodopsin Pro347Leu transgenic pigs. Trans Am Ophthalmol Soc 95: 467–479 discussion 479-483. [PMC free article] [PubMed] [Google Scholar]
- 13. Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, et al. (2010) Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A 107: 5961–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, et al. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kroeger H, Messah C, Ahern K, Gee J, Joseph V, et al. (2012) Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Invest Ophthalmol Vis Sci 53: 7590–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326–332. [DOI] [PubMed] [Google Scholar]
- 17. Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 18. Fribley A, Zhang K, Kaufman RJ (2009) Regulation of apoptosis by the unfolded protein response. Methods Mol Biol 559: 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lai E, Teodoro T, Volchuk A (2007) Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 22: 193–201. [DOI] [PubMed] [Google Scholar]
- 20. Martinez IM, Chrispeels MJ (2003) Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell 15: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao RV, Ellerby HM, Bredesen DE (2004) Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 11: 372–380. [DOI] [PubMed] [Google Scholar]
- 22. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118: 3378–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, et al. (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 22: 1451–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, et al. (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, et al. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L (2008) Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 314: 950–960. [DOI] [PubMed] [Google Scholar]
- 27. Ron D, Habener JF (1992) CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev 6: 439–453. [DOI] [PubMed] [Google Scholar]
- 28. Shen X, Zhang K, Kaufman RJ (2004) The unfolded protein response–a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat 28: 79–92. [DOI] [PubMed] [Google Scholar]
- 29. Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389. [DOI] [PubMed] [Google Scholar]
- 30. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, et al. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275: 90–94. [DOI] [PubMed] [Google Scholar]
- 31. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, et al. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666. [DOI] [PubMed] [Google Scholar]
- 32. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, et al. (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2: 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang XZ, Ron D (1996) Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272: 1347–1349. [DOI] [PubMed] [Google Scholar]
- 34. Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, et al. (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, et al. (2005) CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 95: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, et al. (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ 11: 403–415. [DOI] [PubMed] [Google Scholar]
- 37. Kong L, Zhou X, Li F, Yodoi J, McGinnis J, et al. (2010) Neuroprotective effect of overexpression of thioredoxin on photoreceptor degeneration in Tubby mice. Neurobiol Dis 38: 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choudhury S, Bhootada Y, Gorbatyuk O, Gorbatyuk M (2013) Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis 4: e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Ruther K, et al. (2001) Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci 42: 826–833. [PubMed] [Google Scholar]
- 40.The Jackson Laboratory website. Available: http://jaxmice.jax.org/strain/005530.html#genotype. Accessed: 2014 Jan 11.
- 41. Umino Y, Everhart D, Solessio E, Cusato K, Pan JC, et al. (2006) Hypoglycemia leads to age-related loss of vision. Proc Natl Acad Sci U S A 103: 19541–19545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pennesi ME, Lyubarsky AL, Pugh EN Jr (1998) Extreme responsiveness of the pupil of the dark-adapted mouse to steady retinal illumination. Invest Ophthalmol Vis Sci 39: 2148–2156. [PubMed] [Google Scholar]
- 43. Saszik SM, Robson JG, Frishman LJ (2002) The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol 543: 899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Behrman S, Acosta-Alvear D, Walter P (2011) A CHOP-regulated microRNA controls rhodopsin expression. J Cell Biol 192: 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Penn RD, Hagins WA (1969) Signal transmission along retinal rods and the origin of the electroretinographic a-wave. Nature 223: 201–204. [DOI] [PubMed] [Google Scholar]
- 47. Tian N, Slaughter MM (1995) Correlation of dynamic responses in the ON bipolar neuron and the b-wave of the electroretinogram. Vision Res 35: 1359–1364. [DOI] [PubMed] [Google Scholar]
- 48. Robson JG, Maeda H, Saszik SM, Frishman LJ (2004) In vivo studies of signaling in rod pathways of the mouse using the electroretinogram. Vision Res 44: 3253–3268. [DOI] [PubMed] [Google Scholar]
- 49. Stockton RA, Slaughter MM (1989) B-wave of the electroretinogram. A reflection of ON bipolar cell activity. J Gen Physiol 93: 101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gresh J, Goletz PW, Crouch RK, Rohrer B (2003) Structure-function analysis of rods and cones in juvenile, adult, and aged C57bl/6 and Balb/c mice. Vis Neurosci 20: 211–220. [DOI] [PubMed] [Google Scholar]
- 51. Goto Y, Peachey NS, Ripps H, Naash MI (1995) Functional abnormalities in transgenic mice expressing a mutant rhodopsin gene. Invest Ophthalmol Vis Sci 36: 62–71. [PubMed] [Google Scholar]
- 52. Perlman I (1983) Relationship between the amplitudes of the b wave and the a wave as a useful index for evaluating the electroretinogram. Br J Ophthalmol 67: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Field GD, Rieke F (2002) Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron 34: 773–785. [DOI] [PubMed] [Google Scholar]
- 54. Nashine S, Bhootada Y, Lewin AS, Gorbatyuk M (2013) Ablation of C/EBP Homologous Protein Does Not Protect T17M RHO Mice from Retinal Degeneration. PLoS One 8: e63205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaur J, Mencl S, Sahaboglu A, Farinelli P, van Veen T, et al. (2011) Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PLoS One 6: e22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li T, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, et al. (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17→methionine and proline-347→serine in transgenic mice and in cell cultures. Proc Natl Acad Sci U S A 95: 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Homma K, Katagiri K, Nishitoh H, Ichijo H (2009) Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opin Ther Targets 13: 653–664. [DOI] [PubMed] [Google Scholar]
- 58. Roof DJ, Adamian M, Hayes A (1994) Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest Ophthalmol Vis Sci 35: 4049–4062. [PubMed] [Google Scholar]
- 59. Price BA, Sandoval IM, Chan F, Nichols R, Roman-Sanchez R, et al. (2012) Rhodopsin gene expression determines rod outer segment size and rod cell resistance to a dominant-negative neurodegeneration mutant. PLoS One 7: e49889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, et al. (1997) Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet 15: 216–219. [DOI] [PubMed] [Google Scholar]
- 61. Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, et al. (1999) Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A 96: 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haeri M, Knox BE (2012) Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One 7: e30101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haeri M, Knox BE, Ahmadi A (2013) Modeling the flexural rigidity of rod photoreceptors. Biophys J 104: 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative ERG waveforms from GHL+ and GHL+/Chop−/− mice. Average scotopic ERG a- and b-wave waveforms from (A) 4 and 28 week old C57BL/6 (blue traces), Chop−/− (orange traces) (B) 4–28 week old GHL+ (black traces) and GHL+/Chop−/− (red traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 6–14). Error bars ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent onset of light stimulus.
(EPS)
Representative ERG waveforms from GHL+ and GHL+/Ask1−/− mice. Average scotopic ERG a- and b-wave waveforms from (A) 4 and 28 week old C57BL/6 (blue traces), Ask1−/− (green traces) (B) 4, 16, and 28 week old GHL+ (black traces) and GHL+/Ask1−/− (purple traces) mice in response to 1 ms flashes of increasing intensity, from bottom to top, (n = 7–14). Error bars ± SD. Scale bar: x = 50 ms, y = 200 µV. Arrows represent onset of light stimulus.
(EPS)