

Supplemental digital content is available in the text.
Key Words: HPV integration, Cervical carcinoma, Next generation sequencing
Abstract
Objective
Integration of carcinogenic human papillomaviruses (HPVs) into the host genome is a significant tumorigenic factor in specific cancers including cervical carcinoma. Although major strides have been made with respect to HPV diagnosis and prevention, identification and development of efficacious treatments for cervical cancer patients remains a goal and thus requires additional detailed characterization of both somatic events and HPV integration. Given this need, the goal of this study was to use the next generation sequencing to simultaneously evaluate somatic alterations and expression changes in a patient’s cervical squamous carcinoma lesion metastatic to the lung and to detect and analyze HPV infection in the same sample.
Materials and Methods
We performed tumor and normal exome, tumor and normal shallow whole-genome sequencing, and RNA sequencing of the patient’s lung metastasis.
Results
We generated over 1.2 billion mapped reads and identified 130 somatic point mutations and indels, 21 genic translocations, 16 coding regions demonstrating copy number changes, and over 36 genes demonstrating altered expression in the tumor (corrected P < 0.05). Sequencing also revealed the HPV type 18 (HPV-18) integration in the metastasis. Using both DNA and RNA reads, we pinpointed 3 major events indicating HPV-18 integration into an intronic region of chromosome 6p25.1 in the patient’s tumor and validated these events with Sanger sequencing. This integration site has not been reported for HPV-18.
Conclusions
We demonstrate that DNA and RNA sequencing can be used to concurrently characterize somatic alterations and expression changes in a biopsy and delineate HPV integration at base resolution in cervical cancer. Further sequencing will allow us to better understand the molecular basis of cervical cancer pathogenesis.
Key events that drive cancer are influenced by a multitude of factors that still remain to be understood. One event is viral infection, which is estimated to have a role in 15% to 20% of cancers.1 Cancers that have been found to be associated with viral infection include cervical,1,2 oropharyngeal and hepatocellular carcinomas,1,3 and some leukemias and lymphomas.1,4–6 One well-known association is the role of human papillomavirus (HPV) in cervical carcinoma (CC). This relationship was discovered after isolation and identification of HPV-16 and HPV-18, now designated as high-risk mucosal HPV subtypes, in CC biopsies.7,8 Additional studies show that infection by these and other high-risk HPV types is the primary risk factor for developing CC2,9,10 such that they are identified in at least 99% of CC cases.2,11
In 2013, 12,340 new CC diagnoses, and 4030 CC-associated deaths are estimated.12 Fortunately, early detection and preventative measures have resulted in a decrease in the mortality rate. However, our understanding of HPV integration in CC is still evolving as studies point to both the presence of HPV integration hotspots13,14 and the absence of a correlation between HPV type and integration location.13 Furthermore, despite the well-established association between HPV and CC, identification of effective therapies for patients with CC remains a challenge and emphasizes the need for additional characterization of CC tumors and further evaluation of HPV in CC.15
Given this demand, we applied next generation sequencing (NGS) to characterize somatic alterations in a live metastatic cervical squamous cell carcinoma patient and to detect and analyze HPV integration in the same sample. We performed whole-genome, exome, and RNA sequencing (RNAseq) from DNA and RNA collected from the patient’s tumor biopsy specimen and peripheral blood sample. We describe here the first reported study of combined DNA and RNA sequencing as well as HPV integration and analysis in a live patient with metastatic CC.
MATERIALS AND METHODS
Please refer to the Supplemental data for detailed methods, available at http://links.lww.com/IGC/A194.
Ethics Statement
The patient was treated on protocols approved by the institutional review board of the Mayo Clinic. This study was conducted in accordance with the 1996 Declaration of Helsinki. Written informed consent was obtained from the patient for sequencing analyses and data release.
Patient Clinical History
The patient’s condition was diagnosed with stage IB2 squamous cell carcinoma of the cervix in June 2007. The patient underwent numerous treatments, and a metastatic lung biopsy was acquired in April 2012. This lung biopsy specimen was sent to the CRL (Clinical Reference Laboratory, Lenexa, KS) for nucleic acid isolation, and the DNA/RNA was sent to the Translational Genomics Research Institute for sequencing.
Sample Assessment
The patient’s lung biopsy specimen was preserved as fresh frozen and assessed as 100% tumor squamous carcinoma. Direct visualization of the sample was performed to estimate the tumor content and the extent of tissue heterogeneity by a board-certified pathologist.
DNA and RNA Isolation
Blood leukocytes were isolated from the whole blood and homogenized. Genomic DNA was purified using the Qiagen AllPrep DNA spin column (Valencia, CA). Tumor DNA and RNA isolations were performed by CRL using Qiagen’s AllPrep Kit.
Library Preparation, Sequencing, and Data Analysis
Isolated DNA and RNA were used to generate whole-genome, exome, and RNA sequencing libraries. Libraries were paired-end sequenced on the Illumina HiSeq 2000 (San Diego, CA) and analyzed after Fastq generation and alignment against the human reference genome (build 37) using the Burrows-Wheeler Alignment16 for DNA sequencing data and Bowtie/Tophat17,18 for RNA sequencing data. The dbGaP (database of Genotypes and Phenotypes) accession number for sequencing data from this study is phs000628.v1.p1.
Experimental Validation
To validate HPV-18 integration sites and the presence of episomal HPV-18, primers were designed upstream and downstream of 4 separate junctions. These junctions include the following: (1) E6 to long control region (LCR) (episomal HPV-18), (2) E2/E4 to chr6:4,328,779, (3) E2 to chr6:4,282,640, and (4) E1 to chr6:4,291,973. Polymerase chain reaction (PCR) was performed using each primer set on cDNA that was previously generated from tumor RNA during library construction, and PCR products were Sanger sequenced. Quantitative PCR was performed by CRL to evaluate PIK3CA expression with β-actin as the control gene.
RESULTS AND DISCUSSION
Whole-Genome Sequencing
We performed shallow whole-genome sequencing (WGS) from DNA isolated from the lung biopsy specimen and whole blood sample to identify somatic copy number changes and translocations. Whole-genome sequencing metrics are listed in Table 1, and identified translocations and copy number changes are summarized in Figure 1. Overall, we identified 21 translocations (Table 2) affecting at least 1 gene. Of these events, 1 affected a COSMIC (Catalogue of Somatic Mutations in Cancer)19 gene, TRIM27 (tripartite motif containing 27; RET). Point mutations in TRIM27 have been identified in other cancers,20,21 but no somatic translocations in this gene have been previously reported in CC. We additionally identified 16 genic regions demonstrating copy number variations (CNVs) encompassing 354 genes (Supplemental Digital Content Fig. S1, available at http://links.lww.com/IGC/A194). The 16 regions encompass 11 COSMIC genes (Table 3) including PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α), SOX2 [SRY (sex-determining region Y)-box 2], IL7R (interleukin 7 receptor), and LIFR (leukemia inhibitory factor receptor α). The gain identified on 5p also overlaps with TERT (telomerase reverse transcriptase), for which altered expression has been described in HPV-mediated CC.22 The 2 regions of CNV loss (4q and 11q) did not overlap with COSMIC genes, but these events have been previously detected in CC.23–25
TABLE 1.
Sequencing metrics
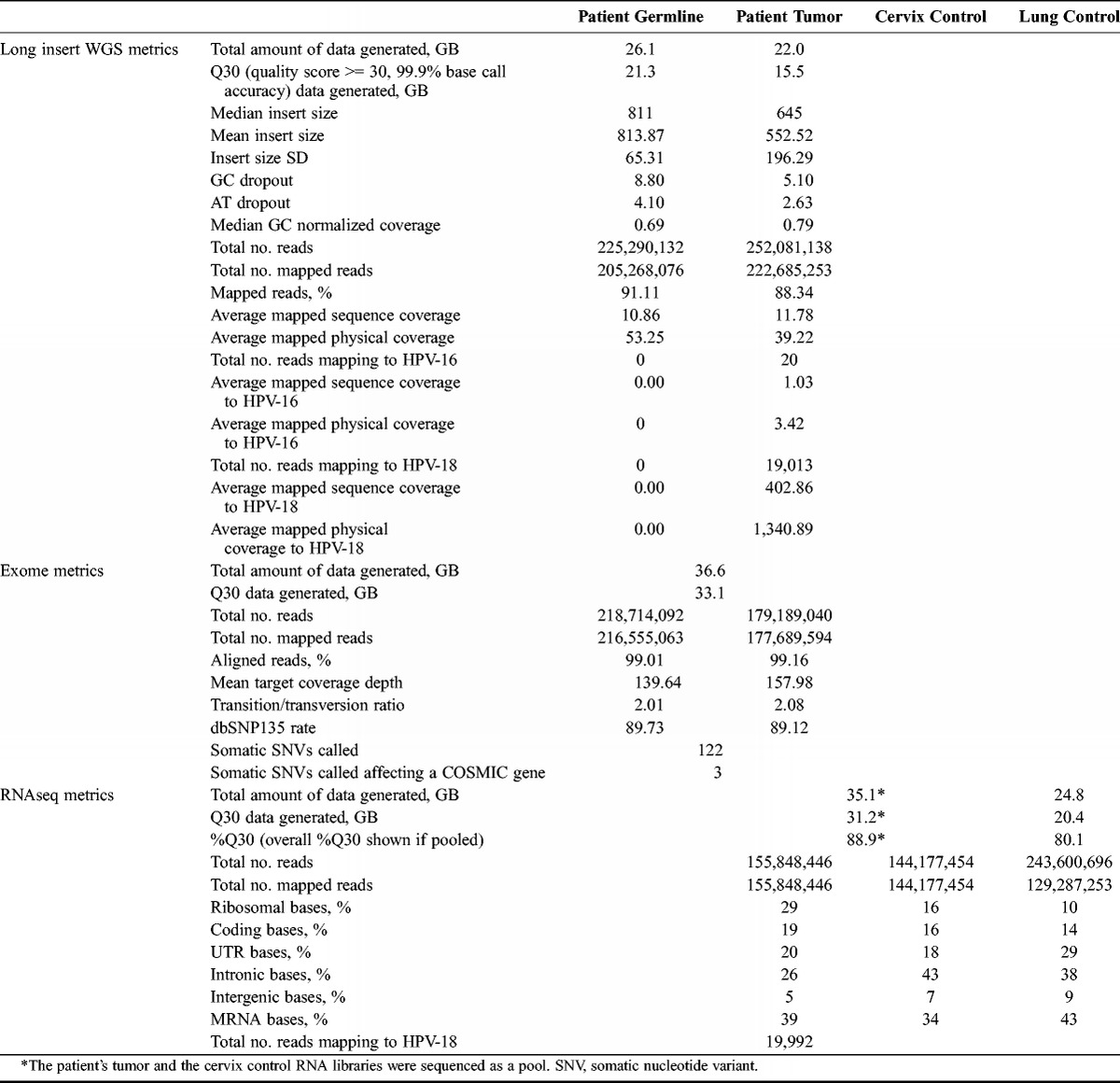
FIGURE 1.
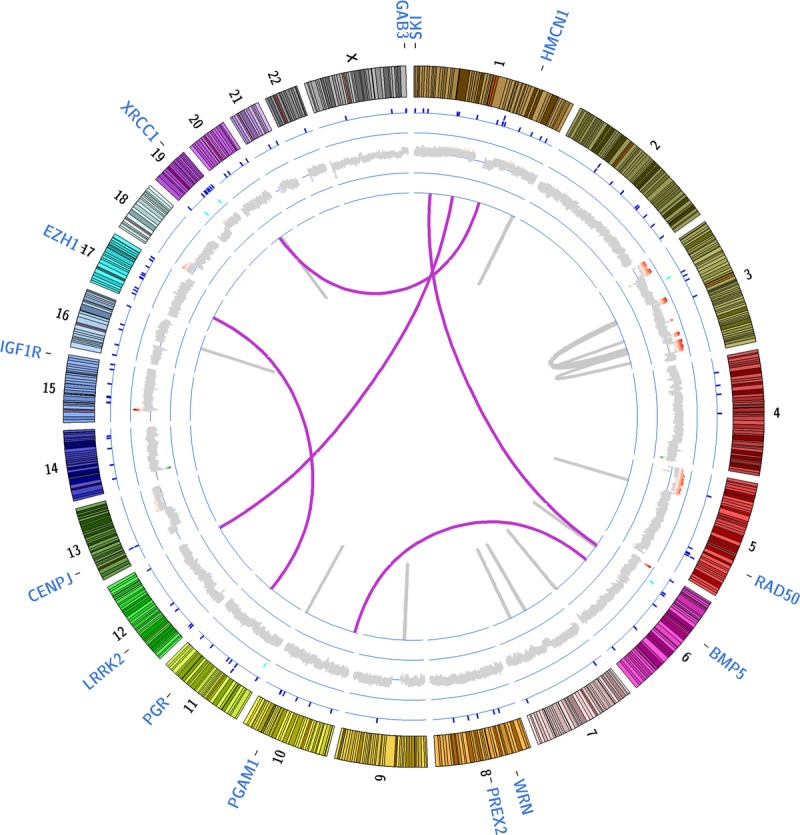
Circos plot summarizing somatic events. A summary of all identified somatic genomic alterations is shown. Translocations are marked by purple (interchromosomal) and gray (intrachromosomal translocations) lines; for intrachromosomal translocations, the gray connecting line may appear as a single line if the joined regions lie within 2000 kb. CNVs are shown along the thick gray ring encircling the translocations (green, regions of loss; red, regions of gain; gray, no change); on the ring encircling CNVs, somatic indels (insertion/deletions) are marked by light blue tick marks and on the ring encircling the indels, somatic point mutations are marked by dark blue tick marks. Because we identified 122 nonsynonymous point mutations, splice site point mutations, and small indels, selected gene labels associated with point mutations are shown along the outermost area of the plot.
TABLE 2.
Identified genic intrachromosomal and interchromosomal translocations
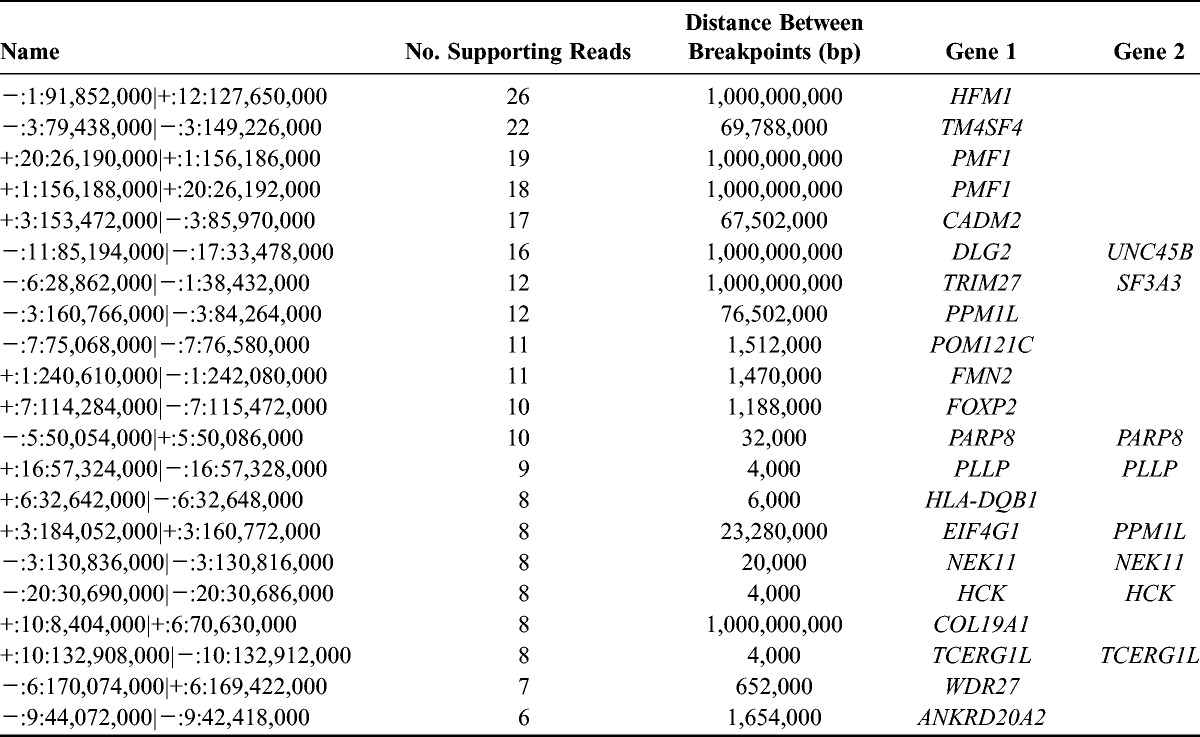
TABLE 3.
Identified copy number alterations affecting coding regions
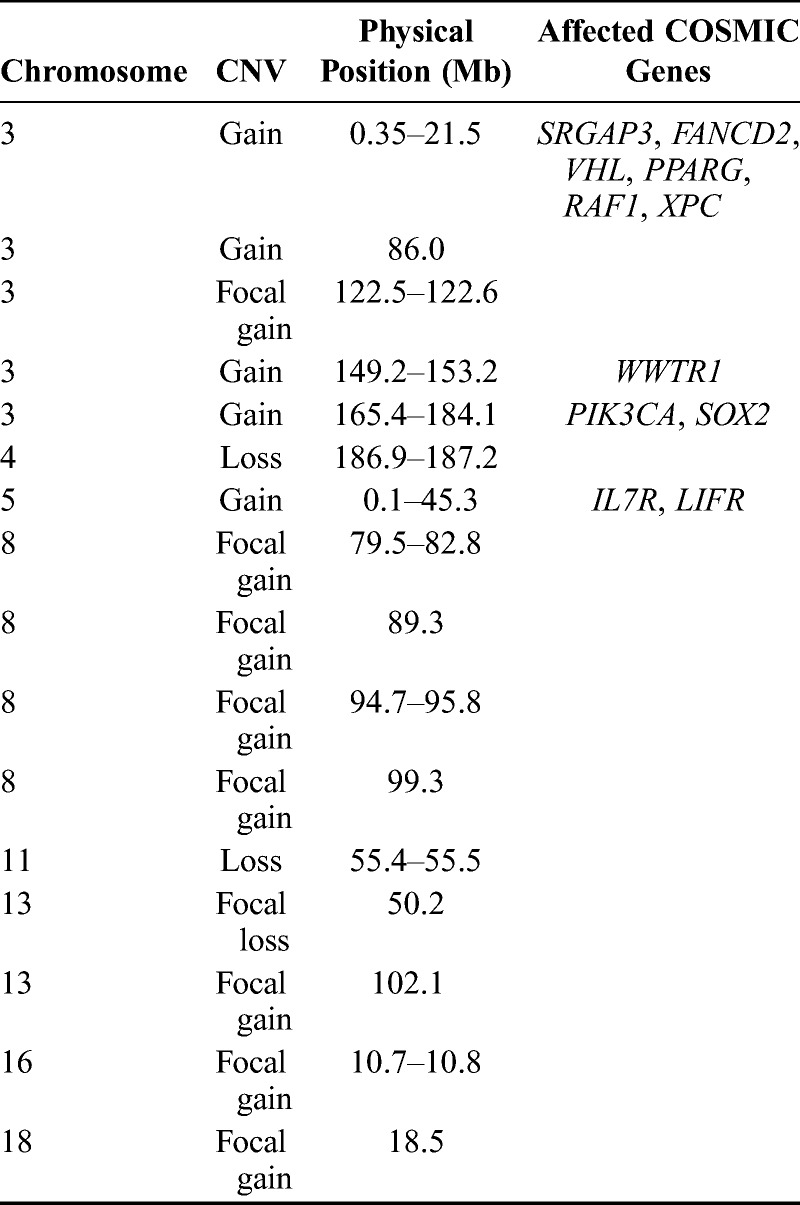
Gains in CC have been reported for regions encompassing IL7R and PIK3CA. A 5p amplification encompassing IL7R was previously identified in a primary cervical adenocarcinoma cell line (COSMIC ID 687509). We also identified multiple gains on 3q, which include a region encompassing PIK3CA. The 3q amplification is considered a frequent somatic alteration in CC.26–30 Cell line studies provide evidence that suggests that amplified PIK3CA, along with increased expression, may activate the PI3K/Akt pathway, lead to apoptosis inhibition, and thereby support tumor cell growth and division.24,27 PIK3CA RNAseq data are detailed later. The International Cancer Genome Consortium25 has also reported events in SOX2 in CC.
Exome Sequencing
Using exome sequencing, we identified 130 nonsynonymous point mutations, splice site point mutations, and small indels (insertion deletions; Fig. 1). Of these events, 3 mutations affected COSMIC genes including WRN (Werner syndrome, RecQ helicase-like) and ASPSCR1 (alveolar soft part sarcoma chromosome region, candidate 1) and have not been previously reported in CC (Table 4). Sorting Tolerant From Intolerant (SIFT)31 and PolyPhen-2 (Polymorphism Phenotyping v2)32 were used to predict potential effects of selected point mutations on protein function (Table 4). We additionally performed allele frequency analysis of all identified mutations to evaluate the extent to which exome reads support a mutation (Supplemental Digital Content Table S1, available at http://links.lww.com/IGC/A194). Ninety-seven of 130 total mutations are supported by all exome reads at the mutation location.
TABLE 4.
Selected SNVs identified through exome sequencing
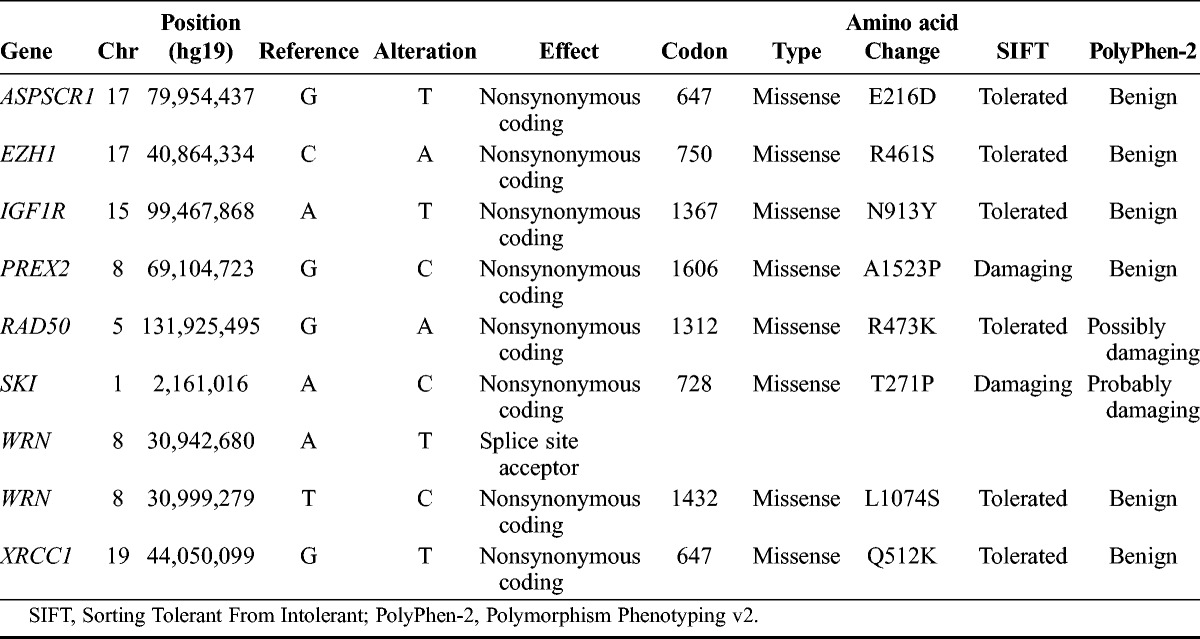
Additional notable nonsynonymous coding mutations that were identified (Table 4) include IGF1R (insulin-like growth factor 1 receptor), SKI (v-ski sarcoma viral oncogene homolog (avian), and RAD50 (RAD50 homolog [S. accharomyces cerevisiae]), and XRCC1 (x-ray repair complementing defective repair in Chinese hamster cells 1). IGF1R is normally involved in initiating signaling after binding of IGF1 (insulin growth factor 1) to activate cell proliferation and inhibit apoptosis,33 and its overexpression has been found in both CC cell lines34 and specimens.35,36 We, however, did not identify significant IGF1R expression changes (corrected P < 0.05) in the tumor. In addition, a recent study showed that resistance to the IGF1R inhibitor figitumamab is associated with the absence of N-linked glycosylation at N913.37 The mutation causing the change of asparagine to tyrosine at this position suggests that the tumor may be resistant to this inhibitor and highlights the benefit of performing WGS analyses to gain insight into therapeutic selection.
In the proto-oncogene SKI, we identified a coding mutation that falls in the SMAD4 (mothers against DPP homolog 4)-binding domain. SKI acts as a corepressor of SMAD proteins and blocks the cell’s ability to stop cell growth and division.14,38 DNA repair genes with missense mutations identified in the patient include RAD50 and XRCC1. RAD50 complexes with MRE11 and NBN (nibrin) and identifies and repairs DNA damage,33 whereas XRCC1 is involved in single-stranded break repair. In XRCC1, we also identified an SNP (Q399R; rs25487) that was reported to be associated with persistent HPV infection.34 Although we do not have records of whether the sequenced patient experienced HPV persistence, this event suggests that the patient may be HPV positive.
RNA Sequencing
Commercially purchased normal cervix and normal lung RNA samples were prepared and sequenced to serve as the control(s) for differential analyses. RNA sequencing metrics are shown in Table 1. Tumor RNA reads were compared against normal cervix RNA reads and also compared against both normal cervix and normal lung RNA reads. Differential analysis of tumor compared with cervix led to the identification of 3468 genes showing expression changes (corrected P < 0.05) of which 83 genes are listed in COSMIC. Analysis of tumor compared with both normal cervix and normal lung led to the identification of 2338 genes (corrected P < 0.05), with 51 genes listed in COSMIC. Differentially expressed COSMIC genes are listed in Supplementary Table S2, available at http://links.lww.com/IGC/A194. In a separate analysis, we additionally identified RNA reads that support mutations identified through exome sequencing (Supplemental Digital Content Table S1). Overall, 35 of 130 mutations identified in exome data are supported by 100% of RNA reads at the respective mutation location. Supplemental data are available at http://links.lww.com/IGC/A194.
Consolidation of both RNAseq analyses led to the identification of 36 common differentially expressed COSMIC genes. Upon evaluation against the Cervical Cancer Gene Database,39 7 of the 36 genes were found to be previously described in CC. These genes include DEK (DEK oncogene), FHIT (fragile histidine triad), GATA3 (GATA-binding protein 3), and KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog).
For COSMIC genes that fell in CNVs, we evaluated if these genes also demonstrated expression changes in the tumor (Supplemental Digital Content Table S3, available at http://links.lww.com/IGC/A194). Overall, FANCD2 (Fanconi anemia, complementation group D2) and SOX2, which both fell in regions of copy number gains, showed increased expression in the tumor (corrected P < 0.05) in both RNAseq comparisons. However, PPARG (peroxisome proliferators–activated receptor γ) and LIFR, which also fell in areas of gain, demonstrated decreased expression in the tumor. Low PPARG expression was also previously reported in CC.36 Given the PIK3CA amplification that was identified, we evaluated PIK3CA, which demonstrated an increased expression in the tumor but was not initially noted as it did not pass multiple testing corrections. In the tumor versus normal cervix analysis, the tumor demonstrated a log2 fold of 1.30 (P = 0.03), whereas in the tumor versus normal cervix and lung analysis, the tumor demonstrated a log2 fold of 1.19 (P = 0.126). Quantitative PCR validation confirmed PIK3CA overexpression in the tumor as compared against β-actin (δ Ct = 8.7; Supplemental Digital Content Table S4, available at http://links.lww.com/IGC/A194).
Pathway analysis was performed on each set of differential analyses using MetaCore GeneGo (corrected P < 0.05). The top 10 affected pathways for each analysis are listed in Supplementary Table S5, available at http://links.lww.com/IGC/A194. Overall, pathways that are likely to be the most affected by identified expression changes include cell cycle processes, DNA methylation, stromal-epithelial interactions, cell adhesion, and DNA damage processes. Although these pathways are commonly affected across cancers, this information may lend additional contextual insight into CC.
HPV Detection
To determine if high-risk HPV integration had occurred in the patient, WGS reads were mapped against all 552 human viral reference sequences posted on NCBI’s (National Center for Biotechnology Information) Entrez Genome Viral Genomes Resource. These 552 references include 42 HPV genomes, including those of HPV-16 and HPV-18. Results from alignment against all 42 HPV references are shown in the Supplementary Table S6, available at http://links.lww.com/IGC/A194. Reads aligning to HPV genomes were only aligned to HPV-16 or HPV-18. Over 19,000 tumor DNA reads were mapped to the HPV-18 genome, whereas no germline reads were mapped (Table 1). In the tumor, 49 reads were mapped to HPV-16, but no germline reads were mapped. Evaluation of HPV-16 reads indicated that mapped reads were discordant such that read pairs were mapped to HPV-18 and the mitochondrial genome. Confidence in these reads is low because of low mapping quality and because of 66% sequence homology between the HPV-16 and HPV-18 genomes. These analyses indicate that HPV-18 integration had occurred in the metastasis.
HPV-18 Integration Analysis
The HPV-18 genome consists of 6 early genes (E1, E2, E4, E5, E6, E7) and 2 late genes (L1, L2). The late genes code for viral capsid proteins, and the early genes include viral oncogenes (E6 and E7) and code for proteins involved in the maintenance of transformation and viral replication. E2 codes for a transcriptional repressor that normally inhibits E6 and E7 expression. Loss of E2 expression occurs after HPV integration into the host genome and allows for E6 and E7 expression.35,37,38 Analysis of whole-genome and RNA data indicated that portions of HPV-18 integrated into multiple locations within a nongenic region of chromosome (chr) 6 (position 4,280,617-4,331,314; 6p25.1), which also overlaps with a region of copy number gain and is a common fragile site (FRA6B).14 This finding correlates with a study that reported that 63% of HPV-18 integrations occur within common fragile sites and that significant structural events often surround integration sites.40 In addition, the chr6p25.1 region that we identified has been reported as an HPV-16 integration site in CC14 but has not been reported for HPV-18.
The evaluation of DNA and RNA reads aligning to HPV-18 indicated that a portion of HPV-18 DNA in the tumor cells remained in the episomal form, a phenotype that has been previously reported for HPV-16 in CC.41 Upon investigating the 3′ end of the noncoding LCR in HPV-18, we found that approximately two thirds of DNA reads were mapped back to the E6 region to indicate that these reads were generated from episomal HPV-18 DNA. The presence of both genomic states of HPV-18 in the sequenced tumor illustrates the complexity of HPV integration and emphasizes that both spatial and temporal analyses are needed to fully understand the role of HPV in CC.
Visual inspection of DNA and RNA reads suggested that multiple integration events occurred. Because of the multicellularity of the sequenced sample, this phenotype suggests that different cells may have undergone different integration events. We assembled RNA reads from HPV-18 and discordant reads from chr6p25.1 that mapped to HPV-18 to pinpoint the major breakage events in expressed transcripts. Figure 2 illustrates the mapped HPV-18 RNA reads (A), assembled contigs generated from the HPV-18 RNA reads (B), and a linearized schematic of the HPV-18 genome and the chr6 region into which HPV-18 integrated (C). Overall, we assembled 155,884 paired reads to generate 20 contigs that ranged in size from 220 to 11,616 base pairs. Contigs that fully or partially aligned to HPV-18 are also shown.
FIGURE 2.
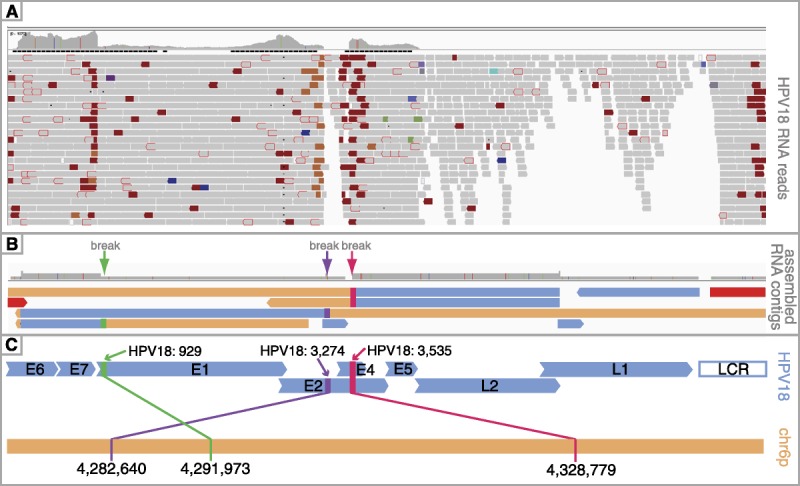
HPV-18 integration analysis. This schematic summarizes the major integration events that were detected using RNA data. Integrated Genomics Viewer was used to visualize RNA reads mapping to HPV-18 (A). A coverage track at the top of (A) illustrates HPV-18 expression levels. Orange and dark red coloring mark discordant reads that fall at breakpoints, whereas gray reads mapped without discordance. B, The figure shows the contigs assembled from the reads such that peach-colored portions of the contigs correspond to chromosome 6 sequences and blue-colored portions of the contigs correspond to HPV-18 sequences. The red contig spans the LCR and E6 regions of HPV-18 to indicate the presence of episomal DNA. The locations of identified breakpoints are shown at the top of panel B with green, purple, and pink arrows, and lineup with breakpoints identified in panel A. C, The figure shows a linearized view of the HPV-18 genome (blue) and the chromosome 6 region (peach) where HPV-18 reads were aligned. As shown in panel B and C, 3 separate events were identified (demarcated by green, purple, and pink arrows in panel B and corresponding green, purple, and pink connecting lines in panel C).
We identified a contig (3B; red) that spanned HPV-18’s E6 and LCR junction to provide additional evidence of the presence of HPV-18 episomes in the sequenced tumor and also demonstrated that transcription across the E6/LCR junction occurs in HPV-18 episomes. Overall, we identified 3 major breaks in the HPV-18 genome (Figs. 2B-C). The breaks shown in Figure 2C colored purple and pink may represent different points (in different cells) where the circular viral genome opened during integration as an early event in HPV-18 integration is the disruption of the E2 gene to allow for genome linearization. A caveat with these analyses is that the contigs are a representation of the most commonly occurring events. Additional events that are less common in the cells of the sequenced biopsy may not be captured using this approach.
Transcriptomic Analysis of HPV-18 Gene Expression
We additionally used the tumor HPV-18 reads to evaluate expression of HPV-18 genes. Because of the absence of a control dataset for the HPV-18 genes, we evaluated the number of reads that were acquired for each gene to determine the level of expression (Supplemental Digital Content Table S7, available at http://links.lww.com/IGC/A194). We identified 4103 reads mapping to E2, but no expression of integrated E2 regions, as is expected because integration disrupts E2 to subsequently allow for increased expression of E6 and E7, which we also identified in the patient’s tumor. We also identified expression of E5—loss of E5 expression is associated with HPV integration42 and thus correlates with the presence of HPV-18 episomes in the biopsied tumor cells. In conjunction with high levels of E6 and E7 expression (3774 and 2922 reads, respectively) and integration of both genes into chr6, we see over 550X mapped reads in the chromosome 6 region affected by HPV-18 integration. This phenotype is likely the result of viral promotion of E6 and E7 expression, which subsequently promotes expression of the intronic chromosome 6 regions situated at the 3′ end of the integrated viral oncogenes.
After HPV integration, HPV proteins acquire control of host cellular pathways. We thus evaluated expression changes of genes whose products are affected by HPV proteins. These genes include TP53 (tumor protein p53), WAF1/CDKN1A (p21; cycle-dependent kinase inhibitor 1A), RB1 (retinoblastoma 1), CCNE2 (cyclin E2), E2F1 (E2F transcription factor 1), CDKN2A (cyclin-dependent kinase inhibitor 2A), BRD4 (bromodomain containing 4), and CDC25A (cell division cycle 25A; Supplemental Digital Content Table S8, available at http://links.lww.com/IGC/A194). These genes have roles in 2 key tumor suppressor pathways affected by HPV-18’s E6 and E7 genes. Overall, we identified increased expression of TP53, CCNE2, E2F1, CDKN2A, and CDC25A (corrected P < 0.05). HPV-18’s E6 protein targets and inhibits p53 such that the up-regulated TP53 expression identified in the tumor may represent a compensatory response. Furthermore, p53 is activated by p14/CDKNA2A such that up-regulated expression of CDKN2A may also represent a response to inhibition of p53’s functions. HPV-18’s E7 protein also degrades RB1 and inhibits the RB1 tumor suppressor pathway causing abnormal gene expression normally controlled by E2F, CDK2 complex activation, and increasing CDC25 protein. The significantly up-regulated expression of CCNE2, E2F1, and CDC25A that we identified thus correlate with RB1 pathway inhibition that would result from E7 expression.
HPV-18 Integration Validation
To validate the presence of the 4 identified junctions resulting from HPV-18 integration and the presence of episomal HPV-18 DNA, we performed PCR of regions spanning the 4 junctions on cDNA generated from tumor RNA. These junctions include HPV-18 LCR-E6 (found in episomal forms of HPV-18), E1-chr6:4,291,973, E2-chr6:4,282,640, and E2/E4-chr6:4,328,779. PCR products were Sanger sequenced, and resulting chromatograms confirmed the presence of the 4 junctions (Supplemental Digital Content Figure S2, available at http://links.lww.com/IGC/A194).
CONCLUSIONS
In this study, we used NGS to characterize somatic alterations in a patient with CC as well as to identify and analyze HPV integration. Although combined DNA and RNA sequencing has been performed for other cancers, this study is the first to apply this approach to a patient with CC and to concurrently analyze HPV integration in the same sequencing data. Before sequencing, the patient’s HPV status was not known but using NGS, we determined that HPV-18 integration had occurred in the patient’s tumor and subsequently identified a novel integration site on chr6p25.1. We also found that although some tumor cells in the biopsy garnered episomal HPV-18, other cells had undergone HPV-18 integration. Although this phenotype has been reported for HPV-16 in CC, this finding reiterates the need to consider both spatial and temporal analyses to fully understand the role of HPV in CC. The approach used in this study is relevant not only to HPV-associated carcinomas but also to other malignancies involving viral infection. Furthermore, using NGS to simultaneously characterize a tumor genome and to analyze HPV integration is relevant for CC because HPV integration is not the sole driving event in CC as additional debilitating events are required for carcinogenesis.43 As we continue to sequence CC tumors, we set the foundation for improving our molecular knowledge of HPV, CC, and HPV in CC.
ACKNOWLEDGMENT
The authors thank the St Joseph’s Foundation and Mayo Clinic Comprehensive Cancer Center for support and the network and computing systems division of the Translational Genomics Research Institute for making available the supercomputing resources funded by National Institutes of Health grant 1S10RR25056-01. We would also like to thank the patient and her family for contributing to this study, Steve Mastrian for staff support, and Waibhav Tembe and Raghu Metpally for assistance with allele frequency analysis.
Footnotes
Address correspondence and reprint requests to Winnie S. Liang, PhD, Collaborative Sequencing Center, Translational Genomics Research Institute, 445 N, Fifth St, Phoenix, AZ 85004. E-mail: wliang@tgen.org.
The last three authors contributed equally to this manuscript.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.ijgc.net).
The authors declare no conflicts of interest.
REFERENCES
- 1. McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta. 2008; 1782: 127– 150 Epub 2007 Dec 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999; 189: 12– 19 [DOI] [PubMed] [Google Scholar]
- 3. Beasley RP, Hwang LY, Lin CC, et al. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981; 2: 1129– 1133 [DOI] [PubMed] [Google Scholar]
- 4. Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980; 77: 7415– 7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. 1982; 79: 2031– 2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964; 1: 702– 703 [DOI] [PubMed] [Google Scholar]
- 7. Boshart M, Gissmann L, Ikenberg H, et al. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984; 3: 1151– 1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Durst M, Gissmann L, Ikenberg H, et al. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983; 80: 3812– 3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer, I.A.f.R.o IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon, France: Sons Presse; 1995 [Google Scholar]
- 10. Dunne EF, Markowitz LE. Genital human papillomavirus infection. Clin Infect Dis. 2006; 43: 624– 629 Epub 2006 Jul 26 [DOI] [PubMed] [Google Scholar]
- 11. Bosch FX, Lorincz A, Munoz N, et al. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002; 55: 244– 265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. \CA Cancer J Clin. 2013; 63: 11– 30 Epub 2013 Jan 17 [DOI] [PubMed] [Google Scholar]
- 13. Kraus I, Driesch C, Vinokurova S, et al. The majority of viral-cellular fusion transcripts in cervical carcinomas cotranscribe cellular sequences of known or predicted genes. Cancer Res. 2008; 68: 2514– 2522 [DOI] [PubMed] [Google Scholar]
- 14. Schmitz M, Driesch C, Jansen L, et al. Non-random integration of the HPV genome in cervical cancer. PLoS One. 2012; 7: e39632 Epub 2012 Jun 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diaz-Padilla I, Monk BJ, Mackay HJ, et al. Treatment of metastatic cervical cancer: future directions involving targeted agents. Crit Rev Oncol Hematol. 2013; 85: 303– 314 Epub 2012 Aug 9 [DOI] [PubMed] [Google Scholar]
- 16. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25: 1754– 1760 Epub 2009 May 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10: R25 Epub 2009 Mar 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7: 562– 578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forbes SA, Tang G, Bindal N, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010; 38: D652– D657 Epub 2009 Nov 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609– 615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012; 44: 1104– 1110 Epub 2012 Sep 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Wilde J, Fernandez-Cuesta L, Sos ML, et al. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer. 2010; 10: 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi CH, Lee KM, Choi JJ, et al. Hypermethylation and loss of heterozygosity of tumor suppressor genes on chromosome 3p in cervical cancer. Cancer Lett. 2007; 255: 26– 33 Epub 2007 Apr 30 [DOI] [PubMed] [Google Scholar]
- 24. Henken FE, Banerjee NS, Snijders PJ, et al. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Mol Cancer. 2011; 10: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J, Baran J, Cros A, et al. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database (Oxford). 2011; 2011: bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertelsen BI, Steine SJ, Sandvei R, et al. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer. 2006; 118: 1877– 1883 [DOI] [PubMed] [Google Scholar]
- 27. Ma YY, Wei SJ, Lin YC, et al. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000; 19: 2739– 2744 [DOI] [PubMed] [Google Scholar]
- 28. Zhang A, Maner S, Betz R, et al. Genetic alterations in cervical carcinomas: frequent low-level amplifications of oncogenes are associated with human papillomavirus infection. Int J Cancer. 2002; 101: 427– 433 [DOI] [PubMed] [Google Scholar]
- 29. Rao PH, Arias-Pulido H, Lu XY, et al. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer. 2004; 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayan G, Murty VV. Integrative genomic approaches in cervical cancer: implications for molecular pathogenesis. Future Oncol. 2010; 6: 1643– 1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 0.For the complete list of references, please contact: wliang@tgen.org