

Abstract
Force production in muscle results from ATP-driven cyclic interactions of myosin with actin. A myosin cross bridge consists of a globular head domain, containing actin and ATP-binding sites, and a neck domain with the associated light chain 1 (LC1) and the regulatory light chain (RLC). The actin polymer serves as a “rail” over which myosin translates. Phosphorylation of the RLC is thought to play a significant role in the regulation of muscle relaxation by increasing the degree of skeletal cross-bridge disorder and increasing muscle ATPase activity. The effect of phosphorylation on skeletal cross-bridge kinetics and the distribution of orientations during steady-state contraction of rabbit muscle is investigated here. Because the kinetics and orientation of an assembly of cross bridges (XBs) can only be studied when an individual XB makes a significant contribution to the overall signal, the number of observed XBs was minimized to ∼20 by limiting the detection volume and concentration of fluorescent XBs. The autofluorescence and photobleaching from an ex vivo sample was reduced by choosing a dye that was excited in the red and observed in the far red. The interference from scattering was eliminated by gating the signal. These techniques decrease large uncertainties associated with determination of the effect of phosphorylation on a few molecules ex vivo with millisecond time resolution. In spite of the remaining uncertainties, we conclude that the state of phosphorylation of RLC had no effect on the rate of dissociation of cross bridges from thin filaments, on the rate of myosin head binding to thin filaments, and on the rate of power stroke. On the other hand, phosphorylation slightly increased the degree of disorder of active cross bridges.
ca2+ binding to thin filaments is a major factor contributing to the initiation of skeletal muscle contraction. But it is thought that in addition to Ca2+, another factor, phosphorylation of the regulatory light chain (RLC) of myosin, may play an important regulatory role. Phosphorylation of skeletal myosin RLC is accomplished by skeletal myosin light-chain kinase (MLCK) with Ca2+-calmodulin acting as a coenzyme. A single twitch or short tetanus induces a small amount of phosphorylation of RLC (10). Phosphorylation and phosphatase-induced dephosphorylation of RLC ensure that the level of phosphorylation during the steady-state contraction is relatively constant. It is well known that the level of phosphorylation increases during heavy use and fatigue (10, 26). The current thinking is that RLC transmits the influence through myosin heavy chain (MHC) and by stabilizing the lever arm. It has been suggested that phosphorylation of RLC may cause charge repulsion with the thick filament core in a heart (46, 72) and, thus, move heads closer to thin filaments. Moving heads closer to actin would increase the likelihood of MHC interacting, which, in turn, is expected to increase the rate of force development. Indeed, Metzger et al. (49) have shown that phosphorylation of RLC in skeletal muscle increased the rate of tension redevelopment at lower [Ca2+] but was unaffected at maximum [Ca2+]. The same was true in in vitro studies of isolated striated (38, 72) thick filaments. Colson et al. (9) used synchrotron low-angle X-ray diffraction to show that phosphorylation of either RLC or cMyBP-C resulted in displacement of cross bridges (XBs) away from the thick filament backbone in cardiac muscle. The other effect of phosphorylation of RLC is the stabilization of the lever arm, which would make it easier to transmit force (63).
Ca2+ withdrawal from the myofilament space is a major contributing factor in relaxation (24). It has been shown to influence order and ATPase of XBs in relaxed muscle (67). Dephosphorylated muscle having low ATPase activity and high degree of XB order has been suggested to exist in a new super-relaxed state (SRX) (11). It applies to both skeletal (67) and cardiac muscle fibers (31). We have recently confirmed and quantified these findings by examining the local order of a few XBs in a relaxed half-sarcomere (51).
In this communication, we examine the influence of RLC phosphorylation on the movement and distribution of the orientations of XBs during contraction at the level of individual molecules. There are three fundamental reasons for examining contraction using a small number of XBs.
Steady-state kinetic information about XBs can be obtained from fluctuations of orientations of XBs (in our case, the polarization of fluorescence). The relative size of fluctuations is large only when the number of XBs is sufficiently small (22, 23). This allows determination of the kinetics of contraction by the so-called “mesoscopic” approach, first applied to biological problems by Elson et al. (22, 23). The reason why it is necessary to observe only a small number of molecules at any time is fully explained by Midde et al. (51). In our experiments, a typical number of molecules was 20, and the corresponding fluctuation was ∼22%. The use of a small number of molecules eliminates problems associated with nonuniformity of contracting sarcomeres (74), as fully explained by Midde et al. (51). Finally, our method recognizes that only when an individual XB makes a significant contribution to the overall signal, i.e., the total number of XBs is small, its contribution can significantly affect the total distribution of orientations.
Since the kinetics and orientation of an assembly of XBs can only be studied when an individual XB makes a significant contribution to the overall signal, we minimized the number of observed XBs by limiting the detection volume and concentration of fluorescent XBs. The ideal number of molecules to observe is n = 1, because then 100% of the signal carries kinetic information.1 We have chosen to investigate ∼20 molecules, because with current technology, the signal from a single molecule in an ex vivo sample is not enough to collect data with millisecond time resolution and overcome complications due to light scattering and autofluorescence.
There has been many measurements of the dynamics of single motor proteins in vitro because they do not suffer from complications due to high-protein concentration, light scattering, autofluorescence, and the presence of physical barriers, which are encountered by motor proteins (65). Thus, important information about the kinetics of heavy meromyosin (80), myosin V, (25) kinesin (42, 82), myosin VI (81), gizzard heavy meromyosin (2, 41, 61), myosin X (69), plant myosin XI (76), and dynein (60) have been gathered. But it is not clear how the in vitro measurements relate to the in vivo situation in which high protein concentrations lead to exclusion effects (53) and in which motor proteins encounter obstacles when actively transporting material throughout the cell (65). In the case of muscle, the situation is further complicated by the high density of muscle proteins (1).
To ensure that only a few XBs contribute to the signal, we genetically produced light chain 1 (LC1). It was labeled at cysteine 178 with the reactive dye SeTau-maleimide and deliberately exchanged inefficiently with the native LC1 of myofibrils. This inefficient exchange ensured that there were only a few XBs in the detection volume (DV). We recorded the parallel (‖) and perpendicular (⊥) components of fluorescent light emitted by the dye and calculated polarization of fluorescence (PF), the normalized ratio of the difference between these two. We show that the dye is immobilized during critical transient states of the contraction cycle, in spite of the fact that it is attached to LC1 at only one point, rather than more firm attachment at two points (12). PF is known to be a sensitive indicator of the orientation of the transition dipole of a fluorophore (16, 17, 32, 33, 54, 57, 66, 77). We show that PF is different in different transient states of contraction cycle, and, hence, its fluctuations reflect transitions between different states. Autocorrelation analysis of fluctuations in the PF signal revealed the kinetics of a single fluorophore. Statistical analysis revealed the distribution of orientations of XBs during contraction. Although we report that phosphorylation of RLC has no statistically significant effect on the kinetics, a small effect on the degree of order (probability distribution of orientations) of cross bridges of contracting rabbit psoas muscle was observed. In particular, phosphorylation has no effect on the rate of dissociation of cross bridges from thin filaments and on the rate of execution of the power stroke.
MATERIALS AND METHODS
Chemicals and solutions.
The glycerinating solution contained 50% glycerol, 150 mM KCl, 10 mM Tris·HCl pH 7.5, 5 mM MgCl2, 5 mM EGTA, 5 mM ATP, 1 mM DTT, 2 mM PMSF, and 0.1% β-mercaptoethanol. The rigor solution contained 50 mM KCl, 10 mM Tris·HCl at pH 7.5, and 2 mM MgCl2. EDTA rigor had the same composition, except Mg was replaced with 5 mM EDTA. Ca rigor was the same as ordinary rigor, except it contained 0.1 mM CaCl2. Contracting solution contained 50 mM KCl, 10 mM Tris·HCl, at pH 7.5, 5 mM MgCl2, 0.1 mM CaCl2, 5 mM ATP, 20 mM creatine phosphate, and 10 units/ml of 1 mg/ml creatine kinase. The relaxing solution was the same as contracting, except Ca was replaced by 2 mM EGTA, and it did not contain ATP-regenerating solution. All chemicals were from Sigma-Aldrich (St. Louis, MO). Labeling dye SeTau-647-mono-maleimide was purchased from SETA BioMedicals (Urbana, IL).
Preparation of myofibrils.
Rabbit psoas muscle bundles were washed three times with ice-cold EDTA-rigor solution for 30 min. The purpose of this step was to wash out ATP present in the glycerinating solution without causing contraction. It was followed by an extensive wash with rigor solution (not EDTA-rigor) and homogenization by the Cole-Palmer LabGen 125 homogenizer for 10 s. After a cool down period of 30 s, it was homogenized for a further 10 s. The reason that it was homogenized in rigor solution was to avoid foam, which forms when the homogenization is carried out in EDTA rigor.
Preparation of phosphorylated and dephosphorylated muscle.
Muscle fibers were naturally dephosphorylated after a few weeks in glycerinating solution. Fibers were phosphorylated by overnight incubation in ice in a solution containing 5 mM ATP, 12.5 mM MgCl2, 0.1 mM CaCl2, 5 μM calmodulin, 0.5 μM MLCK, 20 mM PO4, and 30 mM KCl. Fibers were further dephosphorylated by overnight incubation in ice in solution containing 5 mM ATP, 2.0 mM MgCl2, 0.1 mM CaCl2, 1 mM EGTA, 20 mM PO4, and 30 mM KCl. Phosphorylation by incubation in glycerinating solution with phosphatase enzyme inhibitor [20 mM sodium fluoride and 20 mM phosphate (67) were not used to avoid complications due to the direct effect of fluoride on myosin (45, 62, 64)]. Isoelectric focusing gel of phosphorylated (by the addition of MLCK, lane 2, and dephosphorylated, lane 1) myofibrillar lysate of psoas muscle stained with Pro Q showed that phosphorylated RLC band was 1.52 times stronger than the dephosphorylated band.
Isoelectric focusing.
To assess the degree of phosphorylation, muscle proteins were analyzed by isoeletric focusing. Total protein concentration was analyzed by the Bradford assay after homogenizing phosphorylated and dephosphorylated muscle in 8 M urea. For isolectric focusing, a sample was diluted to 2.5 mg/ml in a buffer [9 M urea, 130 mM DTT, 20% glycerol, 250 μl of 40% carrier ampholines pH 4–6 (Bio-Rad, Hercules, CA), 10% Triton X-100]. Denaturing isoelectric focusing gel (10 M urea, 18% acrylamide/bis-acrylamide, 20% glycerol, 10% Triton-X 100, 1 ml of 40% ampholines, 60 μl of 10% APS, and 30 μl of TEMED) was run overnight at constant 350 V and fixed with 7% TCA, 50% methanol for 2 h following the procedure of Cooke and colleagues (56, 67). The gel was washed with double-distilled water for 10 min and stained with Diamond Pro-Q phosphoprotein gel stain (Invitrogen) for 3 h in the dark. It was destained with 50 mM sodium acetate and 20% acetonitrile by shaking at room temperature for at least 30 min.
Expressing LC1.
Recombinant vector (pQE60) containing LC1 with a single cysteine residue (Cys178) was donated by Dr. Susan Lowey (University of Vermont). It was transfected into Escherichia coli M15-competent cells. Recombinant clones were selected by resistance to ampicillin. That the sequencing was correct was confirmed by an independent company (Iowa State University of Science and Technology). To overexpress LC1, Luria broth containing 100 μg/ml of ampicillin was used, and induction was achieved with isopropylthiogalactoside. His-tagged LC1 was affinity-purified on a Ni-NTA column. The fractions were eluted by imidazole and were run on SDS-PAGE. This was followed by a Western blot analysis with anti-LCN1 antibodies (Abcam, Cambridge, MA). LC1 fractions were pooled and dialyzed against 50 mM KCl and 10 mM phosphate buffer pH 7.0. SDS-PAGE showed that the dialyzed protein exhibited a single ∼25-kDa band. Protein concentration was determined using the Bradford assay. In some experiments, commercial skeletal human LC1 was used (Prospec, Ness Ziona, Israel).
Choice of the dye.
The fluorophore of choice must be excited in the red to bypass most contributions by autofluorescence (37), and, therefore, the following two dyes were suitable choices: SeTau 647 or Alexa Fluor 647. SeTau was selected mainly because it was more resistant to photobleaching and brighter than Alexa Fluor 647. Fig. 1 compares the photobleaching of a solution of 1 μM SeTau 647 and 0.7 μM Alexa Fluor 647. At these concentrations, both dyes have the same extinction coefficients at the excitation wavelength used (640 nm). Further reasons to select SeTau were that it has a large Stokes shift (44 nm) compared with Alexa Fluor 647 (20 nm) and high extinction coefficient and quantum yield (0.65).
Fig. 1.

Comparison of photobleaching of Alexa Fluor 647 (A) and SeTau (B). The photobleaching occurred in two phases: the rate of the first phase, lasting ∼0.5 s was 10.1 s−1 for Alexa Fluor 647. It was 4.2 times slower for SeTau. The rate of the second phase was 0.036 s−1 for Alexa Fluor 647 and ∼3 times slower for SeTau. Note that the mean count rate of SeTau was 4.2 times larger than that of Alexa Fluor 647. The laser (640 nm) power was 0.9 mW before the objective.
The initial rate of photobleaching was 10.1 s−1 for Alexa Fluor 647 and 2.4 s−1 for SeTau. The diffusion of dye molecules were slowed down by the addition of 80% glycerol to increase the time they spent in the laser beam before diffusing out. Photobleaching is more dramatic in the case of muscle, in which molecules are not merely slowed down, but the same XBs are illuminated for the entire experiment (20 s). The protection from photobleaching of SeTau is achieved by nanoencapsulation of the squaraine moiety of the dye chromophore system in a mixed aliphatic-aromatic macrocycle. Although SeTau has a slightly lower extinction coefficient at 640 nm than Alexa Fluor 647, its overall fluorescence is more than compensated by its high quantum yield (0.65) and by the fact that its quantum yield increases 70% beyond that of Alexa Fluor 647 upon protein binding. All in all, after a 1-s exposure to 640 nm excitation, the fluorescence intensity of SeTau was 4.2 times larger than fluorescence intensity of Alexa Fluor 647. All of these attributes are necessary for extracting the maximum number of photons from a few molecules.
Labeling of LC1.
LC1 (and not RLC) was chosen as the site of labeling to ensure that the phosphorylation site is not disturbed by the dye. Labeling RLC can conceivably disturb the extension of the NH2 terminal of RLC and phosphorylation of Ser-15. It was labeled at position 178 with a 5 molar excess of protein over SeTau overnight on ice. After initial purification by dialysis against 50 mM KCL, 10 mM phosphate buffer pH 7.0, labeled LC1, was passed through a Sephadex G50 LP column to eliminate free dye. The dye and protein were complexed in a 0.2:1 ratio.
Cross-linking.
Contracting myofibrils must be completely still while recording polarized fluorescence. Otherwise, fluctuations could be attributed to the “number” fluctuations, i.e., the change in the number of fluorescent XBs in the detection volume as the myofibril moves through the volume. In addition, we want to avoid muscle shortening during LC1 exchange. Although the exchange solution contains ATP and EGTA, it is possible that some troponin C becomes defective during the exchange and some myofibrils become Ca-insensitive. To completely immobilize myofibrils without affecting their ATPase, we treated myofibrils with a water-soluble cross-linker 1-ethyl-3-[3-(dimethylamino)-propyl]-carbodiimide (EDC) (3, 30, 78). 20 mM EDC was added to 1 mg/ml myofibrils in Ca-rigor solution. Myofibrils were incubated for 20 min at room temperature. The reaction was stopped by adding 20 mM DTT. The pH of the solution (7.5) remained unchanged throughout the 20-min reaction. We measured ATPase by malachite green phosphate assay method, and confirmed that under our conditions X-linking had no effect on ATPase. The absence of shortening was verified by labeling the myofibrils with a 10-nM rhodamine-phalloidin and observing contraction in a TIRF microscope. No shortening was observed.
LC1 exchange into myofibrils.
To ensure a small number of XBs in the DV, we incubated myofibrils under very mild conditions [20 min at 30°C (79)] with very low concentration (5–10 nM) of SeTau-LC1 (the usual exchange takes place at 37°C for 1/2 h.). 1 mg/ml of freshly prepared myofibrils were incubated with fluorescently tagged LC1 in the exchange solution [similar to the one used in (39)]: 15 mM KCl, 5 mM EDTA, 5 mM DTT, 10 mM KH2PO4, 5 mM ATP, 1 mM trifluoperazine (TFP), and 10 mM imidazole (pH 7).
Data collection.
Myofibrils are inefficiently exchanged, as described above, with SeTau-LC1. The 640-nm excitation light beam is focused to the diffraction limit on the overlap band of a myofibril. Fluorescence is collected a few nanoseconds after the initial pulse of exciting light is delivered. Fig. 2 explains how this procedure eliminates the contribution of scattered light: Because scattered light has no fluorescence lifetime, this eliminates the contribution of light scattering from our experiment.
Fig. 2.

Fluorescence decay of parallel polarized light from rigor myofibril. A: no gate applied, i.e., the signal collection is started at the peak of the signal (violet line) and continued through the complete fluorescence decay. This includes scattered light (arrow). B: the residuals have significant dip at the time corresponding to scattering of light and are not well fitted by a straight horizontal line. C: gating applied. In TCSPC systems, photons are collected one-by-one. Here, photons detected within 8.8 ns of the start of the measurement (an arbitrarily determined point in time with relation to the excitation pulse) are eliminated from our measurements. All that remains are photons collected 8.8 ns or later from the start of the measurement (indicated by the violet line). D: residuals of the gated signal are quite flat. The same procedure is applied to perpendicular polarized light.
The laser beam is next redirected to a neighboring half-sarcomere, adjusting the laser power to make sure that each sarcomere provides a similar photon rate. If this power adjustment is not done, the differences between FWHMs of PF become statistically uninterpretable (51) (also see discussion below). The process is repeated 20–30 times, i.e., we obtain 40,000–60,000 PF values from each myofibril.
The detection volume was estimated by measuring the FWHM of an image of 20-nm fluorescent beads in the axial and lateral dimensions, which were 700 nm and 400 nm, respectively. The theoretical detection volume (DV) is equal to (π/2)3/2·(0.400 μm)2·(0.700 μm) = 0.6 μm3. Because of edge effects, the actual DV is 2.8 times greater = 1.7 μm3 (7). The concentration of myosin in muscle is 0.1 mM (1); therefore, the DV contains ∼3×105 myosin XBs. The number of observed (fluorescent) XBs in this volume is estimated to be 4 (because of the inefficient exchange) (see results). A PicoQuant MicroTime 200 confocal system (PicoQuant, Berlin, Germany) coupled to an Olympus IX71 microscope was used to acquire the fluorescence data, as described earlier (51). Before each experiment, fluorescence of an isotropic solution of a dye with long fluorescence lifetime (50 nM rhodamine 700) was measured, after first splitting into the parallel and perpendicular channels. Since rhodamine 700 is small (and, hence, its solution is nearly isotropic), the signal into each detector was attenuated to make sure that they gave identical readings. The PicoQuant is the time-resolved instrument capable of lifetime imaging with single-molecule detection sensitivity. Each photon is recorded individually by the time-correlated single photon counting electronics in time-tagged time-resolved mode. A 640-nm pulsed laser provided linearly polarized excitation parallel to the myofibrillar axis, which was always vertical. Fluorescence was collected 7.36 ns after the beginning of the excitation pulse, or 8.8 ns after the arbitrary chosen start point of the measurements (see above). A 60×, 1.2 NA water immersion objective of the Olympus IX71 collected fluorescent light and passed it through a 650-nm-long pass interference filter before going through the confocal pinhole. A birefringent prism separated the vertically and horizontally polarized fluorescent light, and Avalanche Photodiodes measured the emitted light. Figure 3 shows images of a contracting myofibril.
Fig. 3.

Images of contracting phosphorylated myofibril, effect of gating. A, B, E, and F: ungated images. C, D, G, and H: the same images after gating. The color scale (in FLIM images) and contrast scale (in intensity images) are much improved by gating. The green circle pointed to by the green arrow in A is a projection of the confocal aperture on the sample plane. Top row: images obtained with analyzer perpendicular to the myofibrillar axis. Bottom row: images obtained with analyzer parallel to the myofibrillar axis. Notice that perpendicular images are weaker than parallel images, indicating that a sample is anisotropic. A, C, E, G: FLIM images. B, D, F, H: intensity images. The fluorescent lifetime scales are in nanoseconds, with 1.5 ns corresponding to blue and 3.4 ns to red. The intensity scales are in counts with 30 corresponding to black and 150 to white. Native myofibrillar LC1 was exchanged with 10 nM SeTau-LC1. Scale bar = 5 μm, sarcomere length = 2.1 μm. Sarcomere length does not change during contraction because of cross-linking (30, 78). Images were acquired on a PicoQuant Micro Time 200 confocal lifetime microscope. The sample was excited with a 640-nm pulsed laser and observed through a LP 650 filter.
Statistical analysis.
Origin v.8.6 (Northampton, MA) was used to compute autocorrelation functions and fit the data by a nonlinear (Levenberg-Marquardt) algorithm for χ2 minimization. SigmaPlot 11 (Systat Software, San Jose, CA) was used to compute histograms.
RESULTS
The number of observed myosin molecules.
Fluorescence correlation spectroscopy (FCS) analyzes the fluctuations in the signal as fluorescent molecules in solution diffuse through the detection volume. It is well established that the autocorrelation function of fluctuations at delay time 0 is equal to the inverse of the number of molecules contributing to fluctuations n = 1/ACF(0) (19, 21, 44). Six ACF's were obtained from solutions of the fluorophore in the range of 1.29–77.3 nM. Extrapolating the plot of concentration vs. 1/ACF(0) to 1 molecule revealed that a single molecule of SeTau illuminated with 0.2 μW of laser power corresponded to ∼75 counts per channel [i.e., total fluorescence (Itotal = I‖ + 2* I−) from one molecule of SeTau at 0.2 μW laser power was 225 counts/s (51)]. We use this fact to estimate the number of molecules contributing to the signal. Fig. 4A shows the intensities of perpendicular and parallel channels of a typical contracting myofibril. The polarized intensities in ‖ (ch3) and − (ch2) channels were 2.45 and 1.13 counts/ms, respectively, giving total intensity Itotal = I‖ + 2·I− = 4.71 counts/ms. Therefore, with the power of the laser of 0.2 μW, the number of observed molecules is 20. The actual power used varied between 0.2 and 0.4 μW (to keep the count in ch1 at ∼2 counts/ms), so we estimate that there were between 20 and 40 XBs in the DV. However, it should be emphasized that as long as the number of cross bridges is mesoscopic, the exact number does not matter, i.e., 20 molecules should give the same result as 40 molecules.
Fig. 4.

A: typical time course of intensity of contracting MLCK phosphorylated psoas muscle myofibril. Ch3 (red) and Ch2 (black) are the fluorescence intensities polarized parallel (I‖) and perpendicular (I⊥) to the myofibrillar axis, respectively. The direction of excitation polarization is ‖ to the myofibrillar axis. The gate time was set to 0 ns. B: same time course but with the gate set to 7.36 ns. Note the decrease of intensity due to elimination of light scattering. C: polarization of fluorescence of ungated (blue) and gated (green) at 7.36-ns signals. To emphasize the differences, the ungated signal is plotted on top of the gated signal in D. Laser intensity was 0.2–0.4 μW.
Approximately 20 rotating cross bridges imply that the fluctuating signal is 22% of the mean. The precision of measurement is approximately equal to 1/√X, where X is the number of fluctuations detected. Therefore, to get 1% precision we have to analyze 10,000 fluctuations. The characteristic life-time of fluctuation due to rotations of the lever arm is of the order of milliseconds (34). Therefore, 10,000 fluctuations will occur in 10 s. We collected the data over 20 s.
Kinetics.
XBs constantly change orientation while undergoing the mechanochemical cycle, thus causing fluctuations of PF. PF fluctuate about their equilibrium values with certain rate constants. The transitions between various enzymatic and mechanical states of muscle are governed by a Poisson process. A single fluctuation cannot predict a chemical rate constant, but the rates of many fluctuations are the same as rate constants that govern macroscopic dynamic processes (58). For certain processes, the effect of fluctuations on the rate constants were analytically predicted (22). Here, we attempt to predict rate constants from fluctuation caused by a rotating XB. An autocorrelation function, ACF(τ), of FP fluctuations accomplishes this when fluctuations are due only to XBs. By definition, ACF(τ) = <δFP(t)δFP(t+τ)> where < > denote averaging over a long time period (20). The system is in a steady state [dependence of ACF(τ) on t is eliminated]. Fluctuations of PF as in Fig. 3D can arise from 1) random noise, 2) a change in the number of fluorophores in the DV (22, 44), and 3) a change of orientation of the SeTau transition dipole imbedded in LC1. The contribution 1) to fluctuations is eliminated because random noise is uncorrelated. The contribution 2) is not significant because each myofibril is cross-linked and does not change in length, so no fluorophores can enter or leave DV during contraction. In any case, a change in signal can only be caused by rotation, and myofibrils do not twist during contraction. We conclude that the fluctuations arise solely because of a change in orientation of the SeTau transition dipole embedded in LC1. The autocorrelation function is initially large because the average product of a fluctuation amplitude at some time to and at short time later, τ, must be large, because the amount of fluctuation at time to [δF(to)] is not much different than δF(τ). The average of the product [δF(to)δF(t+τ)] is close to its maximal value of [δF(τ)]2. Since the signal average is 0, when τ is large, the amplitude is likely to be the same above and below the average, i.e., on the average δF(to)=−δF(τ). Therefore, the autocorrelation function at long τ tends to decay to 0. The rate of decay of autocorrelation function from maximal value to 0 is a reflection of how quickly the average fluctuation crosses the 0 baseline.
The exact shape of the autocorrelation function depends on the model assumed for the chemical reaction that drives the cycling of the XBs. In the past, we used a simple ON-OFF model, where cross bridges were either immobilized (attached) to thin filaments or mobile (detached) from thin filaments (51). Here, we use a more realistic model. The model must include transitions between three fundamental states as illustrated in Fig. 5. Each state has to be characterized in terms of mobility of LC1-SeTau. This is done by measuring the time 0 anisotropy, steady-state anisotropy, and the lifetime of SeTau in each of the transient states. A FluoTime 200 fluorometer (PicoQuant) was used, and measurements were done at room temperature (50). The sample was excited by a 635-nm pulsed (at 20 MHz) diode laser and observed through a 670-nm monochromator with an additional 650-nm long-pass filter. The time resolution was <10 ps, and the FWHM of the pulse response function was less than 100 ps. The anisotropy decays and lifetimes were analyzed by a multiexponential model using FluoFit software (PicoQuant). Typical records of anisotropy decay of SeTau coupled to LC1 showed that the anisotropy decay was composed entirely of a single exponential decay. Records of SeTau-LC1 coupled to myofibrils gave steady state anisotropy = 0.071.
Fig. 5.
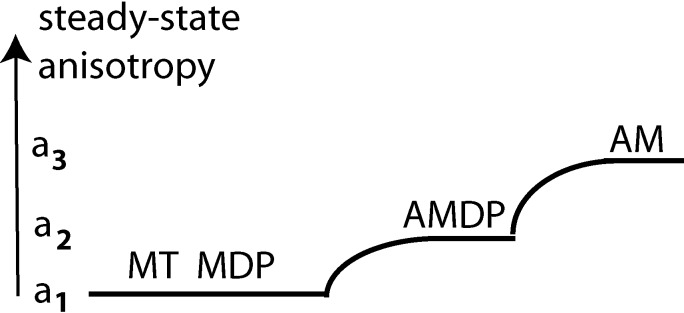
The three-state model of muscle contraction. M, myosin; D, ADP; P, inorganic phosphate; A, F-actin; T, ATP. The polarizations of fluorescence associated with different cross bridge states. a1, a2 and a3 are amplitude of polarizations.
The cycle begins with a prehydrolytic state MT, in which myosin (M) has dissociated from thin filament by binding ATP (T), which is hydrolyzed to assume a posthydrolytic MDP state (43). After hydrolysis, the products remain bound to M. The MDP state is characterized by partially open cleft between upper and lower subdomains of the 50-KDa domain, as well as a lever arm in the UP position (13). This complex, as well as MT, are free to rotate in the myofilament space and have low steady-state anisotropy a1 (Table 1). Next, the XB binds to actin to assume a prepower stroke state AMD, which becomes partially immobilized. Table 1 shows that its steady state anisotropy becomes 0.116,2 which is represented in Fig. 5 by value of a2. The AMDP state is characterized by a partially open cleft between the upper and lower subdomains of a 50-kDa domain and a lever arm in the UP position (13). Next, dissociation of P from AMDP state initializes a XB power stroke involving transition to a rigor-like AMD state and a final rigor (AM) state. This state has the highest steady-state anisotropy a3. AMD and AM states are characterized by a closed cleft and the lever arm in the DOWN position. Table 1 shows that the absence of a nucleotide further increases steady-state anisotropy. Transition from AMDP to AM is a power stroke. This is represented in Fig. 5 as a transition a2 to a3. This may occur in several steps (13, 34). Finally, the XB rapidly dissociates from actin by binding fresh molecule of ATP, (assuming low anisotropy a1), after which the cycle repeats itself.
Table 1.
Anisotropies of intermediate states of XB cycle
| Time zero anisotropy, Ro | Steady-State Anisotropy, a | Correlation Time (ns) | |
|---|---|---|---|
| LC1 | 0.325 | 0.077 | 0.777 |
| AM | 0.225 | 0.210 | 1.484 |
| MT | 0.232 | 0.071 | 0.780 |
| AMDP | 0.236 | 0.116 | 0.999 |
XB, cross bridge; LC1, light chain 1; AM, actin-myosin; MT, myosin with ATP; AMDP, actin-myosin with ADP ATP; ns, not significant.
Table 1 shows that the dye does not need to be attached at two points to the light chain, although the procedure described by Corrie et al. (12) is more attractive. Of course, it needs to have a well-defined PF in different enzymatic and mechanical states of the muscle.
The autocorrelation function of such a system is (47):
| (1) |
| (2) |
| (3) |
Origin 8.6 was used to fit the experimental data (circles) to the R3. The red lines in Fig. 6 are the best nonlinear fit to Eq. 1. Shown are four representative traces from 28 experiments on MLCK-phosphorylated muscle. Fluoride has a strong effect on enzymatic properties of myosin (8, 45, 62, 64). To avoid using fluoride to inhibit dephosphorylation, phosphorylation was achieved by adding MLCK, as described in the materials and methods.
Fig. 6.

Representative traces of normalized autocorrelation functions of polarization of fluorescence of contracting MLCK phosphorylated psoas myofibril. Circles are experimental data, and a red line is the fit to Eq. 1. The fact that the correlation decays in time indicates that the orientation of absorption-emission dipoles change in time. The fact that ACF decays to a value >0 is due to the fact that mean polarization was nonzero (approximately −0.23). Delay time is in seconds.
The data could not be fitted with two-state (ON and OFF) mode. The fit was not so good for dephosphorylated myofibrils. The average values of a and k were determined by experiment. It remains to assign experimental k values to the model. The average values are summarized in Table 2.
Table 2.
Experimental values of the kinetic coefficients from 26 experiments on dephosphorylated and phosphorylated XBs in contracting psoas myofibrils
| Myofibrils | k1, s−1 | k2, s−1 | k3, s−1 |
|---|---|---|---|
| Dephosphorylated | 961 ± 560 | 1.9 ± 2.7 | 0.19 ± 0.14 |
| MLCK phosphorylated | 689 ± 484 | 0.30 ± 0.30 | 0.18 ± 0.20 |
MLCK, myosin light-chain kinase.
The assignment of k1 is obvious: it is the fastest of the processes; therefore, it must be myosin head dissociation from thin filaments: AM → MT. k2 and k3 are symmetrical in Eqs. 1–3. However, the rate of myosin binding to actin is more likely to be higher than the rate in the power stroke. Since the rate k2 is higher (at least for dephosphorylated muscle) than k3, we assign k2 as the rate of myosin head binding to actin and k3 as the power stroke rate.
None of the differences in ks of phosphorylated and dephosphorylated myofibrils were statistically significant, primarily because the SD of measurements was so large. The difference in k1 of −261.782 was not statistically significant (t = −1.625, P = 0.112 with 41 degrees of freedom). The 95% confidence interval for difference of means was −543.615 to 128.311. The difference in k2 of −1.642 was not statistically significant at 95% confidence level. The difference in k3 of −0.0122 was not statistically significant (t = −0.223 with 40 degrees of freedom, P = 0.824). The 95 percent confidence interval for difference of means was −0.123 to 0.0986.
We conclude that phosphorylation of RLC induces no obvious changes in the mechanochemical cycle of myosin. We can only say that, under ex vivo conditions, the mean rate of XB dissociation of actin is 825 s−1, the mean rate of myosin binding to thin filaments is 1.1 s−1, and the mean rate of the power stroke is 0.19 s−1.
Distribution of XB orientations.
The easiest way to characterize spatial distributions of the transition dipole orientations of the dye is to plot polarization values vs. the number of times that a given orientation occurs during a 20-s experiment. Fig. 7 shows representative probability distributions of dephosphorylated myofibrils.
Fig. 7.

Selected examples of the probability distributions of orientations of cross bridges (XBs) of contracting myofibrils containing dephosphorylated RLC. Dephosphorylation was affected by the addition of EGTA to WT myofibrils.
The difference in FWHM, while small in absolute terms (6%), was statistically significant (P = 0.012, t = 2.634 with 45 degrees of freedom). The difference of the mean polarization of −0.107 was also statistically significant (t = −10.209 with 45 degrees of freedom, P = <0.001). 95 percent confidence interval for difference of means was −0.128 to −0.0859. We conclude that phosphorylation makes a small difference to the degree of order of active cross bridges (Table 3).
Table 3.
Comparison of FWHMs of 28 probability distributions of phosphorylated and dephosphorylated myofibrils
| Myofibrils | FWHM | Mean Polarization | Mean Intensity, counts/ms |
|---|---|---|---|
| Dephosphorylated | 0.47 ± 0.04 | −0.13 ± 0.03 | 2.305 ± 0.336 |
| MLCK phosphorylated | 0.50 ± 0.03 | −0.23 ± 0.03 | 1.416 ± 0.387 |
FWHM, full-width half-maximum.
DISCUSSION
We studied the effect of phosphorylation on skeletal cross-bridge kinetics and the distribution of orientations during steady-state contraction of muscle. By sacrificing the idea of measuring the signal from an individual XB and instead concentrating on detecting the signal from a few molecules, we gained the ability to increase the time resolution of ex vivo measurements in muscle to a few milliseconds. To achieve this, we exchanged fluorescent LC1 with native LC1 of myofibrils under exceedingly mild conditions (see materials and methods). Such mild labeling required the use of a microscope with single-molecule sensitivity such as the PicoQuant MicroTime 200. In a typical experiment, we observed about 20 XBs. Minimization of inhomogeneities was possible by focusing the exciting laser light on a single half-sarcomere. The dye of choice was SeTau, which was excited in the red and observed in the far red. Using SeTau dye decreased photobleaching, increased signal intensity, and decreased autofluorescence. The fluorescent signal was gated, assuring no interference from scattering.
Kinetics.
Determination of k1, k2, and k3. Dissociation of the myosin head from the thin filaments is the most rapid process occurring during contraction. Since dissociation is the only rapid process detected by our method, it was natural to assign it to k1. The SD of the means of phoshorylated and dephosphorylated myofibrils was so large as to make the differences between the means statistically insignificant. The average value of k1 was 825 s−1. Similarly, the large value of the SD of the means of k2 of dephosphorylated myofibrils was so large as to make the difference statistically insignificant. The mean rate of myosin binding to thin filaments was 1.1 s−1. Finally, there was no statistical difference between the rates of power strokes in phosphorylated and dephosphorylated myofibrils k3 (the average rate was 0.19 s−1).
The detection of a few myosin molecules in ex vivo ventricle with millisecond time resolution is not technically easy. The main problem is that the signals are weak. Weak Gaussian signals with low signal-to-noise (S/N) ratio have intrinsically large relative FWHM, and this applies as well to the ratio of these signals, such as polarization of fluorescence, as shown by (51). In addition, there are at least five reasons for such a large FWHM or SD of measurements: 1) Nonuniform incorporation of LC1 into sarcomeres (Fig. 3); 2) incomplete degree of phosphorylation of myofibrils (Supplemental Fig. S1); 3) low steady-state anisotropy of SeTau and relatively small differences in anisotropy between different mechochemical steps (Table 1); 4) large variability between samples (Ventricles are prepared in Florida and stored for various periods at −80°C). They are then shipped on ice to Texas and stored for a few days at −20°C before the experiment is performed. Each of these steps imposes some variability on the quality of the sample; and 5) the fitting program must estimate six parameters. This allows it a great freedom in selecting the best fit. Most of these reasons are beyond our control; e.g., nonuniformity of muscle labeling (#1) is caused by extremely low concentration of the dye, #3 is an intrinsic property of SeTau and replacing it with Alexa Fluor 647 would result in even smaller S/N, #5 could have been reduced by enforcing limits on the “a” values, but it would have imposed additional arbitrary elements on the model. More experiments are needed to decrease SD, e.g., to make more measurements at room temperature, perform additional measurements at 37°C, and better control #2 and #4.
No effect of phosphorylation of RLC is consistent with the earlier conclusion of Metzger et al. (49) that phosphorylation of RLC plays no role in the rate of tension redevelopment and maximum isometric tension at saturating concentrations of Ca2+. It is also consistent with the earlier result from this laboratory, which suggested that the duty cycle was unaffected by phosphorylation (51), suggesting that isometric force was not changed by phosphorylation of RLC. Also, no effect on force was observed at saturating Ca2+ concentrations in skeletal (73) and cardiac (55) muscle, consistent with earlier results (4). The fact that the rate was so slow leads to the conclusion that prevention of shortening by cross-linking slows down, but does not prevent XBs from executing power strokes, suggesting that they can reach neighboring target zones. If they were unable to reach neighboring target sites, no fluctuations due to rotation of XBs would have occurred, i.e., ACF would have been flat. The mechanical cycle goes on in spite of cross-linking, like the enzymatic cycle (29). Further, we can conclude that XBs act independently from each other, i.e., they are unsynchronized. If they were synchronized, the ACF would be periodic, because the autocorrelation function of a periodic signal is periodic (5).
Overall, our kinetic results suggest that phosphorylation at saturating [Ca2+] has no effect on maximal isometric tension (59, 68, 71). The situation is different at low [Ca2+] at which phosphorylation is able to increase force because the fraction of XBs in force generating state is low (6, 70). The situation is also different in muscles carrying familiar hypertrophy mutations, which show obvious differences in kinetics compared with wild-type muscles (18, 27, 28, 36, 47, 48, 52) and even between α-and β-cardiac myosins (40).
Distribution.
Comparison of the probability distributions of orientations of XBs containing phosphorylated and dephosphorylated RLC can be misleading unless the means of distributions are the same (51). This is because the relative FWHM of a distribution3 is sharply dependent on the strength of the signal4. Because of the stochastic nature of the Gaussian signal, a small mean is associated with a broad distribution (large relative FWHM) and a large mean is associated with a narrow distribution (small relative FWHM). The same is true for the ratio of two Gaussian signals (like polarization of fluorescence). Therefore, if the means are different, an impression is created that the distributions are different. Meaningful comparison of FWHMs or SDs of the ratio of two Gaussian signals requires that they have nearly equal means. (It is not always possible to achieve exactly the same mean, especially when the signal is as weak as in our case).
The absence of an effect from phosphorylation on the kinetic rate and FWHM is illustrated schematically in Fig. 8. The white, blue, and red spheres symbolize LC1, and phosphorylated and dephosphorylated RLC, respectively. The transition dipole of SeTau is shown as a white arrow. The violet cones represent a three-dimensional Gaussian distribution of SeTau transition moments. The absence of an effect on kinetics is represented by the fact that the left (phosphorylated) XB is in the same transitional state (here dissociated from thin filament) as the right (dephosphorylated) one. The small effect on FWHM is represented by the fact that the violet cone within which phosphorylated XB rotates is slightly (6%) more open.
Fig. 8.

Schematic representation of the fact that phosphorylation of RLC has no effect on kinetics and minimal effect on distribution of XBs. Phosphorylated and dephosphorylated RLC are shown as blue and red spheres, respectively. The white sphere is LC1, and white arrows are the transition dipoles of the dye. The violet cone is the cone of angles within which the transition dipole fluctuates. The thick filament is shown in dark blue, actin filament in yellow, and myosin neck in green.
We have previously found that relaxed XBs of muscle in which RLCs were dephosphorylated were organized more tightly than XBs of muscle in which RLCs were phosphorylated (51). We also found small differences in the kinetics and orientational distribution of XBs. The differences observed in active XBs are likely due to the fact that in earlier work, muscles were phosphorylated by the addition of phosphatase inhibitors. These contain a high concentration of fluoride that may affect the kinetics and distribution of XBs during contraction. Also, the kinetic constants were obtained from a two-state (ON-OFF) model, which may be too great of an oversimplification.
Perspectives and Significance
There is convincing evidence that in cardiac muscle, phosphorylation enrichment increases isometric force and peak power output and both maximum shortening speed and the shortening velocity at peak power. This suggests that reduced RLC phosphorylation is a key aspect of impaired contractile function in the diseased myocardium (e.g., Refs. 15 and 75). The future research on this subject must, therefore, concentrate on the role of phosphorylation of cardiac RLC. Phosphorylation of skeletal RLC is perhaps a remaining vestige of evolution of cardiac myosin. The mechanical changes observed earlier [e.g., (10, 46, 72)] are perhaps inevitable consequences of adding a negative charge to the lever arm of myosin.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.D., J.N., R.R., and K.M. performed experiments; J.N., R.R., R.F., and I.G. edited and revised manuscript; R.R. and J.B. interpreted results of experiments; R.R., R.F., and J.B. prepared figures; K.M., I.G., and J.B. analyzed data; I.G. and J.B. conception and design of research; J.B. drafted manuscript; J.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. B. Maliwal for useful comments. This article was supported by the National Institutes of Health Grants R01AR-048622 (to J. Borejdo) and R01HL-090786 (to J. Borejdo and D. Szczesna).
Footnotes
In principle, kinetic behavior can be deduced from a large number of molecules by synchronizing them by applying transients [e.g., tension transients (14, 34)]. The Onsager principle predicts that observations of the relaxation of macroscopic concentration gradients yields the same chemical rate constants as the statistically averaged time course of the fluctuations as used in mesoscopic measurements, but imposing rapid gradients disturbs steady state and is technically difficult without introducing extra series of elasticity and inhomogeneities.
Notice that time zero anisotropy of AMDP complex is diminished. We do not understand why this is so. Perhaps the electronic structure of the dye has changed.
Since the distribution is normal, FWHM = 2Sqrt(2ln2) = 2.355 SD.
In practice, we ensure that the average photon counts in each series of experiments are similar by keeping the photon rate constant by adjusting the power of the illuminating laser.
REFERENCES
- 1.Bagshaw CR. Muscle Contraction. London: Chapman & Hall, 1982 [Google Scholar]
- 2.Beausang JF, Sun Y, Quinlan ME, Forkey JN, Goldman YE. Orientation and rotational motions of single molecules by polarized total internal reflection fluorescence microscopy (polTIRFM). Cold Spring Harb Protoc 2012, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bershitsky SY, Tsaturyan AK, Bershitskaya ON, Mashanov GI, Brown P, Burns R, Ferenczi MA. Muscle force is generated by myosin heads stereospecifically attached to actin. Nature 388: 186–190, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Borejdo J, Midde K. Rapid measurements of orientation and rotation of a small number of cross-bridges in ex vivo muscle. In: Advanced Fluorescence Microscopy Techniques, edited by Conn M, New York: Elesevier, In press [Google Scholar]
- 5.Bracewell R. The Fourier Transform and Its Applications. New York: McGraw-Hill, 1965 [Google Scholar]
- 6.Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA 85: 3265–3269, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buschmann V, Krämer B, Koberling F. Quantitative FCS: Determination of confocal volume by FCS and bead scanning with MicroTime 200. PicoQuant, Application Note Quantitative FCS v 1.1, 2009 [Google Scholar]
- 8.Chase PB, Martyn DA, Kushmerick MJ, Gordon AM. Effects of inorganic phosphate analogues on stiffness and unloaded shortening of skinned muscle fibres from rabbit. J Physiol 460: 231–246, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colson BA, Locher MR, Bekyarova T, Patel JR, Fitzsimons DP, Irving TC, Moss RL. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J Physiol 588: 981–993, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooke R. Modulation of the actomyosin interaction during fatigue of skeletal muscle. Muscle Nerve 36: 756–777, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Cooke R. The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys Rev 3: 33–45, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrie JE, Craik JS, Munasinghe VR. A homobifunctional rhodamine for labeling proteins with defined orientations of a fluorophore. Bioconjug Chem 9: 160–167, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Coureux PD, Sweeney HL, Houdusse A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J 23: 4527–4537, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dantzig JA, Higuchi H, Goldman YE. Studies of molecular motors using caged compounds. Methods Enzymol 291: 307–348., 1998 [DOI] [PubMed] [Google Scholar]
- 15.Dias FA, Walker LA, Arteaga GM, Walker JS, Vijayan K, Pena JR, Ke Y, Fogaca RT, Sanbe A, Robbins J, Wolska BM. The effect of myosin regulatory light chain phosphorylation on the frequency-dependent regulation of cardiac function. J Mol Cell Cardiol 41: 330–339, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Dos Remedios CG, Millikan RG, Morales MF. Polarization of tryptophan fluorescence from single striated muscle fibers. A molecular probe of contractile state. J Gen Physiol 59: 103–120, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dos Remedios CG, Yount RG, Morales MF. Individual states in the cycle of muscle contraction. Proc Natl Acad Sci USA 69: 2542–2546, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumka D, Talent J, Akopova I, Guzman G, Szczesna-Cordary D, Borejdo J. E22K mutation of RLC that causes familial hypertrophic cardiomyopathy in heterozygous mouse myocardium: effect on cross-bridge kinetics. Am J Physiol Heart Circ Physiol 291: H2098–H2106, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Elson EL. Fluorescence correlataion spectroscopy and photobleaching recovery. Annu Rev Phys Chem 36: 379–406, 1985 [Google Scholar]
- 20.Elson EL. Fluorescence correlation spectroscopy: past, present, future. Biophys J 101: 2855–2870, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elson EL. Introduction to FCS. Fort Worth, TX: University of North Texas, 2007 [Google Scholar]
- 22.Elson EL, Magde D. Fluorescence correlation spectroscopy: conceptual basis and theory. Biopolymers 13: 1–28, 1974 [DOI] [PubMed] [Google Scholar]
- 23.Elson EL, Webb WW. Concentration correlation spectroscopy: a new biophysical probe based on occupation number fluctuations. Annu Rev Biophys Bioeng 4: 311–334, 1975 [DOI] [PubMed] [Google Scholar]
- 24.Endo M. Professor Ebashi's journey toward the discovery of troponin: a personal recollection. Adv Exp Med Biol 592: 7–9, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Forkey JN, Quinlan ME, Shaw MA, Corrie JE, Goldman YE. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature 422: 399–404, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Greenberg MJ, Mealy TR, Jones M, Szczesna-Cordary D, Moore JR. The direct molecular effects of fatigue and myosin regulatory light chain phosphorylation on the actomyosin contractile apparatus. Am J Physiol Regul Integr Comp Physiol 298: R989–R996, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenberg MJ, Watt JD, Jones M, Kazmierczak K, Szczesna-Cordary D, Moore JR. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol 46: 108–115, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, Wang Y, Kerrick WG, Potter JD. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem 280: 37183–37194, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Herrmann C, Lionne C, Travers F, Barman T. Correlation of ActoS1, myofibrillar, and muscle fiber ATPases. Biochemistry 33: 4148–4154, 1994 [DOI] [PubMed] [Google Scholar]
- 30.Herrmann C, Sleep J, Chaussepied P, Travers F, Barman T. A structural and kinetic study on myofibrils prevented from shortening by chemical cross-linking. Biochemistry 32: 7255–7263, 1993 [DOI] [PubMed] [Google Scholar]
- 31.Hooijman P, Stewart MA, Cooke R. A new state of cardiac Myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J 100: 1969–1976, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hopkins SC, Sabido-David C, Corrie JE, Irving M, Goldman YE. Fluorescence polarization transients from rhodamine isomers on the myosin regulatory light chain in skeletal muscle fibers. Biophys J 74: 3093–3110, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopkins SC, Sabido-David C, van der Heide UA, Ferguson RE, Brandmeier BD, Dale RE, Kendrick-Jones J, Corrie JE, Trentham DR, Irving M, Goldman YE. Orientation changes of the myosin light chain domain during filament sliding in active and rigor muscle. J Mol Biol 318: 1275–1291, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Huxley AF, Simmons RM. Proposed mechanism of force generation in striated muscle. Nature 233: 533–538, 1971 [DOI] [PubMed] [Google Scholar]
- 35.James J, Robbins J. Signaling and myosin-binding protein C. J Biol Chem 286: 9913–9919, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerrick WG, Kazmierczak K, Xu Y, Wang Y, Szczesna-Cordary D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J 23: 855–865, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lakowicz JR. Principles of Fluorescence Spectroscopy: New York: Springer, 2006 [Google Scholar]
- 38.Levine RJ, Kensler RW, Yang Z, Sweeney HL. Myosin regulatory light chain phosphorylation and the production of functionally significant changes in myosin head arrangement on striated muscle thick filaments. Biophys J 68: 224S, 1995 [PMC free article] [PubMed] [Google Scholar]
- 39.Ling N, Shrimpton C, Sleep J, Kendrick-Jones J, Irving M. Fluorescent probes of the orientation of myosin regulatory light chains in relaxed, rigor, and contracting muscle. Biophys J 70: 1836–1846, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM. Transgenic mouse alpha-and beta-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. J Biol Chem 288: 14780–14787, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu H, Ali MY, Bookwalter CS, Warshaw DM, Trybus KM. Diffusive movement of processive kinesin-1 on microtubules. Traffic 10: 1429–1438, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H, Kennedy GG, Warshaw DM, Trybus KM. Simultaneous observation of tail and head movements of myosin V during processive motion. J Biol Chem 285: 42068–42074, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lymn RW, Taylor EW. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10: 4617–4624, 1971 [DOI] [PubMed] [Google Scholar]
- 44.Magde D, Elson EL, Webb WW. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13: 29–61, 1974 [DOI] [PubMed] [Google Scholar]
- 45.Maruta S, Homma K, Ohki T. Conformational changes at the highly reactive cystein and lysine regions of skeletal muscle myosin induced by formation of transition state analogues. J Biochem 124: 578–584, 1998 [PubMed] [Google Scholar]
- 46.Matsubara I. X-ray diffraction studies of the heart. Annu Rev Biophys Bioeng 9: 81–105, 1980 [DOI] [PubMed] [Google Scholar]
- 47.Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, Zhao J, Szczesna-Cordary D, Borejdo J. Cross-bridge kinetics in myofibrils containing familial hypertrophic cardiomyopathy R58Q mutation in the regulatory light chain of myosin. J Theor Biol 284: 71–81, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mettikolla P, Luchowski R, Gryczynski I, Gryczynski Z, Szczesna-Cordary D, Borejdo J. Fluorescence lifetime of actin in the FHC transgenic heart. Biochemistry 48: 1264–1271, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metzger JM, Greaser ML, Moss RL. Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibers. Implications for twitch potentiation in intact muscle. J Gen Physiol 93: 855–883, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Midde K, Rich R, Hehreiter V, Raut S, Luchowski R, Hinze C, Fudala R, Gryczynski I, Gryczynski Z, Borejdo J. Rotation of myosin lever arms during isometric contraction of skeletal myofibrils. In: Skeletal Muscle: Physiology, Classification and Disease, edited by Willems M. Chichester, United Kingdom Nova Science Publishers, 2012 [Google Scholar]
- 51.Midde K, Rich R, Marandos P, Fudala R, Li A, Gryczynski I, Borejdo J. Orientation and rotational motion of cross-bridges containing phosphorylated and dephosphorylated myosin regulatory light chain. J Biol Chem 288: 7012–7023, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang Y, Xu Y, Kerrick WG, Potter JD. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem 276: 3743–3755, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem 276: 10577–10580, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Morales MF. Calculation of the polarized fluorescence from a labeled muscle fiber. Proc Nat Acad Sci USA 81: 145–149, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muthu P, Kazmierczak K, Jones M, Szczesna-Cordary D. The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. J Cell Mol Med 10: 1582–4934, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naber N, Cooke R, Pate E. EPR spectroscopy shows oriented myosin heads in relaxed muscle fibers. Biophys J 100: 130a–130a, 2011 [Google Scholar]
- 57.Nihei T, Mendelson RA, Botts J. Use of fluorescence polarization to observe changes in attitude of S1 moieties in muscle fibers. Biophys J 14: 236–242, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Onsager L. Reciprocal relations in irreversible processes. Phys Rev 37: 405–426, 1931 [Google Scholar]
- 59.Persechini A, Stull JT, Cooke R. The effect of myosin phosphorylation on the contractile properties of skinned rabbit skeletal muscle fibers. J Biol Chem 260: 7951–7954, 1985 [PubMed] [Google Scholar]
- 60.Qiu W, Derr ND, Goodman BS, Villa E, Wu D, Shih W, Reck-Peterson SL. Dynein achieves processive motion using both stochastic and coordinated stepping. Nat Struct Mol Biol 19: 193–200, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan ME, Forkey JN, Goldman YE. Orientation of the myosin light chain region by single molecule total internal reflection fluorescence polarization microscopy. Biophys J 89: 1132–1142, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raucher D, Fajer PG. Orientation and dynamics of myosin heads in aluminum fluoride induced pre-power stroke states: an EPR study. Biochemistry 33: 11993–11999, 1994 [DOI] [PubMed] [Google Scholar]
- 63.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan R. Structure of the actin-myosin complex and its implications for muscle contraction. Science 261: 58–65, 1993 [DOI] [PubMed] [Google Scholar]
- 64.Regnier M, Chase PB, Martyn DA. Contractile properties of rabbit psoas muscle fibres inhibited by beryllium fluoride. J Muscle Res Cell Motil 20: 425–432, 1999 [DOI] [PubMed] [Google Scholar]
- 65.Ross JL, Shuman H, Holzbaur EL, Goldman YE. Kinesin and dynein-dynactin at intersecting microtubules: motor density affects dynein function. Biophys J 94: 3115–3125, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sabido-David C, Hopkins SC, Saraswat LD, Lowey S, Goldman YE, Irving M. Orientation changes of fluorescent probes at five sites on the myosin regulatory light chain during contraction of single skeletal muscle fibres. J Mol Biol 279: 387–402, 1998 [DOI] [PubMed] [Google Scholar]
- 67.Stewart MA, Franks-Skiba K, Chen S, Cooke R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci USA 107: 430–435, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stull JT, Kamm KE, Vandenboom R. Myosin light chain kinase and the role of myosin light chain phosphorylation in skeletal muscle. Arch Biochem Biophys 510: 120–128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun Y, Sato O, Ruhnow F, Arsenault ME, Ikebe M, Goldman YE. Single-molecule stepping and structural dynamics of myosin X. Nat Struct Mol Biol 17: 485–491, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sweeney HL, Stull JT. Alteration of cross-bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin-myosin interaction. Proc Natl Acad Sci USA 87: 414–418, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sweeney HL, Stull JT. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am J Physiol Cell Physiol 250: C657–C660, 1986 [DOI] [PubMed] [Google Scholar]
- 72.Sweeney HL, Yang Z, Zhi G, Stull JT, Trybus KM. Charge replacement near the phosphorylatable serine of the myosin regulatory light chain mimics aspects of phosphorylation. Proc Natl Acad Sci USA 91: 1490–1494, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szczesna D, Zhao J, Jones M, Zhi G, Stull J, Potter JD. Phosphorylation of the regulatory light chains of myosin affects Ca2+ sensitivity of skeletal muscle contraction. J Appl Physiol 92: 1661–1670, 2002 [DOI] [PubMed] [Google Scholar]
- 74.Telley IA, Stehle R, Ranatunga KW, Pfitzer G, Stussi E, Denoth J. Dynamic behaviour of half-sarcomeres during and after stretch in activated rabbit psoas myofibrils: sarcomere asymmetry but no ‘sarcomere popping’. J Physiol 573: 173–185, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toepfer C, Caorsi V, Kampourakis T, Sikkel MB, West TG, Leung MC, Al-Saud SA, MacLeod KT, Lyon AR, Marston SB, Sellers JR, Ferenczi MA. Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. J Biol Chem 288: 13446–13454, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tominaga M, Kojima H, Yokota E, Nakamori R, Anson M, Shimmen T, Oiwa K. Calcium-induced mechanical change in the neck domain alters the activity of plant myosin XI. J Biol Chem 287: 30711–30718, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tregear RT, Mendelson RA. Polarization from a helix of fluorophores and its relation to that obtained from muscle. Biophys J 15: 455–467, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsaturyan AK, Bershitsky SY, Burns R, Ferenczi MA. Structural changes in the actin-myosin cross-bridges associated with force generation induced by temperature jump in permeabilized frog muscle fibers. Biophys J 77: 354–372, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ushakov DS, Caorsi V, Ibanez-Garcia D, Manning HB, Konitsiotis AD, West TG, Dunsby C, French PM, Ferenczi MA. Response of rigor cross-bridges to stretch detected by fluorescence lifetime imaging microscopy of myosin essential light chain in skeletal muscle fibers. J Biol Chem 286: 842–850, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warshaw DM, Hayes E, Gaffney D, Lauzon AM, Wu J, Kennedy G, Trybus K, Lowey S, Berger C. Myosin conformational states determined by single fluorophore polarization. Proc Natl Acad Sci USA 95: 8034–8039, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yildiz A, Park H, Safer D, Yang Z, Chen LQ, Selvin PR, Sweeney HL. Myosin VI steps via a hand-over-hand mechanism with its lever arm undergoing fluctuations when attached to actin. J Biol Chem 279: 37223–37226, 2004 [DOI] [PubMed] [Google Scholar]
- 82.Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science 303: 676–678, 2004 [DOI] [PubMed] [Google Scholar]