

Abstract
Recent studies indicate that the lateral subdivision of the central amygdala (CeL) is essential for fear learning. Specifically, fear conditioning induces cell-type-specific synaptic plasticity in CeL neurons that is required for the storage of fear memories. The CeL also controls fear expression by gating the activity of the medial subdivision of the central amygdala (CeM), the canonical amygdala output to areas that mediate defensive responses. In addition to the connection with CeM, the CeL sends long-range projections to innervate extra-amygdala areas. However, the long-range projection CeL neurons have not been well characterized, and their role in fear regulation is unknown. Here we show in mice that a subset of CeL neurons directly project to the midbrain periaqueductal gray (PAG) and the paraventricular nucleus of the thalamus, two brain areas implicated in defensive behavior. These long-range projection CeL neurons are predominantly somatostatin-positive (SOM+) neurons, which can directly inhibit PAG neurons, and some of which innervate both the PAG and paraventricular nucleus of the thalamus. Notably, fear conditioning potentiates excitatory synaptic transmission onto these long-range projection CeL neurons. Thus, our study identifies a subpopulation of SOM+ CeL neurons that may contribute to fear learning and regulate fear expression independent of CeM.
Keywords: central amygdala, connectivity, fear conditioning, synaptic plasticity
Introduction
The central amygdala (CeL) is composed of distinct types of GABA-producing inhibitory neurons (Cassell and Gray, 1989; Cassell et al., 1999; Ehrlich et al., 2009; Haubensak et al., 2010), among which the somatostatin-positive (SOM+) neurons are a major population (Cassell and Gray, 1989; Li et al., 2013). These are intermingled with SOM− cells that are predominantly protein kinase C-δ-expressing (PKC-δ+) neurons (Haubensak et al., 2010; Li et al., 2013). SOM+ neurons and PKC-δ+ neurons collectively constitute >90% of the entire CeL population, and the two cell types are mutually inhibitory (Haubensak et al., 2010; Li et al., 2013). We recently discovered that fear conditioning induces cell-type-specific plasticity in the CeL, with excitatory synapses onto SOM+ neurons being strengthened and excitatory synapses onto SOM− neurons being weakened (Li et al., 2013). Notably, preventing the strengthening of excitatory synapses onto SOM+ neurons is associated with impaired fear memory formation, and activation of SOM+ neurons is necessary for fear memory recall and sufficient to drive fear responses. These observations indicate that experience-dependent synaptic potentiation onto SOM+ CeL neurons stores fear memories and enables the expression of conditioned fear responses (Li et al., 2013).
The CeL presumably gates fear expression by tonically inhibiting medial subdivision of the central amygdala (CeM), the major amygdala output to downstream fear effectors that control defensive responses (LeDoux et al., 1988; Johansen et al., 2011). Interestingly, although the SOM+ CeL neurons can inhibit locally within CeL, strongly inhibiting SOM− neurons, they neither send noticeable projections to the CeM nor inhibit CeM neurons (Li et al., 2013). In sharp contrast, the PKC-δ+ CeL neurons strongly project to and inhibit CeM neurons (Haubensak et al., 2010; Li et al., 2013). These findings suggest that activation of SOM+ neurons promotes fear expression, at least in part, by inhibiting PKC-δ+ (SOM−) neurons, thereby disinhibiting CeM. They also suggest that the fear conditioning-induced synaptic modifications in CeL likely underlie the changes in cell firing in CeL and CeM during fear conditioning (Ciocchi et al., 2010; Duvarci et al., 2011).
Another feature of the CeL circuit organization is that some CeL neurons have long-range projections that innervate extra-amygdala structures (Moga and Gray, 1985; Moga et al., 1990; Sun et al., 1991; Sun and Cassell, 1993; Petrovich and Swanson, 1997; Bourgeais et al., 2001). These findings raise the possibility that, in addition to the CeL–CeM pathway, additional channels exist for CeL to participate in fear processing. However, CeL projections to major extra-amygdala structures within the fear circuitry have not been fully examined. Moreover, the role of the long-range projection CeL neurons in fear regulation is unclear. In this study, we identified two long-range projections originating from SOM+ CeL neurons, the CeL–periaqueductal gray (PAG) and CeL–paraventricular thalamus (PVT) projections, which potentially have important roles in controlling defensive responses.
Materials and Methods
Animals.
Mice were group-housed under a 12 h light-dark cycle (7:00 A.M. to 7:00 P.M. light), with food and water freely available. The SOM-IRES-Cre mice were generated as described previously (Taniguchi et al., 2011). All mice were bred onto C57BL/6J genetic background. Male and female mice of 40–60 d of age were used for all the experiments. All procedures involving animals were approved by the Institute Animal Care and Use Committees of Cold Spring Harbor Laboratory.
Stereotaxic surgery.
The AAV-DIO-ChR2(H134R)-YFP was produced by the University of North Carolina Vector Core Facilities. The retrograde tracer AlexaFluor-488- or 555-conjugated cholera toxin (CTB) was purchased from Invitrogen. Standard surgical procedures were followed for stereotaxic injection (Li et al., 2011; Li et al., 2013). Briefly, animals were anesthetized with ketamine (100 mg/kg) supplemented with dexmedetomidine hydrochloride (0.4 mg/kg) and positioned in a stereotaxic injection frame (www.myNeuroLab.com). A digital mouse brain atlas was linked to the injection frame to guide the identification and targeting of CeL (Angle Two Stereotaxic System, www.myNeuroLab.com). CTB or viruses were delivered with a glass micropipette through a skull window (1–2 mm2) by pressure application (5–12 psi, controlled by a Picrospritzer III, General Valve, Fairfield, NJ, USA). The injections were performed using the following stereotaxic coordinates for CeL: −1.22 mm from bregma, 2.6–2.75 mm lateral from the midline, and 4.6 mm vertical from the cortical surface; for PAG: −4.48 mm from bregma, 0.36 mm lateral from the midline, and 2.83 mm vertical from the cortical surface; and for PVT: −1.34 mm from bregma, 0.05 mm lateral from the midline, and 3.03 mm vertical from the cortical surface. During all surgical procedures, animals were kept on a heating pad and were brought back to their home cages after regaining movement. For postoperative care, mice were hydrated by intraperitoneal injection with 0.3–0.5 ml of lactated Ringer's solution. We used Metacam (meloxicam, 1–2 mg/kg) as an analgesic and to reduce inflammation. For the injection of CTB, we injected 1 μl (0.5% in PBS) and waited 3–15 d to allow the retrograde labeling of projection neurons. For the injection of AAV, we injected 0.2 μl of viral solution (∼1012 virus particles/ml) followed by 2–3 weeks to allow maximal viral expression.
Fear conditioning.
Fear conditioning procedures were performed as previously described (Kopec et al., 2007; Li et al., 2013). Habituation and conditioning were performed in separate days in a Mouse Conditioning Cage (Test-A; 18 cm × 18 cm × 30 cm) with an electrifiable floor connected to a H13–15 shock generator (Coulbourn Instruments). The Test-A cage was situated in a larger sound-attenuated cabinet (H10–24A; Coulbourn Instruments). On day 1, mice were individually habituated in a Test-A cage with five pure tones (4 kHz, 75 dB, 30 s each) delivered at variable intervals (60–120 s). The entire duration of this session was 600 s. On day 2, animals were individually conditioned using a procedure similar to that in day 1, except that each of the five tones coterminated with a 2 s 1 mA foot-shock. The floor and walls of the cage were cleaned with 70% ethanol for each animal. During habituation and conditioning, the cabinet was illuminated. On day 3, animals were tested for fear memory in a testing cage, Test-B, in darkness. Test-B (the testing cage) had a different shape (22 cm × 22 cm × 21 cm) and floor texture compared with Test-A (the conditioning cage). The floor and walls of Test-B were wiped with 0.5% acetic acid for each animal before testing to make the scent distinct from that of Test-A. Behavioral response to two 4 kHz 75 dB tone (the conditioned stimulus) delivered with a 120 s interval was recorded. The entire duration of the test session was 340 s. Freezing behavior in response to the two conditioned stimulus presentations during the test session was scored. The FreezeFrame software (Coulbourn Instruments) was used to control the delivery of tones and foot-shocks. Animal behavior was captured with a monochrome CCD camera (Panasonic WV-BP334) at 4 Hz and stored on a personal computer. Freezing behavior was analyzed offline with the FreezeFrame software (Coulbourn Instruments).
Control animals were subjected to the same procedure except for foot shocks.
Preparation of acute brain slices and electrophysiology.
Experiments were always performed on interleaved naive and fear-conditioned animals. Acute brain slices were prepared 90 min after behavioral assessment. Mice were anesthetized with isoflurane, decapitated, and their brains quickly removed and chilled in ice-cold dissection buffer (110.0 mm choline chloride, 25.0 mm NaHCO3, 1.25 mm NaH2PO4, 2.5 mm KCl, 0.5 mm CaCl2, 7.0 mm MgCl2, 25.0 mm glucose, 11.6 mm ascorbic acid, and 3.1 mm pyruvic acid, gassed with 95% O2 and 5% CO2). Coronal slices (300 μm) containing the amygdala complex were cut in dissection buffer using an HM650 Vibrating Microtome (MICROM International) and subsequently transferred to a storage chamber containing artificial CSF (118 mm NaCl, 2.5 mm KCl, 26.2 mm NaHCO3, 1 mm NaH2PO4, 20 mm glucose, 2 mm MgCl2, and 2 mm CaCl2, at 34°C, pH 7.4, gassed with 95% O2 and 5% CO2). After at least 40 min recovery time, slices were transferred to room temperature and were constantly perfused with artificial CSF.
Whole-cell patch-clamp recordings were obtained with Multiclamp 700B amplifiers (Molecular Devices) low-pass filtered at 1 kHz. Recordings were under visual guidance using an Olympus BX51 microscope equipped with both transmitted light illumination and epifluorescence illumination. Recordings were performed using glass pipettes (open-tip resistance = 4–5 mΩ) filled with internal solution containing 115 mm cesium methanesulphonate, 20 mm CsCl, 10 mm HEPES, 2.5 mm MgCl2, 4 mm Na2-ATP, 0.4 mm Na3GTP, 10 mm Na-phosphocreatine, and 0.6 mm EGTA, pH 7.2. The mEPSCs were recorded at a holding potential of −70 mV in the presence of tetrodotoxin (TTX; 1 ìM) and picrotoxin (100 ìM). Light-evoked ChR2-mediated currents were recorded at a holding potential of –70 mV. The light-evoked IPSCs were recorded at a holding potential of 0 mV in the presence of APV (100 μm) and NBQX (3 μm). Electrophysiological data were acquired and analyzed using pCLAMP 10 software (Molecular Devices). The mEPSCs were analyzed using Mini Analysis Program (Synaptosoft).
To stimulate neurons expressing ChR2, we used a single-wavelength LED system (λ = 470 nm; www.CoolLED.com) connected to the epifluorescence port of the Olympus BX51 microscope. Light pulses triggered by a TTL signal from the Clampex software (Molecular Devices) were used to activate neurons and evoke synaptic transmission.
Immunohistochemistry.
Immunohistochemistry experiments were performed following standard procedures. Briefly, mice were anesthetized and transcardially perfused with PBS, followed by perfusion with 4% PFA. Brains were extracted and further fixed in 4% PFA overnight at 4°C followed by cryoprotection in a 30% PBS-buffered sucrose solution for 36 h. Coronal sections (40 μm) were cut using a freezing microtome (Leica SM 2010R, Leica). Sections were first washed in PBS (3 × 5 min) and then incubated in PBST (0.3% Triton X-100 in PBS) for 30 min at room temperature, followed by washing with PBS (3 × 5 min). Next, sections were blocked in 5% normal goat serum in PBST for 2 h at room temperature, followed by incubation with primary antibodies overnight at 4°C. Sections were then washed with PBS (3 × 10 min) and incubated with fluorescent secondary antibodies at room temperature for 1 h. After washing with PBS (5 × 10 min), sections were mounted onto slides with Fluoromount-G (Beckman Coulter). Images were taken using a LSM 710 laser-scanning confocal microscope (Carl Zeiss). Image analysis and cell counting were performed using ImageJ software. The primary antibodies used were anti-PKC-δ (rabbit, Millipore Bioscience Research Reagents, 1:500) and anti-somatostatin (rabbit, Millipore Bioscience Research Reagents, 1:1000).
Results
Subsets of SOM+ CeL neurons project to PAG or PVT
To determine whether CeL sends direct projections to PAG or PVT, we injected the PAG and PVT with a retrograde tracer CTB. The CTB conjugated to the dye AlexaFluor-488 or AlexaFluor-555 was separately injected into the ventrolateral PAG and the posterior PVT of the same mice (Fig. 1A,B). A substantial number of CeL neurons were labeled by CTB injected into the PAG, whereas fewer were labeled by CTB injected into the PVT, indicating that CeL neurons send long-range projections directly to either PAG or PVT (Fig. 1C–E). Interestingly, a fraction of CeL neurons project to both PAG and PVT, as they were colabeled by CTB injected into these two areas (Fig. 1C; 30.1 ± 4.8% of the identified PAG-projecting neurons coprojected to PVT, whereas 70.3 ± 4.3% of the identified PVT-projecting neurons coprojected to PAG; mean ± SEM, n = 7 mice).
Figure 1.
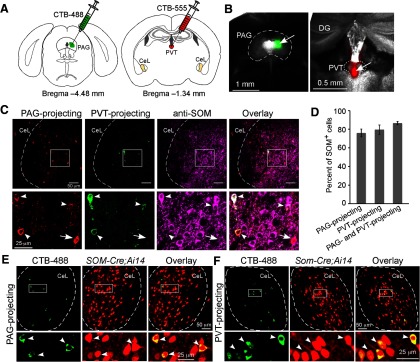
SOM+ CeL neurons project to PAG and PVT. A, Schematics of CTB injection. B, Representative coronal brain sections showing CTB injection into PAG (left) and PVT (right). In this mouse, the PAG and PVT, respectively, were injected with the CTB conjugated to AlexaFluor-488 (CTB-488, green) and CTB conjugated to AlexaFluor-555 (CTB-555, red) (arrows indicate the injection locations). DG, dentate gyrus. C, A representative coronal brain section containing CeL, in which the PAG- and PVT-projecting neurons were labeled with CTB-555 and CTB-488, respectively. SOM+ neurons were recognized by an antibody. Bottom, Higher-magnification images of the boxed area in the corresponding top. Arrowheads indicate the colocalization of CTB-555, CTB-488, and SOM in the same neurons. Arrow indicates the colocalization of CTB555 with SOM, but not with CTB488, in the same neuron. D, Quantification of the percentage of long-range projection CeL neurons that are SOM+ (n = 3 mice for each group; mean ± SEM). E, A coronal brain section from a SOM-Cre;Ai14 mouse that contains CeL, in which the PAG-projecting neurons were labeled with CTB-488 (left). SOM+ neurons were recognized by the red fluorescence from tdTomato (middle). Bottom, Higher-magnification images of the boxed area in the corresponding top. Arrowheads indicate the colocalization of CTB-488 and tdTomato in the same neurons. F, Same as E, except that the PVT-projecting CeL neurons were examined.
The CeL contains two major populations, the SOM+ neurons and PKC-δ+ neurons, which have different connectivity and differing roles in fear regulation (Haubensak et al., 2010; Li et al., 2013). To determine whether the long-range projection CeL neurons belong to either of these cell types, we examined the identity of CTB-labeled neurons by immunohistochemistry. We found that the vast majority (∼80%) of the PAG-projecting, PVT-projecting, and dual-projecting neurons are SOM+ cells (Fig. 1C,D). As detection with antibodies may underestimate the number of SOM+ neurons, we also used the SOM-Cre;Ai14 mice (Madisen et al., 2010; Taniguchi et al., 2011), in which SOM+ neurons are readily identified by their red fluorescence (Li et al., 2013). Using this strategy, ∼94% of PAG-projecting and 95% of PVT-projecting CeL neurons (data from two mice and one mouse, respectively) were determined to be SOM+ (Fig. 1E,F). In contrast, only a small fraction (∼20%) of the long-projection CeL neurons were PKC-δ+ neurons (Fig. 2).
Figure 2.

PKC-δ+ neurons do not appreciably project to PAG or PVT. A, A representative coronal brain section containing CeL, in which the PAG-projecting neurons were labeled with CTB-488 (left). PKC-δ+ neurons were recognized by an antibody (middle). Bottom, Higher-magnification images of the boxed area in the corresponding top. B, Same as A, except that the PVT-projecting CeL neurons were examined. C, Quantification of the percentage of long-range projection CeL neurons that are PKC-δ+ (n = 4 mice for each group; mean ± SEM).
To determine whether the long-range projection CeL neurons directly synapse onto PAG or PVT neurons, we used an optogenetic method whereby channelrhodopsin-2 (ChR2) activates neurons when stimulated by light (Zhang et al., 2006). As these long-range projection neurons are predominantly SOM+ (Fig. 1), we used the SOM-Cre mice (Taniguchi et al., 2011) to target the SOM+ CeL neurons. We injected the CeL of these mice with a Cre-dependent adeno-associated virus AAV-DIO-ChR2 (H134R)-YFP to selectively express ChR2 in SOM+ CeL neurons (Fig. 3A,B). As expected, SOM+ CeL neurons expressing ChR2 could be reliably activated by a blue light (λ = 470 nm) (Fig. 3C). Furthermore, we found that photo-stimulation of axons originating from the SOM+ CeL neurons reliably evoked IPSCs in 3 of 21 neurons (2 mice) recorded in the PAG (Fig. 3D). Notably, responsive cells were restricted to the ventrolateral portion of the PAG. In contrast, we failed to detect any IPSC in neurons recorded in the PVT (22 neurons, 4 mice).
Figure 3.

Direct synaptic connectivity between CeL and PAG. A, Schematic of viral injection. B, Left, Representative image of a coronal section obtained from a SOM-Cre mouse, in which the CeL was injected with the AAV-DIO-ChR2(H134R)-YFP. This section was recovered after electrophysiological recording. Right, Higher magnification of the CeL region, showing that ChR2-YFP is expressed in the SOM+ CeL neurons. C, Left, Schematic recording configuration. Right, Light-evoked excitatory currents (at −70 mV holding potential) recorded from a SOM+ CeL neuron expressing ChR2-YFP. An LED (λ = 470 nm) was used to deliver a train of blue light pulses (2 ms at 5 Hz, denoted by blue bars). D, Left, Schematic recording configuration. Right, IPSCs (at 0 mV holding potential) recorded from a PAG neuron, in response to the photo-stimulation (2 ms at 5 Hz, denoted by blue bars) of axon terminals originating from SOM+ CeL neurons. LA, Lateral amygdala; BLA, basolateral amygdala.
Fear conditioning strengthens excitatory synaptic transmission onto the long-range projection CeL neurons
Our recent study indicates that synaptic plasticity onto SOM+ CeL neurons stores fear memories (Li et al., 2013). To determine whether fear conditioning can induce synaptic plasticity onto the long-range projection CeL neurons, we prepared acute brain slices from fear-conditioned mice and control mice and recorded miniature EPSCs (mEPSCs) from the CeL neurons projecting to PAG or PVT, or to both targets (Fig. 4A–C). Remarkably, fear conditioning increased the frequency of mEPSCs recorded from either the PAG-projecting or the PVT-projecting neurons (Fig. 4D,E) and had a trend toward increasing mEPSC frequency in the dual-projecting neurons (p = 0.08, t test; Fig. 4E). Fear conditioning also increased the amplitude of mEPSCs recorded from these neurons (Fig. 4E). These results indicate that fear conditioning strengthens the excitatory synapses onto long-range projection CeL neurons.
Figure 4.

Fear conditioning potentiates the excitatory synapses onto long-range projection CeL neurons. A, Schematic experimental procedure. B, The behavioral performance of animals used in the electrophysiology experiments, which is measured as the percentage of time animal spent in freezing, during habituation, conditioning, and recall (n = 6 mice, mean ± SEM). C, Schematic recording configuration. Acute slices for electrophysiological recording were prepared 1.5 h after fear memory recall test (see A). D, Representative mEPSC traces recorded from PAG-projection CeL neurons in a fear-conditioned (left) and a control mouse (right). Control mice were treated with the same procedure as fear-conditioned mice, except for foot shocks. E, Quantification of the frequency (left) and amplitude (right) of mEPSCs recorded from the fear-conditioned and control mice (PAG-projecting, fear, n = 28 cells in 6 mice, control, n = 31 cells in 6 mice; PVT-projecting, fear, n = 32 cells in 6 mice, control, n = 21 cells in 6 mice; dual-projecting, fear, n = 18 in 5 mice, control, n = 16 cells in 4 mice; mean ± SEM). *p < 0.05 (t test). ***p < 0.001 (t test). LA, Lateral amygdala; BLA, basolateral amygdala.
Discussion
It is well established that PAG controls defensive responses (LeDoux et al., 1988; Johansen et al., 2011), and the PVT has recently been implicated in the regulation of conditioned fear expression (Padilla-Coreano et al., 2012). By combining retrograde tracing, immunohistochemistry, and genetic labeling, we identified a subset of SOM+ CeL neurons directly projecting to the PAG and/or PVT. The identified fraction of CeL neurons projecting to both PAG and PVT is likely an underestimation, as the probability of dual retrograde labeling depends on the efficiency in targeting axon fibers from CeL to both PAG and PVT.
By using optogenetic techniques, we confirmed that SOM+ CeL neurons synapse onto a subset of neurons located within the ventrolateral portion of PAG. Therefore, activation of SOM+ CeL neurons may, like activation of CeM neurons, acts to disinhibit PAG output, thereby driving fear expression. Importantly, we found that fear conditioning potentiates excitatory synapses onto these long-range projection neurons. This synaptic potentiation is expressed as an increase in both the frequency and amplitude of mEPSCs in fear-conditioned mice compared with control mice. Whereas higher mEPSC frequency may reflect an increase in either presynaptic release probability or the number of active synapses, a change in mEPSC amplitude usually indicates a postsynaptic effect (Kerchner and Nicoll, 2008; Li et al., 2013). Thus, consistent with our previous findings, it is likely that both presynaptic and postsynaptic mechanisms contribute to the fear conditioning-induced synaptic potentiation onto CeL neurons (Li et al., 2013).
Our retrograde tracing results indicate that a fraction of CeL neurons projects to the PVT. However, we were unable to detect CeL–PVT fast synaptic transmission. This could be for the following reasons: (1) the innervation of PVT neurons by CeL is sparse; (2) only a small number of CeL neurons project to the PVT; the AAV-DIO-ChR2(H134R)-YFP failed to target these neurons, as we restricted the size of infection area to avoid contamination of structures adjacent to the CeL; and (3) transmission in the CeL–PVT pathway is mediated by neuropeptides (e.g., somatostatin) or other neuromodulators.
Recent studies strongly indicate that the CeL is critical for fear conditioning (Goosens and Maren, 2003; Wilensky et al., 2006; Ciocchi et al., 2010; Haubensak et al., 2010) and that synaptic potentiation onto SOM+ CeL neurons stores fear memories and is required for the expression of conditioned fear (Li et al., 2013). The present study suggests that a specific population of the SOM+ CeL neurons, the long-range projection neurons, is likely an important contributor to fear conditioning, as these neurons undergo fear conditioning-induced synaptic potentiation. In particular, the CeL–PAG pathway is capable of regulating fear expression by directly controlling the activity of PAG neurons, bypassing the canonical CeL–CeM–PAG pathway. The CeL–PVT pathway, on the other hand, may contribute to different aspects of fear regulation (Padilla-Coreano et al., 2012) or modulate different behavioral states, such as adaptation to chronic stress (Hsu and Price, 2009). The exact role of the long-range projection CeL neurons in fear regulation warrants further investigation.
Footnotes
This work was supported by the National Institutes of Health, Dana Foundation, National Alliance for Research on Schizophrenia and Depression, and Louis Feil Trust (to B.L.) as well as a Harvey L. Karp Discovery Award to M.A.P. We thank members of the B.L. laboratory for discussions.
The authors declare no competing financial interests.
References
- Bourgeais L, Gauriau C, Bernard JF. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci. 2001;14:229–255. doi: 10.1046/j.0953-816x.2001.01640.x. [DOI] [PubMed] [Google Scholar]
- Cassell MD, Gray TS. Morphology of peptide-immunoreactive neurons in the rat central nucleus of the amygdala. J Comp Neurol. 1989;281:320–333. doi: 10.1002/cne.902810212. [DOI] [PubMed] [Google Scholar]
- Cassell MD, Freedman LJ, Shi C. The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci. 1999;877:217–241. doi: 10.1111/j.1749-6632.1999.tb09270.x. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Müller C, Lüthi A. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Popa D, Paré D. Central amygdala activity during fear conditioning. J Neurosci. 2011;31:289–294. doi: 10.1523/JNEUROSCI.4985-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci. 2003;117:738–750. doi: 10.1037/0735-7044.117.4.738. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Lüthi A, Anderson DJ. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DT, Price JL. Paraventricular thalamic nucleus: subcortical connections and innervation by serotonin, orexin, and corticotropin-releasing hormone in macaque monkeys. J Comp Neurol. 2009;512:825–848. doi: 10.1002/cne.21934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Kessels HW, Bush DE, Cain CK, LeDoux JE, Malinow R. A robust automated method to analyze rodent motion during fear conditioning. Neuropharmacology. 2007;52:228–233. doi: 10.1016/j.neuropharm.2006.07.028. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B. Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci. 2013;16:332–339. doi: 10.1038/nn.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moga MM, Gray TS. Evidence for corticotropin-releasing factor, neurotensin, and somatostatin in the neural pathway from the central nucleus of the amygdala to the parabrachial nucleus. J Comp Neurol. 1985;241:275–284. doi: 10.1002/cne.902410304. [DOI] [PubMed] [Google Scholar]
- Moga MM, Herbert H, Hurley KM, Yasui Y, Gray TS, Saper CB. Organization of cortical, basal forebrain, and hypothalamic afferents to the parabrachial nucleus in the rat. J Comp Neurol. 1990;295:624–661. doi: 10.1002/cne.902950408. [DOI] [PubMed] [Google Scholar]
- Padilla-Coreano N, Do-Monte FH, Quirk GJ. A time-dependent role of midline thalamic nuclei in the retrieval of fear memory. Neuropharmacology. 2012;62:457–463. doi: 10.1016/j.neuropharm.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovich GD, Swanson LW. Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Res. 1997;763:247–254. doi: 10.1016/S0006-8993(96)01361-3. [DOI] [PubMed] [Google Scholar]
- Sun N, Cassell MD. Intrinsic GABAergic neurons in the rat central extended amygdala. J Comp Neurol. 1993;330:381–404. doi: 10.1002/cne.903300308. [DOI] [PubMed] [Google Scholar]
- Sun N, Roberts L, Cassell MD. Rat central amygdaloid nucleus projections to the bed nucleus of the stria terminalis. Brain Res Bull. 1991;27:651–662. doi: 10.1016/0361-9230(91)90041-H. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci. 2006;26:12387–12396. doi: 10.1523/JNEUROSCI.4316-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]