

Abstract
The excessive accumulation of soluble amyloid peptides (Aβ) plays a crucial role in the pathogenesis of Alzheimer's disease (AD), particularly in synaptic dysfunction. The role of the two major chaperone proteins, Hsp70 and Hsp90, in clearing misfolded protein aggregates has been established. Despite their abundant presence in synapses, the role of these chaperones in synapses remains elusive. Here, we report that Hsp90 inhibition by 17-AAG elicited not only a heat shock-like response but also upregulated presynaptic and postsynaptic proteins, such as synapsin I, synaptophysin, and PSD95 in neurons. 17-AAG treatment enhanced high-frequency stimulation-evoked LTP and protected neurons from synaptic damage induced by soluble Aβ. In AD transgenic mice, the daily administration of 17-AAG over 7 d resulted in a marked increase in PSD95 expression in hippocampi. 17-AAG treatments in wild-type C57BL/6 mice challenged by soluble Aβ significantly improved contextual fear memory. Further, we demonstrate that 17-AAG activated synaptic protein expression via transcriptional mechanisms through the heat shock transcription factor HSF1. Together, our findings identify a novel function of Hsp90 inhibition in regulating synaptic plasticity, in addition to the known neuroprotective effects of the chaperones against Aβ and tau toxicity, thus further supporting the potential of Hsp90 inhibitors in treating neurodegenerative diseases.
Introduction
Alzheimer's disease (AD) is a devastating neurodegenerative disorder characterized by progressive memory loss and cognitive dysfunction. Alteration of vulnerable dendritic structures is thought to be an early event in AD pathogenesis that precedes synaptic loss. Excessive generation and accumulation of amyloid peptides (Aβ) in neurons are thought to initiate the pathological cascade in AD. Compelling evidence indicates that Aβ, especially the soluble oligomers, selectively targets synapses and disrupts their structures and functions (Palop and Mucke, 2010; Koffie et al., 2011). PSD95 is the major scaffolding protein at excitatory synapses and in postsynaptic densities (PSDs) and is crucial for synapse maturation and plasticity (Sheng and Hoogenraad, 2007). The level of PSD95 protein is reduced in AD brains (Gylys et al., 2004), with the degree of reduction correlating with Aβ oligomer levels and the severity of dementia (Love et al., 2006; Proctor et al., 2010). Aβ oligomers have been reported to directly interact with PSD95 at postsynaptic sites (Pham et al., 2010) with the continuous overproduction of Aβ reducing the number and plasticity of local synapses (Wei et al., 2010).
Molecular chaperones consisting of heat shock proteins (HSPs) are crucial in the quality control of proteins through the ubiquitin proteasomal pathways. Neurodegenerative diseases are frequently characterized by a gain of toxic function by misfolded proteins. Increased chaperone expression, as best exemplified by Hsp70, can suppress the neurotoxicity of these molecules and represents a novel potential therapy. Like Hsp70, the other major HSP Hsp90 facilitates the folding of newly synthesized and denatured proteins by forming large complexes with a variety of cochaperones. Because most of Hsp90's “client proteins” are oncogenic or neurotoxic, it has long been postulated that Hsp90 inhibition exerts therapeutic effects by dissociating these toxic proteins and facilitating their degradation. Therefore, Hsp90 inhibitors were found to hold promise in treating neurodegenerative diseases, as they can clear inclusions, such as PolyQ, α-synuclein, and tau (Luo et al., 2010). In AD, Hsp90 inhibition has been extensively studied in regard to reducing tau pathology (Dickey et al., 2007). However, its effects on synapses are completely unknown.
In this study, we investigated a novel aspect of Hsp90 inhibition in regulating synaptic functions. Herein, we present in vitro and in vivo evidence that 17-(allylamino)geldanamycin (17-AAG), which is being actively tested in numerous clinical cancer trials, induces rapid and sustained expression of multiple HSPs and synaptic proteins in primary neurons via activated heat shock transcription factor HSF1. Additionally, 17-AAG can attenuate synaptic toxicity induced by soluble Aβ as well as memory loss in mice, as determined by contextual fear conditioning test.
Materials and Methods
Chemicals and antibodies.
17-AAG and antibody against β-actin were from Sigma-Aldrich. Antibodies against HSF1, Hsp90, Hsp70, Hsp40, Hsp27, PSD95, synapsin I, Akt, p-Akt, Rab 3a, SNAP25, SAP97, CaMKII, and BDNF were from Cell Signaling Technology. KRIBB11 and antibody against synaptophysin were from EMD Millipore. AlexaFluor-conjugated antibodies were from Invitrogen.
Primary neuron transfection.
Primary cortical and hippocampal neurons were prepared from E17 rat embryos and maintained in neurobasal medium supplemented with 0.8 mm l-glutamine and B27 (Chen et al., 2012). Transient transfections of primary neurons with a constitutive construct pCA-hsf1-S303A as well as hsf1 siRNA (Santa Cruz Biotechnology) were conducted via electroporation using the 4D-Nucleofector Y-unit system (IF-100; Lonza).
Naturally secreted Aβ-containing conditioned medium.
Conditioned medium (CM) containing naturally secreted Aβ oligomers was collected from cultures of 7PA2/CHO cells (Walsh et al., 2002).
Western blot analysis.
Western blot analysis was performed as described previously (Chen et al., 2012).
Immunocytochemistry and dendritic spine quantification.
Cultured neurons were fixed, permeabilized, and immunostained on synaptic proteins and visualized under a fluorescence microscope. Clusters of PSD95 and synapsin I on dendrites were counted, and the densities of clusters were calculated as the average number of clusters per 10 μm of dendritic length. The density of PSD95 clusters proximate to synapsin I was also calculated. To visualize dendritic spines, mature hippocampal neurons (21 DIV) at low density (5 × 103 cells per 35 mm dish) were infected with a Sindbis virus carrying EGFP for 2 d, followed by exposure to control or 7PA2 CM for 24 h. The neurons were then fixed and visualized under a fluorescence microscope. Dendritic spines along primary and secondary basilar dendrites were counted. The density of dendritic spines was calculated as the average number of spines per 10 μm of dendritic length.
Mice.
Male Tg2576 mice and nontransgenic littermates (6 months of age) received intracerebroventricular injections of 17-AAG (0.5 μl of 100 μm) through an implanted cannula daily over 7 contiguous days. Analysis was performed on the eighth day.
In vivo LTP.
Brain surgery and electrophysiological recordings were performed as described previously (Zhu et al., 2007).
Fear conditioning.
The experiments were conducted with conditioned freezing chambers (Coulbourn Instruments) following a similar protocol recently published (Kittelberger et al., 2012). Wild-type C57BL/6 mice (2 months of age) received 2 μl of control (CHO medium), 7PA2 CM, or 7PA2 CM in combination with 17-AAG (100 μm), via an implanted cannula. Animal protocols were approved by the Animal Research Committee at the University of Tennessee Health Science Center.
Statistical analysis.
All data are presented as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by Tukey's post hoc test, or Student's t test. The p value of <0.05 was considered to be statistically significant.
Results
17-AAG rapidly induces HSPs and synaptic proteins in neurons
Geldanamycin and its analog 17-AAG have been shown to initiate a heat shock-like response in multiple cell types via HSF1-mediated induction of HSPs. We show here that 17-AAG treatment also rapidly induced HSF1 activation in primary rat cortical neurons (14 DIV), as evidenced by its nuclear translocation (Fig. 1A). Similar to other cell types, such as cancer cells, 17-AAG treatment also led to rapid induction of multiple HSPs in neurons, in particular the inducible iHsp70 and Hsp27 (Fig. 1B), with a sustained effect lasting for at least 24 h achieved at 100–200 nm concentrations; concentrations >1 μm caused cytotoxicity in neurons. Hsp40 expression was increased to a lesser degree and no change was found in the constitutive cHsp70. Because the Hsp90 inhibitor only disrupts protein interactions between Hsp90 and its clients, it does not change the protein level of Hsp90 itself. The activity of Hsp90 inhibitor was further confirmed by its ability to disrupt a major Hsp90 client protein, protein kinase B/Akt in neurons (Fig. 1B).
Figure 1.
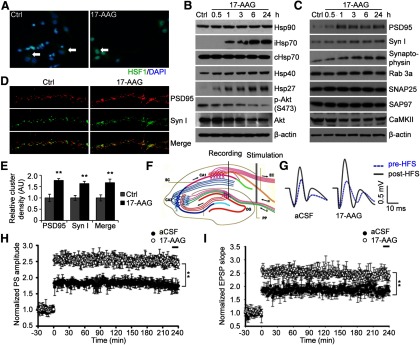
17-AAG induces HSPs and synaptic proteins in neurons and enhances in vivo LTP. A, Immunocytochemistry was conducted using anti-HSF1 antibody and DAPI on primary cortical neurons (14 DIV, >95% purity) 0.5 h after treatment with 200 nm 17-AAG. White arrows indicate cytoplasmic HSF1 (green) in the control neurons and overlap between HSF1 signaling (green) with DAPI (blue) in the nuclei in the majority of treated neurons. Representative images of 6 experiments (original magnification, ×25). B, Western blot analysis of HSPs as well as Akt, p-Akt levels after treatment with 200 nm of 17-AAG. C, Induction of both presynaptic and postsynaptic protein expression by 17-AAG, as determined by Western blot analysis. Syn I, Synapsin I. B, C, Results are representative Western blots of 10 experiments. D, Immunocytochemistry for PSD95 and synapsin I in mature neurons (21 DIV) 4 h after exposure to 17-AAG (200 nm) (original magnification, ×63). E, Quantification of the PSD95 and synapsin I cluster densities. AU, Arbitrary units. **p < 0.01 (Student's t test). n = 6. F–I, In vivo LTP. An ideographic electrophysiology recording setup with the stimulating electrode and recording electrode placed in the perforant path (PP) and dentate gyrus (DG) regions, respectively, and a drug administration system placed near the recording electrode. Electrophysiology recording was started 30 min after adding the 17-AAG (1 μl of 100 μm) or DMSO. At 0 min, the HFS was initiated with the alteration in LTP represented by the normalized amplitude of PS and the slope of EPSP. Data represent mean ± SEM of 6–8 male Sprague Dawley rats. **p < 0.01 (one-way ANOVA).
Furthermore, we found that 17-AAG caused increases in the protein expression of selected presynaptic and postsynaptic proteins (synapsin I, synaptophysin, and PSD95), beginning 1 h after treatment and reaching peaks within 3 h (Fig. 1C). The effect appears to be specific to certain synaptic proteins because the expression of two other PDZ-containing scaffolding proteins, SNAP25 and SAP97, as well as presynaptic Rab3a and the most abundant postsynaptic CaMKII protein was not affected by 17-AAG (Fig. 1C). Immunocytochemistry also revealed marked increases in the expression of both synapsin I and PSD95 (Fig. 1D). Confocal microscopic analysis of the double-labeled immunofluorescence demonstrated that these two vesicle antigens were closely colocalized, showing numerous puncta of synapsin I and PSD95. Further quantification indicates a nearly twofold increase in the overlay signals between these two proteins, similar in degree to the individual proteins (Fig. 1E). These results strongly suggest that 17-AAG induces formation of active synapses because the clusters of these presynaptic and postsynaptic proteins represent functionally active synaptic zones (McAllister, 2007).
17-AAG enhances in vivo LTP
Consistent with its upregulation of synaptic scaffolding proteins, a single administration of 17-AAG 30 min before high-frequency stimulation (HFS) was also found to enhance in vivo LTP recorded from rat medial perforant path-dentate granule cell synapses (Fig. 1F–I), as evidenced by both the increased amplitude of PS and the slope of EPSP. Without HFS, we did not observe any obvious changes in basal synaptic transmission. Moreover, the effect of 17-AAG appeared to be on the induction phase, as no enhancement was observed when 17-AAG was added 30 min after HFS. We chose to measure LTP in DG region because DG plasticity was severely defective whereas CA1 plasticity was relatively spared in several familial and neurotrophic-deprived AD mouse models (Houeland et al., 2010).
Effect of 17-AAG against Aβ-induced damage of synaptic proteins and structure
To examine the effect of 17-AAG on established synapses, we cultured rat hippocampal neurons for 3 weeks before experiments. We used soluble Aβ derived from a CHO cell line (7PA2) that expresses APP carrying the V717F mutation (Walsh et al., 2002). This cell line secretes high levels of monomeric and small oligomeric Aβ, without larger insoluble aggregates. Low levels of soluble Aβ (∼1 nm), equivalent to the concentrations in human AD brains, rapidly alter dendritic spine morphology and uncouple dendritic spines from their nerve terminals (Calabrese et al., 2007; Shankar and Walsh, 2009). We achieved these concentrations of soluble Aβ using twofold diluted conditioned media collected from 7PA2 (denoted as Aβ). In matured neurons (21 DIV), soluble Aβ treatment significantly reduced PSD95 and synapsin I at synapses. This was largely restored by 17-AAG cotreatment (Fig. 2A–C). Consistent with the Western data, immunocytochemistry suggested the same conclusion: 17-AAG not only prevented the Aβ-induced decrease in individual protein expression, it also rescued functional synapses, as measured by PSD95 clusters and proximate PSD95-synapsin I cluster complexes (Fig. 2D–G).
Figure 2.
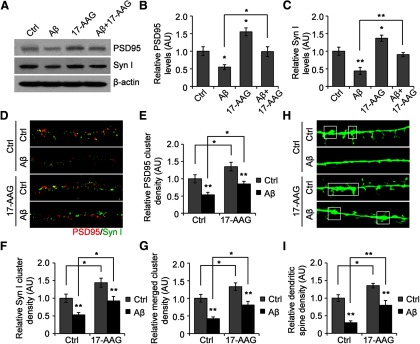
17-AAG protects against Aβ-induced damage to dendritic spine structures in neurons. A–C, Rescue of Aβ-impaired presynaptic and postsynaptic proteins by 17-AAG. Mature hippocampal neurons (21 DIV) were exposed to 1 nm soluble Aβ for 4 h, with or without 17-AAG cotreatment, followed by Western blot analysis 4 h after treatment. *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). n = 4. D–G, Representative micrographs of the immunofluorescent labeling of PSD95 and synapsin I 24 h after the treatment and quantification of presynaptic and postsynaptic clusters, as well as PSD95/synapsin I merged clusters (original magnification, ×63). *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). n = 6. H, Representative spine images denoted by EGFP expressed from Sindbis virus infection. White boxes represent examples of stubby mushroom-like mature spine structures (original magnification, ×63). n = 6. I, Quantification of dendritic spines. *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). n = 6.
We detected Aβ-induced spine loss, which could be completely rescued by cotreatment with 17-AAG, as determined by quantification of dendritic spine density (Fig. 2H,I), with the maximum protective effect observed at 24 h. Dendritic spines represent the structure in the CNS that allows for multiple inputs and is the highly dynamic site of excitatory synapses. Spines are classified as “filopodia-like,” “thin” (immature), or “mushroom like” (mature) (Alvarez and Sabatini, 2007). After 24 h of Aβ treatment, spines were devoid of any mushroom-like structures, with only filopodia-like and thin synapses remaining, compared with control conditions (DMSO). Strikingly, 17-AAG treatment significantly enhanced the number of mushroom-like structures and cotreatment completely rescued the Aβ-induced synaptic structure impairment. Because the mushroom spines are the primary site of glutamate receptors and synaptic transmission, our findings regarding 17-AAG's potent effect in counteracting the Aβ-induced reduction in mature spine density suggest a role for 17-AAG in the functional rescue of Aβ-impaired synaptic transmission.
17-AAG prevents the Aβ-induced synapse and memory loss in vivo
To test the effect of 17-AAG in vivo, we first demonstrated that only injections at dosages equivalent to 200–500 nm in the CSF of wild-type C57BL/6 mice, via intracerebroventricular injection, not a peritoneal route, induced Hsp70 and further augmented PSD95 expression in the hippocampus (Fig. 3A). This was consistent with the non–CNS-permeable nature of this compound. We then tested the effect of 17-AAG on hippocampus-dependent contextual memory. Wild-type C57BL/6 mice received 2 μl of control, 7PA2 CM, or 7PA2 CM in combination with 17-AAG (100 μm), via an implanted cannula. Both context and tone retrieval tests were conducted at 24, 48, and 72 h after fear conditioning training (Fig. 3B). The results showed that 17-AAG rescued Aβ-induced memory impairment at 72 h time point in a contextual memory test (Fig. 3C,D). Interestingly, the amygdala-dependent fear retention to the tone CS was not affected by Aβ (data not shown). Consistent with the in vitro data, expression levels of synaptic protein PSD95 as well as BDNF were significantly increased in the hippocampal region of Aβ mice after 17-AAG treatment (Fig. 3E–G). Furthermore, daily injection of 17-AAG for 7 d via the intracerebroventricular route dramatically increased the protein expression of iHsp70, PSD95, and synapsin I in the hippocampi of 6-month-old Tg2576 transgenic mice (Fig. 3H).
Figure 3.

17-AAG prevents synapse and memory loss induced by Aβ in vivo. A, In vivo induction of iHsp70 and PSD95 in mouse hippocampi 24 h after a single intracerebroventricular injection of 17-AAG. Injection of 1 mm of 17-AAG in a 0.5 μl volume resulted in a ∼10 μm concentration of the compound in the CSF. Considering the diffusion effect from the CSF into hippocampal and cortical tissue, we estimate the local concentration may approximate the effective concentrations used in vitro (200–500 nm). Representative Western blot of three experiments. B, Experimental design of the fear conditioning assay to test the effect of 17-AAG in Aβ-induced contextual memory impairment. 17-AAG was mixed with Aβ for the three injections via intracerebroventricular route through implanted cannula. C, Fear acquisition during the first day training. CS, “Conditioned stimulus” for context; Shk, shock. D, Recorded contextual fear retention. *p < 0.05 (one-way ANOVA). n = 14 animals each group. E, Protein expression levels of PSD95 and BDNF from the mouse hippocampi after the fear conditioning experiment. Representative Western blot is shown on 4 of 14 mice each group. F, G, Quantification of the Western blot images by densitometry. *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). n = 14 animals each group. H, Induction of PSD95 and synapsin I in Tg2576 mice. 17-AAG (0.5 μl of 100 μm) was given to transgenic (Tg) and littermate Non-Tg mice via intracerebroventricular injection through an implanted cannula daily for 7 contiguous days, followed by Western blot analysis. Representative Western blot is shown on 3 of 6 mice each group.
17-AAG upregulates PSD95 transcription via activated HSF1
Because the 17-AAG-induced expression of PSD95 and synapsin I occurs within the first 2 h, the responsible mechanism is more likely transcriptional activation. Indeed, 17-AAG rapidly increased the mRNA levels of PSD95, synapsin I, and synaptophysin by approximately twofold, as determined as early as 0.5 h after treatment by qRT-PCR (Fig. 4A). Multiple sites for HSF1-responsive elements were predicted in the promoter regions of these synaptic genes using the Transcription Element Search System (only PSD95 promoter is shown in Fig. 4B). We therefore asked the question of whether the transcriptional activation of the synaptic protein genes can be mediated directly by the activated HSF1 upon Hsp90 inhibition, a classic action of an Hsp90 inhibitor. When HSF1 activity was blocked by a specific inhibitor KRIBB11 (Yoon et al., 2011) in a dose-dependent manner (Fig. 4C), 17-AAG's effect on PSD95 induction was completely abolished (Fig. 4D,E). Furthermore, overexpression of a construct to constitutively activate HSF1 or introduction of the pooled siRNAs specific to rat hsf1 (100 nm) also resulted in the opposite effect on the protein levels of PSD95 (Fig. 4F,G). Quantitative RT-PCR confirmed that HSF1 overexpression can induce PSD95 gene transcription (data not shown).
Figure 4.
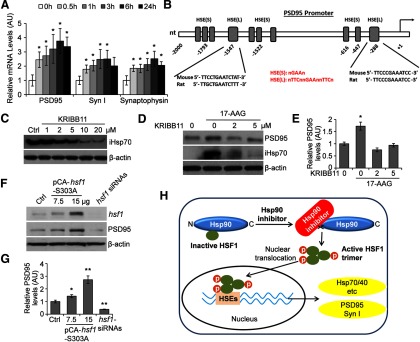
17-AAG activates PSD95 transcription via activated HSF1 in neurons. A, 17-AAG upregulates the transcriptional levels of PSD95, synapsin I, and synaptophysin, as determined by qRT-PCR. *p < 0.05 (Student's t test). n = 3. B, Schematic diagram showing the predicted heat shock-responsive elements (HSEs) in the promoter region of PSD95 gene. C, Dose–response of KRIBB11 in iHsp70 in treated SH-SY5Y neuroblastoma cells as determined 24 h after treatment. Representative Western blot of three experiments. D, E, Cotreatment with KRIBB11 abolishes 17-AAG's effect in inducing PSD95 and iHsp70. *p < 0.05 (one-way ANOVA). n = 3. F, G, Opposing effects of overexpression of constitutively active HSF1 and downregulation of HSF1 in PSD95 protein levels. *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). n = 3. H, Schematic illustration of how an Hsp90 inhibitor activates HSF1, ultimately leading to gene transcriptional activation of both HSPs and synaptic proteins.
Discussion
Hsp90, like Hsp70, is one of the most abundant cytosolic proteins (2%), although it differs from Hsp70 and other HSPs in its mechanism of action. It functions through two major mechanisms, regulating at both levels of gene transcription and protein. There are numerous (>800) proteins identified as Hsp90's client proteins. At protein levels, Hsp90 inhibitors bind to the ATP binding site in the N terminus, usually resulting in dissociation of Hsp90 from its client proteins, resulting in subsequent degradation of its clients via ubiquitin proteasomal pathway. We also speculate that an Hsp90 inhibitor can modulate synapses at both the levels of gene transcription (via HSF1) and via protein–protein dissociation/degradation, assuming that some of the synaptic proteins can be Hsp90's clients. Interestingly, the only reported function of Hsp90 in synapses is to control neurotransmitter release by mediating the continuous cycling of synaptic AMPA receptors (Gerges et al., 2004).
HSF1 is one of Hsp90's major client proteins and the most important transcription factor in mediating heat shock-like response through transcriptional upregulation of selective HSPs (Westerheide and Morimoto, 2005). Upon Hsp90 inhibition via a pharmacological inhibitor, the inactivated HSF1 proteins are dissociated from Hsp90, phosphorylated, and form the active trimers that undergo nuclear translocation to induce both multiple HSPs and the heat shock response (Fig. 4H). Therefore, a conventionally nominated Hsp90 inhibitor, instead of inhibiting Hsp90 (protein or function), acts as HSF1 activator. Perhaps, it is more suitable to call 17-AAG as an Hsp90 modulator.
In primary neurons, we found that 17-AAG, a geldanamycin derivative, cannot only induce multiple HSPs but can also selectively upregulate the expression of several synaptic proteins (synapsin I, synaptophysin, PSD95). Furthermore, we found that this inhibitor completely rescued synapses from the detrimental effects of soluble Aβ in C57BL/6 mice, reversing deterioration in hippocampus-dependent contextual fear conditioning. These findings led to a plausible hypothesis that heat shock-like stress response can protect synaptic functions via HSF1. The remaining fundamental question is how a heat shock-like stress response can regulate synaptic protein expression and subsequent synaptic function. We discover that, in neurons, Hsp90 inhibition by 17-AAG rapidly upregulates the expression of the three synaptic proteins, primarily through a transcriptional mechanism dependent on HSF1 activation. To our knowledge, this is the first report showing that these molecules can be regulated at the transcriptional level; the Aβ-induced PSD95 reduction was previously reported to be mediated by protein degradation via the ubiquitin proteasomal pathway (Roselli et al., 2005). We also observed increased BDNF levels upon 17-AAG treatment (Fig. 3E), and our unpublished data show that increased BDNF mRNAs correlated with increased CREB-P as a result of Hsp90 inhibition in neurons. This suggests a potential role of the PKA/CERB/BDNF pathway in mediating 17-AAG's effect on synaptic plasticity. It remains to be determined, the interplay between the transcriptional events mediated by PKA/CREB/BDNF and HSF1 in neurons upon Hsp90 inhibition.
Despite only a partial understanding of the molecular mechanisms that underlie the multiple beneficial effects of Hsp90 inhibition upon synapses, our strongest finding from this work is the rapid and long-lasting induction of both multiple HSPs and the synaptic proteins via an HSF1-dependent mechanism. Our combined in vitro and in vivo findings imply a therapeutic potential of Hsp90 inhibitors in treating AD. The primary pharmaceutical focus in the search for novel treatments for AD thus far have been on reducing the level of Aβ by inhibiting β- and γ-secretase activities; however, this approach has had little success. Individuals with early-stage AD suffer from a profound failure to form new memories (anterograde amnesia). The ability to enhance synaptic density in AD mice at a symptomatic age strongly supports the therapeutic potential of Hsp90 inhibitors in reversing synaptic loss and cognitive deficits in AD. Although 17-AAG and its derivative 17-DMAG have limited blood–brain barrier permeability, the work presented here demonstrates a proof of principle for use in development of more feasible CNS-permeable Hsp90 inhibitors as AD therapeutics, which is being actively pursued by both academia and industry.
Footnotes
This work was supported by National Institutes of Health Grants R01NS054880, R01AG031893, and R21AG041934 to F.-F.L., National Institutes of Health Grants R01AG021173, R01NS046673, R01AG030197, R01AG038710, and R03AG034366 to H.X., and Alzheimer's Association Grant IIRG-11-204030 to F.-F.L. We thank Drs. Young-Don Kwak, Shiyong Diao, and Tao Ma for assisting in some experiments; Dr. Dennis Selkoe (Harvard Medical School) for providing 7PA2 cells; and Dr. Dennis Thiele (Duke University) for providing pCA-hsf1-S303A construct.
The authors declare no competing financial interests.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S. Rapid, concurrent alterations in pre- and post-synaptic structure induced by soluble natural amyloid-β protein. Mol Cell Neurosci. 2007;35:183–193. doi: 10.1016/j.mcn.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Huang X, Zhang YW, Rockenstein E, Bu G, Golde TE, Masliah E, Xu H. Alzheimer's β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of β-amyloid. J Neurosci. 2012;32:11390–11395. doi: 10.1523/JNEUROSCI.0757-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges NZ, Tran IC, Backos DS, Harrell JM, Chinkers M, Pratt WB, Esteban JA. Independent functions of hsp90 in neurotransmitter release and in the continuous synaptic cycling of AMPA receptors. J Neurosci. 2004;24:4758–4766. doi: 10.1523/JNEUROSCI.0594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/S0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houeland G, Romani A, Marchetti C, Amato G, Capsoni S, Cattaneo A, Marie H. Transgenic mice with chronic NGF deprivation and Alzheimer's disease-like pathology display hippocampal region-specific impairments in short- and long-term plasticities. J Neurosci. 2010;30:13089–13094. doi: 10.1523/JNEUROSCI.0457-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelberger KA, Piazza F, Tesco G, Reijmers LG. Natural amyloid-beta oligomers acutely impair the formation of a contextual fear memory in mice. PLoS One. 2012;7:e29940. doi: 10.1371/journal.pone.0029940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27:797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, Yates JR, Glabe C, Masliah E. Progressive accumulation of amyloid-β oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DT, Coulson EJ, Dodd PR. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis. 2010;21:795–811. doi: 10.3233/JAD-2010-100090. [DOI] [PubMed] [Google Scholar]
- Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF. Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;48:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Aβ. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of the Alzheimer amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J Biol Chem. 2011;286:1737–1747. doi: 10.1074/jbc.M110.179440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, Wang Q, Chen JG, Wang JZ. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci. 2007;27:12211–12220. doi: 10.1523/JNEUROSCI.3321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]