

Abstract
Mutations of MECP2 cause Rett syndrome (RTT), a neurodevelopmental disorder leading to loss of motor and cognitive functions, impaired social interactions, and seizure at young ages. Defects of neuronal circuit development and function are thought to be responsible for the symptoms of RTT. The majority of RTT patients show recurrent seizures, indicating that neuronal hyperexcitation is a common feature of RTT. However, mechanisms underlying hyperexcitation in RTT are poorly understood. Here we show that deletion of Mecp2 from cortical excitatory neurons but not forebrain inhibitory neurons in the mouse leads to spontaneous seizures. Selective deletion of Mecp2 from excitatory but not inhibitory neurons in the forebrain reduces GABAergic transmission in layer 5 pyramidal neurons in the prefrontal and somatosensory cortices. Loss of MeCP2 from cortical excitatory neurons reduces the number of GABAergic synapses in the cortex, and enhances the excitability of layer 5 pyramidal neurons. Using single-cell deletion of Mecp2 in layer 2/3 pyramidal neurons, we show that GABAergic transmission is reduced in neurons without MeCP2, but is normal in neighboring neurons with MeCP2. Together, these results suggest that MeCP2 in cortical excitatory neurons plays a critical role in the regulation of GABAergic transmission and cortical excitability.
Keywords: GABA, hyperexcitation, neocortex, pyramidal neuron, Rett syndrome, seizure
Introduction
Loss-of-function mutations of X-linked MECP2 (methyl-CpG binding protein 2) cause the pervasive Rett syndrome (RTT; Chahrour and Zoghbi, 2007). RTT patients show progressive loss of motor and cognitive functions, impaired social interactions, anxiety, and seizure at young ages. Studies using mouse models of RTT have demonstrated that loss of MeCP2 from the brain is responsible for the neurological symptoms of RTT (Chen et al., 2001). However, the underlying mechanisms are largely unknown. Despite a reduction in brain size in RTT patients, there is no obvious change in the number of neurons or general pattern of brain organization (Armstrong, 2005); similar results have been found in mouse models of RTT (Chen et al., 2001; Guy et al., 2001; Shahbazian et al., 2002). It has been hypothesized that defects of neuronal circuit development and function are a major mechanism of RTT (Chahrour and Zoghbi, 2007). This hypothesis is supported by several studies demonstrating synaptic defects in MeCP2-deficient mice during development and in adulthood. Excitatory synaptic transmission is reduced in hippocampal cultures from Mecp2 knock-out mice (Nelson et al., 2006; Chao et al., 2007), but increased in those from mice overexpressing Mecp2 (Chao et al., 2007; Na et al., 2012). Long-term plasticity of excitatory synapses is also impaired in hippocampal slices of MeCP2-deficient mice (Asaka et al., 2006; Moretti et al., 2006; Guy et al., 2007). Changes of inhibitory transmission have been reported in the cortex, brainstem, hippocampus, and thalamus of Mecp2 knock-out mice (Dani et al., 2005; Medrihan et al., 2008; Zhang et al., 2008, 2010).
Clinical studies have shown that 70–80% of RTT patients have recurrent seizures, with the onset typically between 2 and 5 years of age (Jian et al., 2007; Nissenkorn et al., 2010; Pintaudi et al., 2010; Cardoza et al., 2011). Both partial and generalized seizures are common, and electroencephalographic (EEG) data reveal focal and generalized epileptic discharges in RTT patients (Nissenkorn et al., 2010; Vignoli et al., 2010). Furthermore, abnormal EEG activity in RTT patients is negatively correlated with their cognitive and communication abilities (Vignoli et al., 2010). These studies indicate that in addition to seizures, cognitive and behavioral disabilities in RTT may be caused by a hyperactivity of the neocortex and associated brain regions. How MeCP2 deficiency leads to cortical hyperexcitation is poorly understood. Mecp2 knock-out mice display seizures and hyperexcitability discharges, indicating that as in human, deficiency of MeCP2 causes network hyperexcitation in the brain (Chao et al., 2010; D'Cruz et al., 2010; Calfa et al., 2011). A concern with this model, however, is that there are widespread neuronal defects in the brain of Mecp2 knock-out mice (Wang et al., 2006; Medrihan et al., 2008; Taneja et al., 2009; Zhang et al., 2010; Noutel et al., 2011), therefore any changes observed in the cortex may be caused by defects in other brain regions. Furthermore, MeCP2 may have distinct roles in different types of cells. To address these issues, we examined mice with forebrain-restricted and cell-type-specific deletion of Mecp2.
Materials and Methods
Mice.
Mice carrying the Mecp2 conditional allele (B6.129S4-Mecp2tm1Jae/Mmucd; Mecp2F; Chen et al., 2001) were obtained from the Mutant Mouse Regional Center (MMRRC; stock No. 011918-UCD), and backcrossed to C57BL/6J (B6) for >10 generations. Cre driver strains Emx1-Cre (B6.129S2-Emx1tm1(cre)Krj/J; Gorski et al., 2002), Dlx5/6-Cre (Tg(Dlx6a-Cre)1Mekk/J; Monory et al., 2006), and Rosa-tdTomato reporter (Ai14; Madisen et al., 2010) were obtained from JAX (stock No. 5628, 8199, and 7914, respectively). The Emx1-Cre strain was on a B6 background; the Dlx5/6-Cre strain was backcrossed to B6 for eight generations. The serotonin transporter (SERT) Cre strain (Tg(Slc6a4-Cre)ET124Gsat, or SERT-Cre), generated by GENSAT (Gong et al., 2007), was obtained from the MMRRC (stock No. 017261-UCD). Originally, on a mixed FVB/B6/129/Swiss background, this strain was backcrossed to B6 for six generations before the experiments.
Genotypes were determined by PCR using tail DNA. For the Mecp2F strain, the primers were 5′-CTAGGTAAGAGCTCTTGTTGA-3′ and 5′-CACCACAGAAGTACTATGATC-3′, which gave products of 280 and 180 bp for the mutant and wild-type alleles, respectively. Cre driver strains were genotyped using primers 5′-GCATTACCGGTCGATGCAACGAGTGATGAG-3′ and 5′-GAGTGAACGAACCTGGTCGAAATCAGTGCG-3′, which amplify a 410 bp fragment.
All procedures are in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and have been approved by The Jackson Laboratory Animal Care and Use Committee.
EEG recording.
EEG was recorded from male mice of 6–8 weeks of age using procedures that we described previously (Beyer et al., 2008). Briefly, mice were anesthetized with tribromoethanol (400 mg/kg i.p.). Four small holes were drilled on the skull (1 mm anterior and 2 mm posterior to the bregma, 2 mm lateral to the midline). One Teflon-coated silver wire was inserted into each hole and placed between the dura and skull. The wires were soldered onto a microconnector that was fixed on the skull using dental cement. The mice were given a 48 h recovery period before recordings were made. EEG was recorded for a 2 h period on each of the following 2 d (Stellate Harmonie, Stellate Systems).
Slice preparation.
Male mice aged P17–P20 and P42–P50 were anesthetized with tribromo-ethanol and decapitated. Coronal sections 300 μm thick were prepared with a vibratome (Leica VT1200). Slices were kept in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3.0 KCl, 1.5 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26 NaHCO3, and 20 glucose, saturated with 95% O2 and 5% CO2 at room temperature (21–23°C).
Patch-clamp recording.
Layer 5 pyramidal neurons in the medial prefrontal cortex (mPFC) and the barrel cortex (S1) were recorded using methods that we described previously (Zhang, 2004). A slice was transferred to a submersion type chamber where it was continuously perfused with ACSF saturated with 95% O2 and 5% CO2 at 32–33°C. The slice was first viewed with a 4× objective, and the mPFC was identified as the area between the forceps minor corpus callosum and the midline. The S1 barrel cortex was identified based on characteristic of the layer 4. Pyramidal neurons were identified with a 40× objective and Nomarski optics. For IPSC recording, the pipette solution contained the following (in mm): 120 CsCl, 20 TEA-Cl, 4 ATP-Mg, 0.3 GTP, 0.5 EGTA, 10 HEPES, and 4.0 QX-314 (pH 7.2, 270–280 mOsm with sucrose). For EPSC recording, the pipette solution contained the following (in mm): 110 Cs methylsulfate, 15 CsCl, 20 TEA-Cl, 4 ATP-Mg, 0.3 GTP, 0.5 EGTA, 10 HEPES, and 4.0 QX-314 (pH 7.2, 270–280 mOsm with sucrose). Electrodes had resistances between 2 and 4 MΩ. Whole-cell recordings were made at the soma with a multiclamp 700B amplifier (Molecular Devices). The series resistance, usually between 8 and 14 MΩ, was not compensated in voltage-clamp experiments. Data were discarded when series resistance was >16 MΩ. In some experiments, bipolar electrodes made of insulated microwires (30 μm outside diameter) were placed in layer 5 to apply stimuli (100 μs, 20–300 μA). Experiments were conducted using AxoGraph X (AxoGraph Scientific). Data were filtered at 4 kHz and digitized at 20 kHz. Spontaneous synaptic events were analyzed using the event detection function of AxoGraph.
For current-clamp recording, the pipette solution contained the following (in mm): 120 K-gluconate, 10 KCl, 4 ATP-Mg, 0.3 GTP, 10 HEPES, and 0.5 EGTA (pH 7.2, 270–280 mOsm with sucrose). Series resistance was fully compensated using the bridge circuit of the amplifier MultiClamp 700B. Intrinsic properties were analyzed using methods that we described previously (Zhang, 2004; Zhang and Arsenault, 2005). Briefly, resting potentials of neurons were measured within 20 s of establishing whole-cell configuration. Input resistance was estimated from voltage responses to 400 ms current pulses at −100 pA. Action potentials were evoked using positive current pulses. Action potential threshold was estimated as the point when the slope of rising membrane potential exceeds 50 mV/ms, and for each cell, measurements of 4–6 action potentials were averaged.
Immunostaining.
Mice were anesthetized with tribromoethanol, perfused with 4% paraformaldehyde in 0.1 m PBS, and postfixed overnight. Coronal sections of 60 μm thick were cut on a vibratome. Sections containing mPFC were incubated with 1% BSA, 0.3% Triton X-100, and 3% normal goat serum for 2 h at room temperature, then incubated with polyclonal antibodies of vesicular GABA transporter (VGAT; 1:2000, Synaptic Systems), monoclonal anti-NeuN (1:1000, Millipore), polyclonal RFP (1:2000, Rockland), polyclonal anti-GABA (1:4000, Immunostar), or monoclonal anti-MeCP2 (1:1000, Cell Signaling Technology) for 48 h at 4°C. After wash, sections were incubated with goat AlexaFluor-conjugated secondary antibodies (1:500, Invitrogen) for 2 h at room temperature. Sections were mounted in DPX medium (Sigma-Aldrich).
Confocal images were taken on a Leica SP5 microscope. For VGAT straining, confocal stacks were taken with a 63× objective (NA 1.3) and 4× optical zoom. The pixel size was 60.3 nm. The z-step size was 130 nm, and each stack had 15 steps. Three stacks were obtained from each mouse, and the same laser and microscope setting was used for control and mutant samples. Confocal stacks were analyzed blind to the genotypes using the spot analysis tool of Imaris (Bitplane). The detection threshold was set using images that showed the strongest VGAT staining; the same threshold was applied to all stacks from control and mutant mice of the same experiment. The minimal spot size was set at 0.4 μm (X or Y). VGAT puncta was detected for the volume of the stack (6425 μm3 without shrinkage correction), and the results were expressed as the number of VGAT puncta per 1000 μm3 of tissue.
Statistics.
Statistics was performed using IgorPro (WaveMetrics), InStat (GraphPad), and JMP (SAS). Throughout, means are given as ±SEM. Unless specified otherwise, means were compared using Mann–Whitney test or one-way ANOVA. Distributions of synaptic events were compared with Kolmogorov–Smirnov test (K–S test).
Results
Spontaneous seizures in mice lacking MeCP2 from the forebrain
EEG recordings revealed cortical hyperexcitation and seizures in MeCP2-deficient mice (Shahbazian et al., 2002; Zhang et al., 2008). This increase of cortical discharges may be caused by the loss of MeCP2 from excitatory or inhibitory neurons, or glia in the cortex and subcortical regions. To determine the cellular origin of cortical hyperexcitation in Mecp2 mutant mice, we performed EEG recordings from mice lacking MeCP2 from specific cell types in the forebrain. The Emx1-Cre driver (Gorski et al., 2002) and a conditional Mecp2 strain (Chen et al., 2001) were used to delete Mecp2 from excitatory neurons and glia but not inhibitory neurons in the neocortex and hippocampus. A total of five Emx1-Mecp2 (Mecp2F/y; Emx1-Cre) mutant mice were recorded between 6 and 8 weeks of age. We found that all Emx1-Mecp2 mutant mice showed absence seizures as characterized by spike-wave discharges in animals that were awake (Fig. 1A). Spike-wave discharges occurred at 36 ± 7 episodes/h (n = 5 mice), and the mean duration of episode was 1.3 ± 0.1 s. In contrast, all four control mice that we recorded did not show any spike-wave discharges (Fig. 1B). We also recorded five mice lacking MeCP2 from inhibitory neurons in the forebrain using the Dlx6a-Cre driver (Monory et al., 2006). None of the Dlx6a-Mecp2 (Mecp2F/y; Dlx6a-Cre) mutant mice showed any spike-wave discharges (Fig. 1C). These results indicate that loss of MeCP2 from excitatory neurons and glia in the forebrain is sufficient to cause hyperexcitation and seizures.
Figure 1.

Conditional deletion of Mecp2 with Emx1-Cre leads to spike-wave discharges. A, Seizure activity in an Emx1-Mecp2 mutant mouse at 7 weeks of age. EEG was recorded from a wake Emx1-Mecp2 mouse using four electrodes placed over the right frontal (RF), left frontal (LF), right back (RB), and left back (LB) surface of the neocortex. Each trace is the differential between signals recorded from two of the electrodes. B, EEG recording from a control mouse (Mecp2wt/y; Emx1-Cre) at 7 weeks of age. C, EEG recording from a Dlx6a-Mecp2 mutant mouse at 7 weeks of age.
GABAergic transmission is reduced in the cortex of mice lacking MeCP2 from the forebrain excitatory neurons
The seizure phenotype observed in Emx1-Mecp2 mice suggests that the balance of excitation and inhibition (E/I balance) is disrupted in these mice. In addition to seizures, shift in E/I balance is also implicated in other RTT symptoms, such as cognitive defects, anxiety, and impaired social behaviors (Yizhar et al., 2011). To elucidate the mechanisms underlying the shift in E/I balance in MeCP2-deficient brains, we examined the effect of Mecp2 deletion on synaptic transmission in the mPFC, a region that plays an important role in cognitive functions, anxiety and social behaviors (Holmes and Wellman, 2009; Etkin et al., 2011; Yizhar et al., 2011). We performed whole-cell patch-clamp recording in acute brain slices from Emx1-Mecp2 mutant mice (Mecp2F/y; Emx1-Cre) and two littermate control groups (Mecp2wt/y; Emx1-Cre and Mecp2F/y) at P17–P20. We first assessed inhibitory synaptic transmission by recording spontaneous IPSCs (sIPSCs) from layer 5 pyramidal neurons in the mPFC. Excitatory synaptic transmission was blocked by DNQX (10 μm) and kynurenic acid (1 mm). Emx1-Mecp2 mutant neurons showed fewer sIPSCs than control neurons at P17–P20 (Fig. 2A,B,C), whereas no difference was found between mutant and control neurons in the mean peak amplitude or kinetics of sIPSCs (Fig. 2D). Because sIPSCs may contain both quantal IPSCs (mIPSCs) and spontaneous spike-driven IPSCs, we selectively recorded mIPSCs in the presence of tetrodotoxin (0.5 μm). Mutant (Mecp2F/y; Emx1-Cre) neurons showed fewer quantal events than control (Mecp2wt/y; Emx1-Cre) neurons at P17–P20 (Fig. 2E). In mutant neurons the mean frequency of mIPSCs was reduced by 30%, without any change in the amplitude or kinetics of mIPSCs (Fig. 2F,G).
Figure 2.

Deletion of Mecp2 with Emx1-Cre caused reduction in the frequency of sIPSCs and mIPSCs in layer 5 pyramidal neurons in the mPFC at P17–P20. A, sIPSCs recorded from a control (top trace) and mutant (bottom trace) neurons at P18. B, Distributions of sIPSC intervals for mutant neurons (Mecp2F/y; Emx1-Cre, line in gray, n = 25 from 4 mice) and control neurons (Mecp2F/y, dashed line, n = 15 from 2 mice; Mecp2wt/y; Emx1-Cre, line in black, n = 27 from 4 mice). For each group, the distribution was established using 200 consecutive events from each of the recorded cells; p = 0.0007, K–S test. C, The mean frequency of sIPSCs for the three groups (4.9 ± 0.3 Hz for Mecp2F/y;Emx1-Cre; 6.7 ± 0.4 Hz for Mecp2wt/y;Emx1-Cre; 7.1 ± 0.3 Hz for Mecp2F/y; ***p < 0.001). D, The mean peak amplitude of sIPSCs for the three groups (34.2 ± 1.1 pA for Mecp2F/y;Emx1-Cre; 34.6 ± 1.4 pA for Mecp2wt/y;Emx1-Cre; and 36.7 ± 2.2 pA for Mecp2F/y; p > 0.3). The mean decay constant of sIPSC was 5.6 ± 0.3 ms for mutant neurons (Mecp2F/y; Emx1-Cre), 5.5 ± 0.2 ms for Mecp2F/y control, and 5.8 ± 0.2 ms for Mecp2wt/y; Emx1-Cre control (p = 0.53, one-way ANOVA). E, Distributions of mIPSC intervals for mutant neurons (Mecp2F/y; Emx1-Cre, gray) and control neurons (Mecp2wt/y; Emx1-Cre, black). p = 0.0003, K–S test. The mean frequency of mIPSC was 4.5 ± 0.2 Hz (n = 22 from 3 mice) for mutant neurons, and 6.4 ± 0.5 Hz (n = 24 from 3 mice) for control neurons (p = 0.001). F, Distributions of mIPSC amplitude for mutant neurons (Mecp2F/y; Emx1-Cre, gray) and control neurons (Mecp2wt/y; Emx1-Cre, black). The mean peak amplitude of mIPSC was 28.1 ± 1.2 pA for mutant neurons, and 28.5 ± 1.3 pA for control neurons (p = 0.68). G, Averaged mIPSCs for mutant and control neurons. The mean decay constant of mIPSC was 6.1 ± 0.2 ms for mutant neurons, and 5.7 ± 0.2 ms for control neurons (p = 0.08).
To determine whether GABAergic defects are present in young adult mutant mice, we recorded from layer 5 pyramidal neurons in mPFC of mice aged 6–7 weeks. The frequency of mIPSCs was significantly reduced in Emx1-Mecp2 mutant neurons (5.8 ± 0.4 Hz, n = 15 cells from 3 mutant mice vs 7.3 ± 0.4 Hz, n = 17 cells from 3 control mice; p = 0.008), whereas the peak amplitude of mIPSCs was not different between mutant and control neurons (30.9 ± 1.6 pA for mutant vs 31.4 ± 2.0 pA for control; p = 0.88). These results are consistent with those obtained at 3 weeks of age, suggesting that deletion of Mecp2 from excitatory neurons in the forebrain causes a significant reduction of GABAergic transmission in young adult mice.
To examine whether deletion of Mecp2 alters excitatory transmission, we recorded mEPSCs from layer 5 pyramidal neurons in the mPFC of Emx1-Mecp2 mutant and control mice at P17–P18 and 6–7 weeks of age. At P17–P18, there was no difference between mutant and control neurons in the frequency, amplitude, or decay time of mEPSCs (Fig. 3). Similar results were obtained at 6–7 weeks of age. At 6–7 weeks of age, the mean frequency of mEPSCs was 4.8 ± 0.8 Hz (n = 15 from 3 mice) for control neurons, and 5.8 ± 0.7 Hz (n = 14 from 3 mice) for mutant neurons (p = 0.25); the mean peak amplitude of mEPSCs was 14.2 ± 1.1 pA for control neurons, 12.9 ± 0.8 for mutant neurons (p = 0.51); decay constant of mEPSC was 4.2 ± 0.2 ms for control neurons, and 4.1 ± 0.1 ms for mutant neurons (p = 0.39). These results indicate a selective reduction of inhibitory synaptic transmission in mPFC of Emx1-Mecp2 mice.
Figure 3.

Deletion of Mecp2 with Emx1-Cre had no effect on excitatory transmission in the prefrontal cortex. A, mEPSCs recorded from a control (top trace, black) and mutant (bottom trace, gray) layer 5 pyramidal neurons in mPFC at P18 in presence of TTX (0.3 μm) and picrotoxin (100 μm). B, Distributions of mEPSC intervals for mutant (black) and mutant (gray) neurons. The mean frequency was 6.3 ± 0.6 Hz for control neurons (n = 24 from 3 mice) and 6.4 ± 0.6 Hz for mutant neurons (n = 21 from 3 mice; p = 0.71). C, Distributions of mEPSC peak amplitude for control (black) and mutant (gray) neurons. The mean peak amplitude was 11.8 ± 0.4 pA for control and 12.0 ± 0.3 pA for mutant neurons (p = 0.99). Inset, Averaged mEPSCs for control (black) and mutant (gray) neurons. Scale bars, 2 pA and 2 ms. The mean decay constant of mEPSCs was 3.7 ± 0.1 ms for control and 3.8 ± 0.1 ms for mutant neurons (p = 0.89).
Although the mPFC is critically implicated in cognitive functions, its role in absence seizures is unclear. Previous studies have shown that in a rat model of absence seizure spike-wave discharges are initiated in the deep layers of the somatosensory cortex (Polack et al., 2007). Therefore we recorded from layer 5 pyramidal neurons in the somatosensory cortex (S1; Fig. 4). Similar to what we found in the mPFC, the frequency of sIPSCs was reduced by 25% in S1 neurons of Emx1-Mecp2 mutant mice (Fig. 4B), without significant changes in the amplitude or decay time of sIPSCs (Fig. 4C,D). Similar results were obtained for mIPSCs. The mean frequency of mIPSCs was 7.1 ± 0.3 Hz (n = 18 cells from 3 mice) for mutant and 9.7 ± 0.4 Hz (n = 23 from 3 mice) for control (p = 0.0001); the mean peak amplitude of mIPSCs was 31.5 ± 1.4 pA for mutant and 35.3 ± 1.7 pA for control (p = 0.124); the decay constant was 5.4 ± 0.1 ms for mutant and 5.6 ± 0.1 ms for control (p = 0.854). We also recorded mEPSCs from layer 5 pyramidal neurons in S1 of mutant and control mice. There was no significant difference between mutant and control neurons in the frequency (6.2 ± 0.6 Hz, n = 16 for mutant vs 5.6 ± 0.4 Hz, n = 18 for control; p = 0.40), amplitude (15.5 ± 0.3 pA for mutant vs 14.6 ± 0.4 pA for control; p = 0.18), or decay time (3.0 ± 0.1 ms for mutant vs 3.2 ± 0.1 ms for control; p = 0.06) of mEPSCs. Together, these results suggest that deletion of Mecp2 from the cortical neurons selectively reduces GABAergic transmission in the cortex.
Figure 4.

Deletion of Mecp2 with Emx1-Cre reduced GABAergic transmission in the somatosensory cortex. A, sIPSCs recorded from a control (top trace, black) and mutant (bottom trace, gray) layer 5 pyramidal neurons in S1 at P18. B, Distributions of sIPSC intervals for mutant neurons (Mecp2F/y; Emx1-Cre, gray) and control neurons (Mecp2wt/y; Emx1-Cre, black); **p < 0.001, K–S test. The mean frequency of sIPSCs for control (8.7 ± 0.4 Hz, n = 17 cells from 3 mice) and mutant neurons (6.5 ± 0.6 Hz, n = 19 cells from 3 mice; **p = 0.004). C, Distributions of sIPSC amplitude for mutant neurons (Mecp2F/y; Emx1-Cre, gray) and control neurons (Mecp2wt/y; Emx1-Cre, black). The peak amplitude of sIPSCs for control (37.8 ± 2.3 pA) and mutant neurons (34.3 ± 1.5 pA; p = 0.13). D, Averaged sIPSCs for control and mutant neurons. The mean decay constant of sIPSCs was 5.6 ± 0.2 ms for control neurons, and 4.9 ± 0.1 ms for mutant neurons (p = 0.04).
Deletion of Mecp2 from forebrain inhibitory neurons has no effects on GABAergic transmission in layer 5 pyramidal neurons
Recent studies have shown that deletion of Mecp2 from all inhibitory neurons in the brain reduces GABAergic transmission in the cortex (Chao et al., 2010). To determine whether MeCP2 in cortical inhibitory neurons plays a role in GABAergic transmission in the cortex, we examined GABAergic transmission in the cortex of conditional Dlx6a-Mecp2 mice. In the Dlx6a-Cre strain, Cre recombinase is selectively expressed in GABAergic neurons in the forebrain (Monory et al., 2006). To ascertain the efficiency of deletion, we first performed GABA immunostaining on brain sections from mice that carried the Dlx6a-Cre and Rosa-tdTomato reporter. All GABA-positive neurons in the mPFC also expressed tdTomato (Fig. 4A), indicating that all GABAergic neurons in the mPFC of the Dlx6a-Cre mice express Cre recombinase. Approximately 10% of tdTomato-expressing cells in the mPFC of Dlx6a-Cre;Ai14 mice at P17–P18 did not show GABA immunoreactivity. These cells were smaller than GABA-positive ones, and presumably were immature interneurons. Next, we performed MeCP2 immunostaining on sections from conditional Dlx6a-Mecp2 mutant mice (Mecp2F/y;Dlx6a-Cre) carrying the Rosa-tdTomato reporter. All tdTomato-expressing cells were MeCP2-negative, whereas neighboring tdTomato-negative cells showed normal MeCP2 immunoreactivity (Fig. 5B). These results indicate that in Dlx6a-Mecp2 mutant mice, Mecp2 was deleted from all GABAergic neurons in the mPFC. Spontaneous GABAergic transmission was analyzed in layer 5 pyramidal neurons from Dlx6a-Mecp2 mutant and control littermates at P17–P20. We found no difference between mutant and control neurons in the frequency, amplitude, or decay time of sIPSCs (Fig. 5C–E).
Figure 5.

Selective deletion of Mecp2 from forebrain GABAergic neurons had no effect on spontaneous GABAergic transmission. A, tdTomato signal and GABA immunostaining in the mPFC of a Dlx6a-Cre; Ai14 mouse. Scale bar, 20 μm. B, tdTomato signal and MeCP2 immunostaining in the mPFC of a Mecp2F/y; Dlx6a-Cre; Ai14 mouse. Scale bar, 20 μm. C, sIPSCs recorded from a control (Mecp2F/y) and mutant (Mecp2F/y; Dlx6a-Cre) neurons at P18. D, Distributions of sIPSC intervals for control (black) and mutant (gray) neurons. The mean frequency of sIPSCs was 6.3 ± 0.4 Hz (n = 15 from 3 mice) for control neurons, and 6.0 ± 0.3 Hz (n = 19 from 3 mice) for mutant neurons (p = 0.82). E, Distributions of sIPSC amplitude for control and mutant neurons. Inset, Averaged sIPSCs from control and mutant neurons. Scale bars, 10 ms and 10 pA. The mean amplitude of sIPSCs was 38.1 ± 3.0 pA for control neurons, and 36.9 ± 1.9 pA for mutant neurons (p = 0.8). The mean decay constant was 6.2 ± 0.3 ms for control neurons, and 5.7 ± 0.2 ms for mutant neurons (p = 0.2).
Similar results were obtained from layer 5 pyramidal neurons in the somatosensory cortex at P18–P20. The mean frequency of sIPSCs was 8.5 ± 0.3 Hz (n = 17 cells from 3 mice) for mutant, and 8.2 ± 0.4 Hz (n = 20 cells from 3 mice) for control (p = 0.65); the mean peak amplitude was 38.9 ± 2.7 pA for mutant, and 40.2 ± 2.1 pA for control (p = 0.46); the mean decay time was 5.5 ± 0.2 ms for mutant, and 5.1 ± 0.3 ms for control (p = 0.43). Together, these findings suggest that MeCP2 expression by cortical interneurons is not essential for the formation and function of GABAergic synapses in layer 5 pyramidal neurons.
Defect of GABAergic transmission is restricted to neurons lacking MeCP2
In Emx1-Mecp2 mutant mice, Mecp2 was deleted from all neurons in the cortex. This large-scale removal of MeCP2 in the cortex may have indirect effects on GABAergic transmission. For example, Mecp2 deletion may alter cortical activity, which in turn leads to widespread changes in GABAergic transmission. In addition, Cre recombinase is also expressed in glia cells in the cortex of Emx1-Cre mice (Gorski et al., 2002), and deletion of Mecp2 from glia cells may contribute to GABAergic defects. To examine these possibilities, we took advantage of the sparse expression of Cre recombinase in layer 2/3 of S1 of SERT-Cre mice. Using the Ai14 reporter (Madisen et al., 2010) and NeuN staining, we found that 19% (258 of 1339) of neurons in upper layer 2/3 of S1 in SERT-Cre mice were Cre+ (Fig. 6A,B). Consistent with our previous results (Zhang et al., 2013), all Cre+ cells in the cortex of SERT-cre mice were NeuN+, indicating that Cre expression in these mice was restricted to neurons. Immunostaining for GABA indicated that 94% of Cre+ neurons in layer 2/3 were pyramidal neurons and negative for GABA (Fig. 6C); in contrast, only 16% of GABA-positive neurons were Cre+. We recorded from layer 2/3 pyramidal neurons in S1 of SERT-Mecp2 mutant mice carrying the Ai14 reporter (SERT-Mecp2-Ai14, or Mecp2F/y; SERT-Cre; Ai14). Cre+ and Cre− neurons were identified by epifluorescence of tdTomato. GABAergic transmission was significantly reduced in Cre+ layer 2/3 pyramidal neurons compared with Cre− ones in the same animals (Fig. 6D–G). The frequency of sIPSCs was reduced by 57% in mutant neurons (Fig. 6E, red and black bars), with a small reduction (20%) in sIPSC amplitude (Fig. 6F, red and black bars). To examine any side effects of Cre and tdTomato expression, we also recorded from Cre+ and Cre− neurons in SERT-Ai14 mice wild-type for Mecp2 (Mecp2wt/y; SERT-Cre; Ai14); there was no difference in sIPSCs between Cre+ and Cre− neurons of control mice (Fig. 6E,F, blue and purple bars). Together, these results demonstrate that the lack of MeCP2 in postsynaptic neurons causes a reduction of GABAergic transmission.
Figure 6.
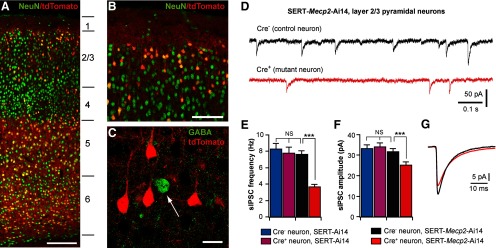
Mosaic deletion of Mecp2 in the cortex selectively disrupts GABAergic transmission in neurons deficient in MeCP2. A, confocal image of the S1 in coronal section of a SERT-Ai14 mouse at P21. Neurons were labeled with anti-NeuN (green); Cre+ cells were identified with tdTomato signal (red). The lines and numbers on the side indicate the locations of the six cortical layers. Scale bar, 200 μm. B, Layer 1 and upper layer 2/3 of S1 at a higher-magnification. Scale bar, 100 μm. Approximately 19% of neurons in upper layer 2/3 were Cre+. C, Layer 2/3 of S1 immunostained for GABA (green). The vast majority of Cre+ neurons (red) in layer 2/3 were pyramidal neurons and negative for GABA. Scale bar, 20 μm. D, sIPSCs recorded from a Cre− (top trace, black) and a Cre+ (bottom trace, red) pyramidal neurons in S1 layer 2/3 of a SERT-Mecp2-Ai14 mutant mouse at P18. E, Mean frequency of sIPSCs from Cre− and Cre+ layer 2/3 pyramidal neurons in SERT-Mecp2-Ai14 mutant and SERT-Ai14 control mice at P17–P18. For mutant mice, the mean frequency was 7.6 ± 0.4 Hz (n = 18, for Cre− neurons, black) and 3.6 ± 0.3 Hz (n = 17, for Cre+ neurons, red; ***p < 0.0005). For control mice, the mean frequency was 8.3 ± 0.7 Hz (n = 17, for Cre− neurons, blue) and 7.8 ± 0.7 Hz (n = 17, for Cre+ neurons, purple). F, Mean peak amplitude of sIPSCs for the four groups. The mean peak amplitude was 31.6 ± 1.6 pA (Cre− neurons in mutant mice), 25.0 ± 1.6 pA (Cre+ neurons in mutant mice), 33.3 ± 1.8 pA (Cre− neurons in control mice), and 34.1 ± 1.9 pA (Cre+ neurons in control mice); ***p < 0.001. G, Averaged sIPSCs for Cre− (in black) and Cre+ (in red) neurons in mutant mice. The decay time constant of sIPSCs was 7.5 ± 0.2 ms (Cre− neurons in mutant mice), 8.4 ± 0.2 ms (Cre+ neurons in mutant mice), 7.4 ± 0.1 ms (Cre− neurons in control mice), and 7.6 ± 0.2 ms (Cre+ neurons in control mice). The decay time of Cre+ neurons in mutant mice was significantly slower than the other three groups (**p = 0.002).
Forebrain-restricted deletion of Mecp2 reduces the number of GABAergic synapses in the cortex
We further examined synaptic mechanisms by which MeCP2 regulates GABAergic transmission. The lack of change in the amplitude or kinetics of sIPSCs in Emx1-Mecp2 mutant neurons indicates that alterations in presynaptic function or synapse number are the possible causes. We recorded evoked IPSCs in neurons from Emx1-Mecp2 mutant and littermate control mice at P18–P20. Stimulation electrodes made of twisted microwires were placed 200 μm away from the recorded cell, and current pulses were applied at 1/15 s. Monosynaptic IPSCs were recorded from mutant and control neurons in the presence of DNQX and kynurenic acid (Fig. 7A). For each cell, the input–output relationship was determined by stimuli at a range of intensity. Mutant neurons showed similar IPSC threshold as control ones (Fig. 7B). However, peak amplitudes of IPSCs were significantly smaller in mutant neurons than control ones (Fig. 7B). The maximal IPSC, reached at about the same stimulus intensities in mutant and control neurons, was 2.6 ± 0.2 nA (n = 14) for mutant neurons, and 3.5 ± 0.3 nA (n = 13) for control neurons (p = 0.02). We recorded paired pulse responses at intensities that produced half-maximal IPSCs. There was no difference between mutant and control neurons in paired pulse ratio (Fig. 7C). These results indicate that a decrease in the number of GABAergic synapses may be the primary cause of the reduction of GABAergic transmission in mutant neurons.
Figure 7.

Evoked GABAergic transmission and the number of GABAergic synapses were reduced in the mPFC of Emx1-Mecp2 mutant mice. A, Monosynaptic IPSCs recorded from control and mutant neurons evoked by local stimulation. B, The input–output curve of IPSCs for control (n = 13 from 3 mice) and mutant (n = 14 from 3 mice) neurons. C, Paired pulse responses for IPSCs from control and mutant neurons. The averaged IPSCs from control and mutant neurons were normalized to the peak of the first IPSC. The paired pulse ratio at 100 ms interval was 0.89 ± 0.02 (n = 10 from 3 mice) for mutant neurons, and 0.85 ± 0.03 (n = 11 from 3 mice) for control neurons (p = 0.14). D, Double immunostaining of VGAT (red) and NeuN (green) in the mPFC of control and mutant mice at P18 (top) and 7 weeks of age (bottom). Scale bar, 10 μm. E, Numbers of VGAT puncta per 1000 μm3 volume in layer 5 of mPFC of mutant and control mice at P18–P19 and 7–8 weeks of age. Data were obtained from four pairs of mice at P18–P19 and 7–8 weeks of age, respectively; three measurements were performed for each mouse. P values were obtained with Mann–Whitney test.
To analyze changes in the number of GABAergic synapses, we performed immunofluorescence staining of VGAT on brain sections from Emx1-Mecp2 mutant and littermate control mice. At P18–P19, Emx1-Mecp2 mutant mice showed significantly fewer VGAT-positive puncta in layer 5 of mPFC than control mice (Fig. 7D, top). The density of VGAT-positive puncta was reduced by 42% in layer 5 of mutant mice (Fig. 7E). Similar reductions (37%) were observed in young adult mutant mice at 7–8 weeks of age (Fig. 7D, bottom, E). These results indicate that conditional deletion of Mecp2 with Emx1-Cre leads to reductions in the number of GABAergic synapses in the mPFC.
Deletion of Mecp2 enhances neuronal discharges through disinhibition
Our results so far suggest that selective deletion of Mecp2 from cortical neurons reduces GABAergic transmission. To determine the effect of Mecp2 deletion on neuronal activity, we performed current-clamp recordings from layer 5 pyramidal neurons of the mPFC in slices obtained from Emx1-Mecp2 mutant mice and control littermates at P18–P20. Neuronal firing was examined first at the resting potential (typically ∼−59 mV), and then at −53 mV by applying a small depolarizing current to adjust the interspike membrane potential. At the resting potential, neither mutant nor control neurons showed any spontaneous firing (Fig. 8A). At −53 mV, more mutant neurons (24/26) fired action potential than control (15/22; p = 0.033, Fisher's exact test). The average firing rate was significantly higher in mutant neurons (Fig. 8B).
Figure 8.

Hyperexcitation of cortical neurons in Emx1-Mecp2 mutant mice. A, Neuronal activity at the resting potential (−58, −59 mV) and at −53 mV in a control (Mecp2wt/y; Emx1-Cre) and mutant (Mecp2F/y; Emx1-Cre) neurons. B, Frequency of action potential of control and mutant neurons at the resting potential and at −53 mV. The mean firing frequency at −53 mV was 0.3 ± 0.2 Hz for control (n = 21 from 3 mice), and 1.6 ± 0.4 Hz for mutant (n = 26 from 3 mice; ***p < 0.001). C, Spontaneous IPSCs recorded from wild-type neurons without and with picrotoxin (0.5 mm) in the patch pipette. These recordings were obtained at 0 mV with the pipette solution containing Cs-methylsulfate ([Cl-]i of 35 mm). D, Firing recorded from a control and mutant neurons at −53 mV with picrotoxin in the patch pipettes. E, Firing frequency of control and mutant neurons at −60 and −53 mV with intracellular picrotoxin. The mean firing frequency at −53 mV was 3.0 ± 0.6 Hz for control (n = 24 from 3 mice), and 2.7 ± 0.5 Hz for mutant (n = 23 from 3 mice; p = 0.86). F, Membrane potential changes in response to current steps in control and mutant neurons. Synaptic transmission was blocked by bath perfusion of DNQX, kynurenic acid, and picrotoxin. G, Plot of spike frequency versus injected current for control (n = 27 from 3 mice) and mutant neurons (n = 31 from 3 mice).
The increased firing observed in mutant neurons could be due to changes in either intrinsic excitability of neurons or synaptic activity; these two possibilities are not mutually exclusive. Our results on synaptic activity suggest that a reduction of GABAergic transmission contributes to the hyperexcitation of Mecp2 mutant neurons. We reasoned that if a reduction in GABAergic transmission is the cause of hyperexcitation in mutant neurons, blocking GABAergic transmission in both control and mutant neurons should abolish the difference between them. Because bath application of GABAA antagonists causes epileptiform activity in cortical slices, we used intracellular application of picrotoxin to block GABAergic transmission in single cells (Inomata et al., 1988). Picrotoxin (0.5 mm) included in the patch pipette blocked sIPSCs in pyramidal neurons (Fig. 8C). Picrotoxin abolished the difference between Emx1-Mecp2 mutant and control neurons in firing, and enhanced firing in both mutant and control neurons (Fig. 8D,E). These results suggest that conditional deletion of Mecp2 with Emx1-Cre causes hyperexcitation of layer 5 pyramidal neurons by reducing GABAergic transmission in the cortex.
We next assessed intrinsic excitability of layer 5 pyramidal neurons in the mPFC in the presence of synaptic blockers picrotoxin (100 μm), DNQX (10 μm), and kynurenic acid (1 mm). Firing was evoked by a series of depolarizing current steps (Fig. 8F). The curve of the mean firing rate vs injected current was not different between mutant and control neurons (Fig. 8G). Furthermore, there was no difference between mutant and control neurons in resting potential (−60.8 ± 0.4 mV, n = 27, for control; −59.9 ± 0.4 mV, n = 31 for mutant; p = 0.08), input resistance (125 ± 8 MΩ for control; 113 ± 7 MΩ for mutant; p = 0.19), or spike threshold (−38.1 ± 0.4 mV for control; −38.6 ± 0.5 mV for mutant; p = 0.41). These results indicate that intrinsic excitability of layer 5 pyramidal neurons was not altered by Mecp2 deletion.
Discussion
Our results suggest that MeCP2 in cortical excitatory neurons has a key role in regulating GABAergic transmission and excitability of cortical circuits. Deletion of Mecp2 from excitatory but not inhibitory neurons in the forebrain leads to seizures, hyperexcitation, and a reduction of GABAergic transmission. Using pharmacological manipulation at the single-cell level, we provide evidence that loss of MeCP2 from cortical neurons leads to enhanced excitability through a reduction of GABAergic transmission.
Seizure and cortical excitability in MeCP2-deficient brain
Studies from several patient groups have shown that seizure is a common feature of RTT (Jian et al., 2007; Nissenkorn et al., 2010; Pintaudi et al., 2010; Cardoza et al., 2011). Both partial and generalized seizures are common among RTT patients (Cardoza et al., 2011). The types of generalized seizures at the onset showed a wide spectrum; the largest group had generalized tonic-clonic seizures, and this is followed by the group showing atypical absence seizures (Pintaudi et al., 2010). Mouse models of RTT recapitulate the seizure phenotype. Recurrent seizures were found in both male (Mecp2−/y) and female heterozygote animals (Shahbazian et al., 2002; Chao et al., 2010; D'Cruz et al., 2010). In female Mecp2 heterozygote mice, spike-wave discharges of 1–2 s in duration were detected in the cortex (D'Cruz et al., 2010). These spontaneous discharges occurred in wake immobile mice, and were sensitive to ethosuximide. Overall, EEG results obtained from RTT patients and Mecp2 mutant mice indicate a hyperexcitation of the cortex and associated regions in MeCP2-deficient brains.
In contrast to in vivo data, early studies using brain slices showed a reduction of firing in cortical pyramidal neurons of germline Mecp2 knock-out mice, and the effect was associated with a reduction of excitatory transmission (Dani et al., 2005). In the present study, we obtained different results: cortical pyramidal neurons in Emx1-Mecp2 mice showed an increase of firing and the effect was associated with a reduction of inhibitory transmission without any change in quantal excitatory transmission. The reason for this discrepancy is unknown. One possibility is the differences in experimental methods. The previous study used low concentrations of divalent cations (0.5 mm Mg2+ and 1.0 mm Ca2+) for the recording of cell firing and synaptic currents (Dani et al., 2005), whereas we used physiological concentrations of divalent cations (1.3 mm Mg2+ and 1.5 mm Ca2+). Neurons and synapses in MeCP2-deficient brains may be different from wild-type neurons in response to low concentrations of Mg2+ and Ca2+. Although no change in intrinsic excitability was found in the previous study, those experiments were conducted in ACSF containing higher concentrations of divalent cations (2 mm Mg2+ and 2 mm Ca2+). It is also unclear whether the reduction in excitatory transmission observed in the previous study significantly contributed to the change in cell firing. There was a 15% reduction in mEPSC amplitude without significant change in mEPSC frequency, and no significant change in the amplitude or frequency of mIPSCs was observed (Dani et al., 2005). In hippocampal neurons, deletion of Mecp2 reduced the frequency but not amplitude of mEPSCs, and the effect was associated with a reduction of the number of glutamatergic synapses (Chao et al., 2007). However, the difference in the number of glutamatergic synapses was observed at 2 weeks of age, but not at 5 weeks of age. It should be noted that our analysis of excitatory transmission was limited to mEPSCs. Therefore, we cannot exclude the possibility that MeCP2 may regulate some aspects of presynaptic function at cortical excitatory synapses. A recent study has shown reductions of visual acuity and neuronal activity in visual cortical neurons of adult Mecp2 knock-out mice (Durand et al., 2012). These effects appeared to be restricted to layer 4 neurons in the visual cortex, and may be related to defects of synaptic transmission observed at retinogeniculate synapses in Mecp2 knock-out mice (Noutel et al., 2011).
Another possibility is the difference in genetic background. Although the previous study used mice on a mixed 129/B6/BALB background (Dani et al., 2005), our studies were conducted in mice on a predominantly B6 background. Because of the complex role of MeCP2 in transcriptional regulation (Chahrour et al., 2008; Guy et al., 2011), genetic background may have a strong effect on disease phenotypes. Indeed results from a recent study using female Mecp2 heterozygous mutant mice indicate that genetic background has significant effects on behavioral phenotypes including anxiety-like behavior and prepulse inhibition (Samaco et al., 2013). Previous studies have reported that MeCP2-deficient mice on a B6 background exhibit hyperexcitability discharges and seizures (Chao et al., 2010; D'Cruz et al., 2010). Our results are consistent with these findings.
GABAergic dysfunction in MeCP2-deficient brain
Several studies reported reductions of GABAergic transmission in MeCP2-deficient brain. In both the brainstem and thalamus of Mecp2 knock-out mice, the reduction of GABAergic transmission was associated with a decrease in the number of GABAergic synapses (Medrihan et al., 2008; Zhang et al., 2010). Our results indicate that similar changes occur in the cortex. Emx1-Mecp2 mutant mice showed reductions in mIPSC frequency and the number of VGAT-positive puncta in the cortex. There was little change in the amplitude or kinetics of mIPSC, and the reduction in mIPSC frequency was comparable to the change in the number of GABAergic synaptic terminals. These results suggest that a major consequence of Mecp2 deletion is a downregulation of GABAergic innervation.
A recent study showed that deletion of Mecp2 from all GABAergic neurons in the brain using a Viaat-Cre driver led to cortical hyperexcitation, but selective deletion of Mecp2 from forebrain GABAergic neurons using Dlx5/6-Cre did not cause hyperexcitation (Chao et al., 2010). This latter observation is confirmed by our results using the same Cre driver with a different nomenclature Dlx6a-Cre. Dlx6a-Mecp2 mutant mice did not show any spike-wave discharges, and our immunostaining data demonstrated that Mecp2 was deleted from all cortical GABAergic neurons in Dlx6a-Mecp2 mice. Spontaneous GABAergic transmission was not altered in cortical neurons in Dlx6a-Mecp2 mice. In contrast, Viaat-Mecp2 mutant mice showed reductions of GAD and GABA levels in the cortex and striatum, and smaller mIPSCs (Chao et al., 2010). It is possible that loss of MeCP2 from hindbrain inhibitory neurons may have a broad impact on GABAergic function in the brain.
It remains unclear how the loss of MeCP2 causes dysfunction of GABAergic transmission. MeCP2 is expressed in all neuronal tissues by neurons, glia, and microglia (Ballas et al., 2009; Maezawa et al., 2009; Maezawa and Jin, 2010; Lioy et al., 2011). Although MeCP2 in glia has been implicated in changes in dendritic morphology and level of vesicular glutamate transporter in Mecp2-knock-out mice, the role of glial MeCP2 in GABAergic transmission is unknown. Our single-cell deletion analysis in SERT-Mecp2 mutant mice suggests that glial MeCP2 is not essential for GABAergic defects in cortical pyramidal neurons in mutant mice. However, we cannot exclude the possibility that MeCP2 in glia is implicated in GABAergic transmission in other brain regions.
Deletion of Mecp2 from all excitatory neurons in the cortex may alter GABAergic transmission through activity-dependent network effects. However, our results of single-cell deletion showed that the loss of MeCP2 from a small number of cortical pyramidal neurons caused a selective reduction of GABAergic transmission in these neurons. Our finding suggests a model where MeCP2 in postsynaptic neurons regulates retrograde signaling at GABAergic synapses. As a transcriptional regulator, MeCP2 has a large number of target genes (Chahrour et al., 2008), and therefore may regulate GABAergic innervation through multiple pathways. MeCP2 has been implicated in the regulation of brain-derived neurotrophic factor (BDNF; Wang et al., 2006; Zhou et al., 2006; Li et al., 2012); loss of BDNF from single neurons leads to a reduction of GABAergic innervation (Kohara et al., 2007); increasing BDNF level in the brain of Mecp2 mutant mice rescues some neurological defects (Chang et al., 2006; Kline et al., 2010). However, the level of BDNF was not altered in the brain of Mecp2 knock-out mice at 2 weeks of age, and was only slightly reduced in the cortex of mutant mice at 6–8 weeks of age (Chang et al., 2006). BDNF signaling was impaired in hippocampal pyramidal neurons in Mecp2 knock-out mice (Li et al., 2012). Future studies are needed to determine whether MeCP2 regulates BDNF signaling in GABAergic neurons.
So far GABAergic defects have been reported in mice with either germline Mecp2 deletion or conditional Mecp2 deletion using early onset Cre drivers. In our case, both Emx1-Cre and SERT-Cre are expressed by birth in the cortex. Together with other studies in the brainstem and thalamus (Medrihan et al., 2008; Zhang et al., 2010), our results suggest that MeCP2 plays an important role in the maturation of GABAergic synapses in the developing brain. On the other hand, MeCP2 is required in the adult for normal brain function (Guy et al., 2007; McGraw et al., 2011; Cheval et al., 2012). It may be interesting and important to know whether MeCP2 plays a role in the maintenance and plasticity of GABAergic synapses in adult brain.
Footnotes
This work was supported by NIH Grants NS082658 (Z.-w.Z.), NS064013 (Z.-w.Z.), and NS031348 (W.N.F.). We thank Da-Ting Lin and Kevin Seburn for comments on the paper, and Verity Letts and Hong Liu for expert assistance.
References
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer B, Deleuze C, Letts VA, Mahaffey CL, Boumil RM, Lew TA, Huguenard JR, Frankel WN. Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Hum Mol Genet. 2008;17:1738–1749. doi: 10.1093/hmg/ddn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfa G, Hablitz JJ, Pozzo-Miller L. Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J Neurophysiol. 2011;105:1768–1784. doi: 10.1152/jn.00800.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoza B, Clarke A, Wilcox J, Gibbon F, Smith PE, Archer H, Hryniewiecka-Jaworska A, Kerr M. Epilepsy in Rett syndrome: association between phenotype and genotype, and implications for practice. Seizure. 2011;20:646–649. doi: 10.1016/j.seizure.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamate synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet. 2012;21:3806–3814. doi: 10.1093/hmg/dds208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol Dis. 2010;38:8–16. doi: 10.1016/j.nbd.2009.12.018. [DOI] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, Carninci P, Hensch TK, Fagiolini M. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 2012;76:1078–1090. doi: 10.1016/j.neuron.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Egner T, Kalisch R. Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn Sci. 2011;15:85–93. doi: 10.1016/j.tics.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neurosci Biobehav Rev. 2009;33:773–783. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inomata N, Tokutomi N, Oyama Y, Akaike N. Intracellular picrotoxin blocks pentobarbital-gated Cl− conductance. Neurosci Res. 1988;6:72–75. doi: 10.1016/0168-0102(88)90007-7. [DOI] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Christodoulou J, Leonard H. Seizures in Rett syndrome: an overview from a one-year calendar study. Eur J Paediatr Neurol. 2007;11:310–317. doi: 10.1016/j.ejpn.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Calfa G, Larimore J, Pozzo-Miller L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc Natl Acad Sci U S A. 2012;109:17087–17092. doi: 10.1073/pnas.1205271109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, Mackie K, Elphick MR, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Adachi M, Autry AE, Mahgoub MA, Kavalali ET, Monteggia LM. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci. 2012;32:3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nissenkorn A, Gak E, Vecsler M, Reznik H, Menascu S, Ben Zeev B. Epilepsy in Rett syndrome: the experience of a national Rett center. Epilepsia. 2010;51:1252–1258. doi: 10.1111/j.1528-1167.2010.02597.x. [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, Chen C. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron. 2011;70:35–42. doi: 10.1016/j.neuron.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintaudi M, Calevo MG, Vignoli A, Parodi E, Aiello F, Baglietto MG, Hayek Y, Buoni S, Renieri A, Russo S, Cogliati F, Giordano L, Canevini M, Veneselli E. Epilepsy in Rett syndrome: clinical and genetic features. Epilepsy Behav. 2010;19:296–300. doi: 10.1016/j.yebeh.2010.06.051. [DOI] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, McGraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2(±) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet. 2013;22:96–109. doi: 10.1093/hmg/dds406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/S0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignoli A, Fabio RA, La Briola F, Giannatiempo S, Antonietti A, Maggiolini S, Canevini MP. Correlations between neurophysiological, behavioral, and cognitive function in Rett syndrome. Epilepsy Behav. 2010;17:489–496. doi: 10.1016/j.yebeh.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18:294–309. doi: 10.1002/hipo.20389. [DOI] [PubMed] [Google Scholar]
- Zhang ZW. Maturation of layer V pyramidal neurons in the rat prefrontal cortex: intrinsic properties and synaptic function. J Neurophysiol. 2004;91:1171–1182. doi: 10.1152/jn.00855.2003. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Arsenault D. Gain modulation by serotonin in pyramidal neurons of the rat prefrontal cortex. J Physiol. 2005;566:379–394. doi: 10.1113/jphysiol.2005.086066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol. 2010;103:2470–2481. doi: 10.1152/jn.00601.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZW, Peterson M, Liu H. Essential role of postsynaptic NMDA receptors in developmental refinement of excitatory synapses. Proc Natl Acad Sci U S A. 2013;110:1095–1100. doi: 10.1073/pnas.1212971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]