

Abstract
AIM: To evaluate the effects of the proteasome inhibitor bortezomib (BZB) on E2Fs and related genes in hepatocellular carcinoma (HCC) cells.
METHODS: The mRNA levels of the E2F family members (pro-proliferative: E2F1-3 and anti-proliferative: E2F4-8) and of their related genes cyclins and cyclin-dependent kinases (cdks) were evaluated in two HCC cell lines following a single BZB administration. mRNA levels of the epithelial-mesenchymal transition (EMT) genes were also measured in both cell lines after BZB treatment. The BZB concentration (40 nmol/L) used was chosen to stay well below the maximal amount/cm2 recommended for in vivo application, and 2 d incubation was chosen as this time point has been found optimal to detect BZB effects in our previous studies. The HCC cell lines, HepG2 and JHH6, were chosen as they display different phenotypes, hepatocyte-like for HepG2 and undifferentiated for JHH6, thus representing an in vitro model of low and high aggressive forms of HCC, respectively. The mRNA levels of the target genes were measured by two-color microarray-based gene expression analysis, performed according to Agilent Technologies protocol and using an Agilent Scan B. For the E2F family members, mRNA levels were quantified by real-time reverse transcription polymerase chain reaction (RT-PCR). Using small interfering RNA’s, the effects of E2F8 depletion on cell number was also evaluated.
RESULTS: After BZB treatment, microarray analysis of the undifferentiated JHH6 revealed a significant decrease in the expression of the pro-proliferative E2F member E2F2. Quantitative RT-PCR data were in keeping with the microarray analysis, and showed a significant increase and decrease in E2F8 and E2F2 mRNA levels, respectively. In contrast, BZB treatment of the hepatocyte-like HCC cell line HepG2 had a significant impact on mRNA levels of 5 of the 8 E2F members. In particular, mRNA levels of the pro-proliferative E2F members E2F1, E2F2, and of the anti-proliferative member E2F8, decreased over 80%. Notably, a reduction in E2F8 expression in HepG2 and JHH6 cells following siRNA treatment had no impact on cell proliferation. As observed with JHH6, BZB treatment of HepG2 cells induced a significant increase in mRNA levels of an anti-proliferative E2F member, E2F6 in this case. As was observed with E2F’s, more dramatic changes in mRNA levels of the E2F related genes cyclins and Cdks and EMT genes were observed after BZB treatment of HepG2 compared to JHH6.
CONCLUSION: The differential expression of E2Fs and related genes induced by BZB in diverse HCC cell phenotypes contribute to bortezomib’s mechanism of action in hepatocellular carcinoma.
Keywords: Bortezomib, Cyclins, E2F family, Hepatocellular carcinoma, Liver, Microarray, 26S proteasome
Core tip: The 26S proteasome inhibitor bortezomib has been proposed as a novel therapeutic molecule for hepatocellular carcinoma (HCC), being able to reduce cell growth. Little information is available on the effect of bortezomib (BZB) on many of E2Fs, a family of transcription factors regulating normal and tumor cell proliferation. Our data show, for the first time, the BZB effect on expression of E2F family members in HCC cell lines is not limited to the most studied E2F1, but, it extends also to other E2F members, in particular E2F2, E2F8 and E2F6, and the effect is phenotypic dependent.
INTRODUCTION
Hepatocellular carcinoma (HCC), accounting for more than 90% of primary liver cancers, is a global health problem[1-4] as it represents the sixth most common cancer and the third cause of cancer related death worldwide. The HCC incidence is age-related with a peak at 70 years. However, variation in different populations has been observed. For example, in Japan the highest incidence is between 70-79 years, whereas in Chinese and black African populations the age of appearance is younger. Males are more often affected than women with an estimated ratio of 2.4. HCC occurrence is highest in East Asia, sub-Saharan Africa, and Melanesia (85% of cases); in developed countries the frequency is lower with the exception of Southern Europe which has a significantly higher frequency compared to other developed countries. The most common risk factors for HCC development are represented by chronic viral hepatitis (types B and C), alcohol intake and aflatoxins exposure.
HCC is usually diagnosed at an advanced stage when the affected patients are often no longer eligible for curative treatments, such as liver resection, liver transplant, or local radiofrequency ablation. The efficacy of systemic chemotherapy is also limited due to the resistance of this disease to anticancer agents[5]. At the moment, the only systemic therapy showing a significant prolonged patient survival is based on the use of sorafenib. This drug is able to inhibit a number of kinases including Raf-1, c-Kit, and the pro-angiogenic receptor tyrosine kinases vascular endothelial growth factor receptor, platelet derived growth factor receptor and fibroblast growth factor receptor 1, all involved in HCC progression and overall prognosis[6]. However, sorafenib only modestly improves patient survival prolonging life span approximately of three months[7]. Thus, the development of novel therapeutic approaches to treat HCC-affected patients is urgently required.
In the last decade the drug bortezomib (BZB) has been studied as a possible novel therapeutic treatment for HCC. BZB is a boronic acid dipeptide derivative able to inhibit the 26S proteasome[8]. In particular, the boron atom present in BZB is responsible for the specific and efficient binding to the catalytic site of the 26S proteasome. This molecular machine is responsible for the degradation, via the ubiquitin proteasome pathway, of proteins involved in cell differentiation, apoptosis and cell cycle regulation including cyclins, cyclin-dependent kinase inhibitors and tumor suppressor proteins. BZB induces the inhibition of the 26S proteasome leading to the increase in levels of various proteins which lead to the generation of confounding signals that promote cell cycle arrest and the activation of the apoptotic program. So far the use of BZB is indicated for the treatment of multiple myeloma and relapsed mantle cell lymphoma[8].
BZB use in HCC is under evaluation as shown by a recent phase II clinical trial[9]. Interestingly, normal hepatocyte function is largely unaffected by BZB treatment, opening the possibility that this drug may not have important side effects in patients, at least when administered locally[10,11]. The mechanisms underlying BZB’s actions are complex and not completely understood. BZB is able to down-regulate HCC cell migration and invasion[12] by suppressing focal adhesion kinase expression[13]. It also promotes apoptosis[10,14] by reducing p-Akt levels[15-17] and it can induce autophagy via proteasome independent mechanisms[18]. With regard to the cell cycle, BZB has been shown to down-regulate HCC cell proliferation by increasing the levels of the cell cycle inhibitors p27/p21[12,19,20], and reducing the levels of cyclin D1[11,20], the phosphorylated form of the retinoblastoma protein pRB, and the transcription factor E2F1[11,20].
E2F1 belongs to a family of transcription factors (reviewed in[21]). The E2F family is currently divided into pro-proliferative (E2F1-E2F3) and anti-proliferative (E2F4-E2F8) members. In quiescent cells, the binding of E2F1-3 to the pocket protein pRb blocks the cell proliferation effects. In the presence of proliferative stimuli, pRB undergoes phosphorylation by cyclin-dependent kinases in complex with their cyclin partners, which in turn allows the release of E2Fs from pRB. Free E2Fs can induce the transcription of many cell cycle-related proteins including cyclin E, which when bound to its cyclin-dependent kinase (cdk) further phosphorylates pRB. This last event increases the amount of free E2Fs which can promote cell cycle progress by inducing the transcription of many S-phase genes, such as cyclin A and cdk 2.
With regard to the anti-proliferative E2Fs, E2F4 exerts its effect when bound to pRb or one of the other two pocket protein members p107 and p130. In contrast, E2F5 associates preferentially with p130. E2F6-8 seem to down-modulate the expression of E2F-responsive genes, and thus cell proliferation, independently of pocket protein binding.
E2F1 has been implicated in HCC cell growth[22-24] and we have observed that it is also involved in the BZB-induced down-modulation of cell growth in HCC cell lines[20]. With regard to the other E2F members, E2F3-E2F5-E2F8 have been shown to be up-regulated in HCC samples and to play a role in HCC cell growth[25-28].
With the exception of E2F1, little information is available with regard to the possible role of the other E2F family members in the BZB-induced inhibition of proliferation in HCC cell lines. This study investigated the effects of BZB treatment on E2F family members in the HCC cell lines HepG2 and JHH6.
MATERIALS AND METHODS
Cell lines and BZB treatment
The HCC cell lines JHH6 and HepG2 were cultured as reported in[20,24,29]. These cell lines were chosen as they display a different phenotype, hepatocyte-like for HepG2[30] (see also ATCC, catalogue No. HB-8065) and undifferentiated for JHH6[31] (see also Japanese Collection of Research Bioresources (JCRB), catalogue No.: JCRB1030), thus representing suitable in vitro models of low and high aggressive forms of HCC, respectively.
BZB was administered as described in[20]; briefly cells were seeded at 3.8 × 103 cells/cm2 in 6-wells plate, allowed adhering 24 h, cultured for two days in the presence of complete medium and 40 nmol/L BZB. The BZB concentration used was chosen to stay well below the maximal amount/cm2 recommended dose for in vivo application[32]. Moreover, two days of incubation was found to be optimal to study BZB effects on HepG2 and JHH6[20].
Two-color microarray-based gene expression analysis
Two-color microarray-based gene expression analysis was performed according to Agilent Technologies protocol, using an Agilent Scan B (supported with GenePix 4000B scanner and Feature Extraction software, version 9.5.3. – Agilent Technologies, United States).
Briefly, total RNA was extracted using the RNeasy Mini kit (Qiagen GmbH, Germany). The quality, integrity and quantification of total RNA was evaluated by spectrophotometric determination using a Lab-on-Chip-System Bioanalyzer 2100 (Applera Corporation, United States) and a NanoDrop ND-1000 (CelBio, Euroclone, Italy). Fluorescent cRNA (complementary RNA) was created using Agilent’s Quick Amp Labeling Kit (Agilent Technologies, United States) with a RNA sample input of 200 ng. For both JHH6 and HepG2, cRNAs were prepared from total RNAs obtained from non BZB-treated cells (NT) and BZB-treated cells (40 nmol/L). The fluorescent dye used for the labelling of NT cRNA was cyanine 3-CTP, (fluorescence emission wavelength of 570 nm) while for BZB-treated cells it was cyanine 5-CTP (fluorescence emission wavelength of 670 nm).
Complementary RNAs were then purified by RNeasy Mini kit (Qiagen S p A., Italy) and quantified using a NanoDrop ND-1000. According to Agilent protocol, for a 4 × 44 K microarray, 825 ng of each cRNAs were used. The Cy-labelled cRNA samples (CY-3 and CY-5 for NT and 40 nmol/L-BZB treated cells, respectively) were mixed and hybridized to a single microarray that was then scanned in a microarray scanner to visualize fluorescence of the two fluorophores. Relative intensities of each fluorophore were used in ratio-based analysis to identify up-regulated and down-regulated genes. Normalization of the data was conducted with RNA-spike ins; i.e., calibrating RNA transcripts. For each mRNA quantified, the Agilent microarray provides a minimum of three and up to ten hybridization oligonucletide probes which matches with different regions of the target mRNA. This means that for any given target mRNA, multiple evaluations were obtained in each of the two independent experiments performed. Microarray analysis were performed 48 h after BZB administration as our previous data[20] indicated this time point to be optimal for BZB effect evaluation.
Quantitative real-time reverse transcription polymerase chain reaction
Part of the total RNAs used for two-color microarray analysis and total RNAs obtained from independent experiments were used to perform quantitative real-time reverse transcription polymerase chain reaction validation. E2F1-8 RT conditions were previously described in[20]; PCR cycles were conducted as follows: pre-denaturation at 95 °C for 10 min, 40 cycles of amplification with denaturation at 95 °C for 15 s, annealing at proper temperature (Table 1) for 60 s and extension at 72 °C for 30 s. A final extension at 72 °C for 10-min and a dissociation stage (95 °C/60 °C/95 °C for 15 s each) was then added. GAPDH house-keeping gene was used to normalize data[20].
Table 1.
Primer sequences performed quantitative real-time reverse transcription polymerase chain reaction
| GenBank Number | Protein | Primer pair | TA(°C) | Amplif.Region | Length (bp) |
| NM_005225 | E2F1 | (F) 5’-CCAGGAAAAGGTGTGAAATC-3’ (R) 5’-AAGCGCTTGGTGGTCAGATT-3’ | 62 | 466-539 | 74 |
| NM_004091 | E2F2 | (F) 5’-CAAGTTGTGCGATGCCTGC-3’ (R) 5’-TCCCAATCCCCTCCAGATC-3’ | 65 | 645-714 | 80 |
| NM_001949 | E2F3 | (F) 5’-AAGTGCCTGACTCAATAGAGAGCC-3’ (R) 5’-AGTCTCTTCTGGACATAAGTAAACCTCA-3’ | 62 | 1307-1392 | 86 |
| NM_001950 | E2F4 | (F) 5’-GCAGACCCCACAGGTGTTTT-3’ (R) 5’-GCTCCGAGCTCATGCACTCT-3’ | 62 | 1081-1162 | 82 |
| NM_001951 | E2F5 | (F) 5’-TTGCTTTAATGGTGATACACTTTTGG-3’ (R) 5’-TCTGACCCATTTCTGGAATGG-3’ | 62 | 577-659 | 83 |
| NM_198256 | E2F6 | (F) 5’-GAAAATGAAAGACTAGCATATGTGACCT-3’ (R) 5’-CTTTAACTGCAATGACGATCTGTTC-3’ | 62 | 818-902 | 85 |
| NM_203394 | E2F7 | (F) 5’-AGGGATGGAGGTAAATTGTTTAACACT-3’ (R) 5’-TTTCCCCATCTTCAACTGCAA-3’ | 65 | 233-318 | 86 |
| NM_024680 | E2F8 | (F) 5’-CTGATCTGCGAACAGGATATTAAAAC-3’ (R) 5’-AAAATGAAAAATCTGGAGTTCCTCC-3’ | 65 | 399-492 | 94 |
| NM_002046 | GAPDH | (F) 5’-CCCATCACCATCTTCCAGGAG-3’ (R) 5’-CTTCTCCATGGTGGTGAAGACG-3’ | 62 | 319-423 | 105 |
E2F8 depletion by siRNA
The sequence of the anti E2F8 siRNA (siE2F8, Eurogentec S.A., Belgium) was previously described in[26]. As control (sense 5’-CGUACGCGGAAUACUUCGA-3’, antisense: 5’-UCGAAGUAUUCCGCGUACG-3’) a siRNA directed against the luciferase gene (siGL2) was analyzed in parallel. Transfections were performed as described in[24] using a weight ratio liposome (Lipofectamin2000 - 1 mg/mL, Invitrogen)/siRNA of 3:1 for three hours at a final siRNA concentration of 220 nmol/L. E2F8 mRNA levels were measured 2 and 4 d after transfection in HepG2 and JHH6, respectively.
Statistical analysis
P values were calculated by the GraphPad InStat tools (GraphPad Software, Inc., La Jolla, CA, United States) using the unpaired t test with or without Welch correction and the Mann-Whitney Test, as appropriate. P values < 0.05 were considered to be statistically significant.
RESULTS
Effects of BZB on E2F family members
We have evidence that E2F1 is dramatically down-regulated upon BZB treatment in the differentiated HCC cell line HepG2[20]. In contrast, in the undifferentiated HCC cell line JHH6 E2F1 down-regulation is only modest. This observation prompted us to verify whether other E2F members are involved in the BZB induced inhibition of proliferation in JHH6. Microarray analysis performed in JHH6 (Figure 1A) following 48 h treatment by BZB at 40 nmol/L, revealed a decrease in the expression of the pro-proliferative E2F member E2F2 and confirmed the modest decrease of E2F1 we previously observed[20]. With regard to the anti-proliferative E2F members, we observed a tendency of BTZ treatment to increase mRNA levels of most E2F members. Quantitative reverse transcription polymerase chain reaction (RT-PCR) data (Figure 1B) confirmed a significant decrease and increase in E2F2 and E2F8 expression.
Figure 1.
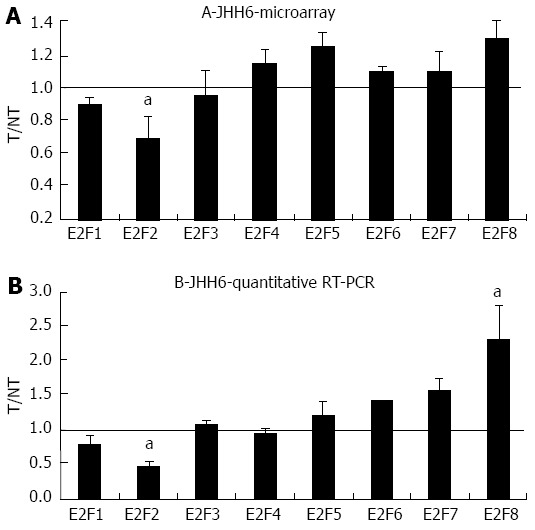
Microarray and quantitative real-time reverse transcription polymerase chain reaction assays in JHH6 following bortezomib treatment. A: The mRNA levels of the indicated E2Fs were evaluated by microarray analysis two days after JHH6 treatment with 40 nmol/L bortezomib. Data are reported as ratio between treated cells (T) and non-treated cells (NT); data are shown as means ± SEM; aP < 0.05 vs controls; depending on the E2F member the number of evaluation ranged from three up to ten; B: The mRNA levels of the indicated E2Fs were evaluated by quantitative real-time reverse transcription polymerase chain reaction assay; data, normalized to GAPDH mRNA, are reported as ratio between T and NT; data are reported as means ± SEM; aP < 0.05 vs controls, n = 5. RT-PCR: Reverse transcription polymerase chain reaction.
Compared to the undifferentiated JHH6, the hepatocyte-like HCC cell line HepG2, displayed a more marked effect of BZB on the differential expression of E2F members, as shown by microarray analysis and confirmed by quantitative RT-PCR (Figure 2). In the HepG2 cell line we detected a stronger down-regulation of the expression of the pro-proliferative E2F members E2F1 and E2F2 (Figure 2B) and of the anti-proliferative member E2F8, with E2F4 and E2F5 being less affected. As with JHH6, BZB treatment of HepG2 cells induced the significant up-regulation of the expression of an anti-proliferative E2F member which, in this case, it was E2F6.
Figure 2.
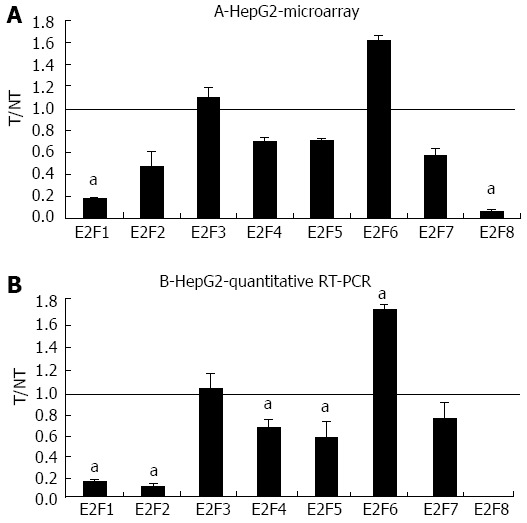
Microarray and quantitative real-time reverse transcription polymerase chain reaction assays in HepG2 following bortezomib treatment. A: The mRNA levels of the indicated E2Fs were evaluated by microarray analysis two days after JHH6 treatment with 40 nmol/L bortezomib. Data are reported as ratio between treated cells (T) and non-treated cells (NT); data are shown as means ± SEM aP < 0.05 vs controls; depending on the E2F member the number of evaluation ranged from three up to ten. B: The mRNA levels of the indicated E2Fs were evaluated by quantitative real-time reverse transcription polymerase chain assay; data, normalized to GAPDH mRNA, are shown as ratio between T and NT; data are reported as means ± SEM; aP < 0.05 vs controls, n = 5. RT-PCR: Reverse transcription polymerase chain reaction.
The marked down-regulation of the expression of the anti-proliferative E2F8 transcription factor in the HepG2 cell line would suggest a pro-proliferative effect of BZB rather than an anti-proliferative effect. However, recently[26] it has been observed that E2F8 depletion by siRNA resulted in the down-regulation of cell proliferation in HCC cell lines. Thus, we verified whether this could have been the case in the HepG2 cell line. Using the siRNA previously proposed[26], we observed that whereas E2F8 siRNA targeting could significantly reduce E2F8 mRNA levels (Figure 3A), no major effects on cell number (Figure 3B) were detected 2 d after a single siRNA transfection. Notably, the siRNA mediated depletion of E2F8 in JHH6 also did not result in a significant decrease in cell number (Figure 3AI and BI). In this last case cell counting was performed four days after siRNA transfection as at this time point the siRNA effect on E2F8 mRNA level was maximum.
Figure 3.
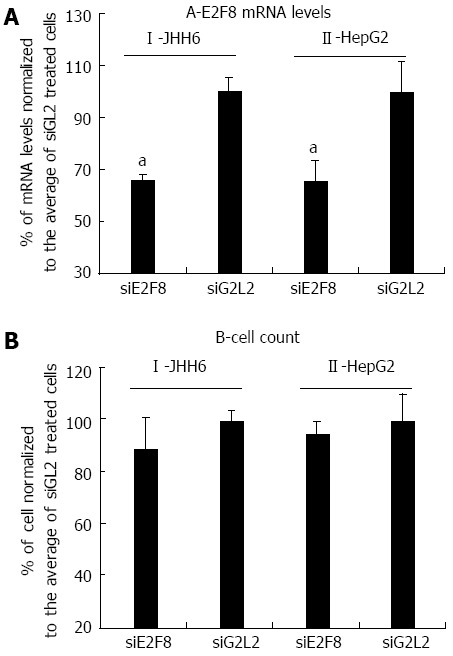
Effects of E2F8 depletion by siRNA in HepG2 and JHH6. A: Four and 2 d after siRNA administration, the reductions of E2F8 mRNA in JHH6 and HepG2, respectively, are reported; the data, normalized to GAPDH levels, are shown as mean ± SEM; aP < 0.05 vs controls, n = 6; B: the effects of siE2F8 on cell number 4 and 2 d after siRNA administration in JHH6 and HepG2, respectively, are reported; the data, normalized to siGL2 treated cells, are shown as mean ± SEM, n = 6. siE2F8: siRNA against E2F8 mRNA; siGL2: Control siRNA against the luciferase mRNA.
Effects of BZB on other cell cycle gene products
The more marked effect of BZB on the differential expression of E2F members in the HepG2 cell line compared to JHH6 prompted us to verify whether this behaviour was restricted to the E2F family or it was a more general feature. For this reason we compared, by microarray analysis, the effects on the differential expression of cyclins (Figure 4) and cdks (data not shown). In the case of cyclin E, cyclin D, cyclin A and cdk2 the microarray data have been previously validated by quantitative RT-PCR[20]. From the analysis of BZB effects on the expression levels of cyclin and cdk gene products, little change was observed with the undifferentiated JHH6 compared to the more differentiated HepG2 cell line. Notably, this behaviour was not restricted to cell cycle genes, and was also seen with the expression of epithelial-mesenchymal transition (EMT) genes; these genes allow the cancer cells to gain migratory and invasive properties, thus resulting in the possibility of metastasis. In JHH6, the impact of BZB on EMT gene expression was definitively less pronounced than in the HepG2 cell line (Figure 5). The reduced BZB impact on gene expression in JHH6 compared to HepG2 cells is also shown by the fact that BZB treatment of HepG2 cells modified the signal coming from about 12000 gene probes, whereas in JHH6 this number was of about 2000.
Figure 4.
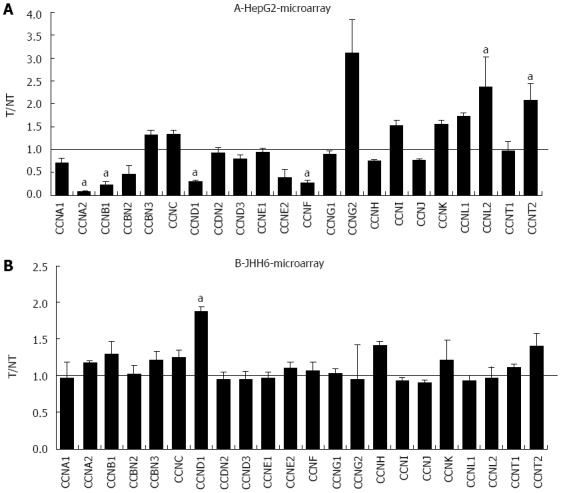
Microarray assay in HepG2 and JHH6 following Bortezomib treatment. The mRNA levels of the indicated cyclins were evaluated by microarray analysis two days after HepG2 (A) or JHH6 (B) treatment by 40 nmol/L Bortezomib. Data are reported as ratio between treated cells (T) and non-treated cells (NT); data are shown as means ± SEM; aP < 0.05 vs controls; depending on the cyclin member, the number of microarray evaluation ranged from three up to ten.
Figure 5.
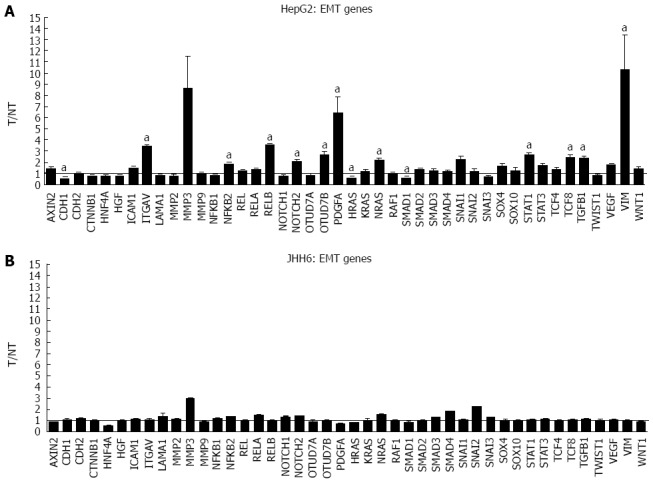
Microarray assays in HepG2 and JHH6 following bortezomib treatment. The mRNA levels of the indicated epithelial-mesenchymal transition (EMT) genes were evaluated by microarray analysis two days after HepG2 (A) or JHH6 (B) treatment by 40 nmol/L Bortezomib. Data are reported as ratio between treated cells (T) and non-treated cells (NT); data are shown as means ± SEM; aP < 0.05 vs controls; depending on the EMT member, the number of microarray evaluation ranged from three up to ten.
DISCUSSION
Several studies suggest that BZB is a potent 26S proteasome inhibitor with the potential to be of therapeutic value for HCC treatment. We have previously observed[20] that BZB can effectively inhibit the growth of the differentiated HCC cell line HepG2 and, to a lesser extent, also that of the undifferentiated HCC cell line JHH6. We can exclude that this difference arises from a reduced BZB-mediated inhibition of the 26S proteasome in JHH6 cells compared to HepG2 as a similar reduction of proteasome activity was previously shown in these two cell lines[20]. It follows that also the effects on gene expression reported in this work unlikely depend on a differential inhibition of the 26S proteasome by BZB in the two cell lines.
The role of E2F family members in HCC has been mainly demonstrated for E2F1[20,22] whose up-regulation, considered an unfavorable prognostic factor[23], has been shown in HCC[33]. The other E2F members studied in the literature are E2F3, E2F5 and E2F8 for which indication of the up-regulation in HCC samples and implication in HCC cell growth were reported[25-28]. The lack of information about the other E2F family members in HCC development in general and in relation to BZB effects, prompted us to start a novel investigation in this field.
In addition to confirming in JHH6 our previous data of a modest effect on E2F1 mRNA by BZB[20], this study suggests that the differential expression of other E2Fs contributes to BZB’s action in JHH6. Interestingly, the down-regulation of the pro-proliferative E2F2 (Figure 1B) and the up-regulation of the anti-proliferative E2F8 suggest a net anti-proliferative effect of BZB. The differential expression of these two E2Fs is not dramatic. It is thus possible that BZB’s anti-proliferative effect stems from the sum of multiple “minor” anti-proliferative signals. In this sense the slight increase of the other anti-proliferative E2F members E2F5, E2F6 and E2F7 may contribute to the global anti-proliferative effect. Future functional investigations will further clarify the contribution of each of the differentially expressed E2F members in the down-regulation of JHH6 proliferation.
In the differentiated HCC cell line HepG2, BZB effects on the expression of the pro-proliferative E2Fs, although qualitatively similar to JHH6 (E2F1 and E2F2 down-regulation with E2F3 substantially unaffected; Figure 2B), are quantitatively more pronounced. The reasons for this quantitative difference deserve additional investigation. These observations are in line with preliminary studies conducted in another HCC cell line HuH7 (Dapas et al, unpublished results), which display an intermediate differentiation grade, showing an effect on E2Fs intermediate between JHH6 and HepG2.
In the HepG2 cell line, BZB effects on the anti-proliferative E2Fs is, in contrast, substantially different from that observed in JHH6 (compare Figure 2B with 1B), characterized by the up-regulation of E2F6 and by the marked down-regulation of E2F8. Particularly intriguing is the dramatic down-regulation of E2F8. Despite being known as an anti-proliferative E2F[21], E2F8 has been recently suggested by Deng et al[26] to promote HCC cell proliferation. Our data (Figure 3) indicate that E2F8 depletion does not seem to have a major effect on HepG2 growth; this event is not limited to HepG2 cells as the same occurs with JHH6 (Figure 3). These contrasting data with Deng et al[26] may firstly depend on the fact that we have used different HCC cell lines (HepG2/JHH6 vs YY-8103/Focus cells). In addition, we measured cell growth at day two and four in HepG2 and JHH6, respectively, following E2F8 depletion; in contrast, Deng et al[26] prolonged the analysis up to day 6, a time point at which the anti-proliferative effect was most evident. We limited the analysis at day two/four in HepG2 and JHH6, respectively, as at these time points the effect was maximal with a single siE2F8 administration. Additionally, the overgrowth of control cells (non-treated cells and control siRNA-treated cells) at longer time points made cell growth evaluation (by cell counting) cumbersome. Together, our observations suggest that, at least in the HCC cell lines considered and under our experimental conditions, E2F8 does not seem to be a major promoter of cell proliferation.
The decrease of most of the anti-proliferative E2F members in the HepG2 cell line treated by BZB would favor the concept of a pro-proliferative effect. However, it should be considered that the prediction of the net effect of the E2Fs deregulation is not so straightforward. This is due to the redundancies of E2F functions[21] which allow one member to take the part of another one. It is thus possible that E2F6 takes the place of the down-regulated E2F4, E2F5 and E2F8, favoring the anti-proliferative effect. This, together with the down-regulation of E2F1 and E2F2, may explain the net anti-proliferative effect.
The substantial reduced quantitative impact of BZB on the expression of E2F family members in JHH6 when compared to HepG2 occurs also for gene products related and unrelated to E2Fs, such as cyclins/cdks and EMT genes, respectively. As remarked above, the differences in BZB effects in JHH6 and HepG2 cells do not seem to be related to proteasome-dependent mechanisms, but possibly to proteasome-independent mechanisms. Despite the reasons for this behavior, these findings are in line with the more contained impact of BZB in the undifferentiated JHH6 when compared to the more differentiated HepG2 cell line. This observation, together with our previous data[20], strongly suggests that not all the HCC types may respond, from the quantitative point of view, in a similar manner to BZB treatment. In this regard, it is interesting to note the negligible effects of BZB on the expression of EMT genes in JHH6. As these genes are involved with metastasis, it is possible that BZB treatment does not effectively prevent metastasis, at least in some HCC cell phenotypes.
In summary, we reported for the first time data indicating that in the HCC cell lines tested, BZB effects on the expression of E2F family members are not limited to E2F1 but are extended to other members. In particular, in both JHH6 and HepG2 the expression of the pro-proliferative E2F2 is down-regulated. Specific to JHH6 is the up-regulation of the expression of the anti-proliferative E2F8, while in the HepG2 cell line it is the up-regulation of the anti-proliferative E2F6 and the marked repression of E2F8 expression. Together these data expand our knowledge on the molecular basis of BZB action in inhibiting the proliferation of HCC cells, strengthening the rationale for its future use in HCC-affected patients.
COMMENTS
Background
In the last decade the drug bortezomib (BZB) has been studied as a possible novel therapeutic treatment for hepatocellular carcinoma (HCC). BZB is a boronic acid dipeptide derivative able to inhibit the 26S proteasome.
Research frontiers
The other E2F members studied in the literature are E2F3, E2F5 and E2F8 for which indication of the up-regulation in HCC samples and implication in HCC cell growth were reported.
Innovations and breakthroughs
The differential expression of E2Fs and related genes induced by BZB in diverse HCC cell phenotypes contribute to bortezomib’s mechanism of action in hepatocellular carcinoma.
Applications
The authors reported for the first time data indicating that in the HCC cell lines tested, BZB effects on the expression of E2F family members are not limited to E2F1 but are extended to other members.
Peer review
The article is an original research paper focusing on the proteasome inhibitor BZB effects on E2Fs transcription factors and related genes in HCC. The study is well structured, the subject is actual and interesting, providing a rationale for performing the research.
Footnotes
Supported by The “Fondazione Cassa di Risparmio of Trieste; the “Fondazione Benefica Kathleen Foreman Casali of Trieste”; the Italian Minister of Instruction, University and Research (MIUR), PRIN 2010-11, No. 20109PLMH2 (in part)
P- Reviewers: Tanase CP, Tiniakos DG, Wang TH S- Editor: Zhai HH L- Editor: A E- Editor: Wu HL
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42 Suppl 3:S206–S214. doi: 10.1016/S1590-8658(10)60507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 4.European Association For The Study Of The Liver, European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943. doi: 10.1016/j.jhep.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Colombo M. Multidisciplinary approach to hepatocellular carcinoma. Preface. Dig Liver Dis. 2010;42 Suppl 3:S205. doi: 10.1016/S1590-8658(10)00205-7. [DOI] [PubMed] [Google Scholar]
- 6.Shin JW, Chung YH. Molecular targeted therapy for hepatocellular carcinoma: current and future. World J Gastroenterol. 2013;19:6144–6155. doi: 10.3748/wjg.v19.i37.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 8.Zavrski I, Jakob C, Schmid P, Krebbel H, Kaiser M, Fleissner C, Rosche M, Possinger K, Sezer O. Proteasome: an emerging target for cancer therapy. Anticancer Drugs. 2005;16:475–481. doi: 10.1097/00001813-200506000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Kim GP, Mahoney MR, Szydlo D, Mok TS, Marshke R, Holen K, Picus J, Boyer M, Pitot HC, Rubin J, et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Invest New Drugs. 2012;30:387–394. doi: 10.1007/s10637-010-9532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahl K, Siegemund M, Lehner F, Vondran F, Nüssler A, Länger F, Krech T, Kontermann R, Manns MP, Schulze-Osthoff K, et al. Increased apoptosis induction in hepatocellular carcinoma by a novel tumor-targeted TRAIL fusion protein combined with bortezomib. Hepatology. 2013;57:625–636. doi: 10.1002/hep.26082. [DOI] [PubMed] [Google Scholar]
- 11.Saeki I, Terai S, Fujisawa K, Takami T, Yamamoto N, Matsumoto T, Hirose Y, Murata Y, Yamasaki T, Sakaida I. Bortezomib induces tumor-specific cell death and growth inhibition in hepatocellular carcinoma and improves liver fibrosis. J Gastroenterol. 2013;48:738–750. doi: 10.1007/s00535-012-0675-z. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Gao D, Guo K, Kang X, Jiang K, Sun C, Li Y, Sun L, Shu H, Jin G, et al. Novel synergistic antitumor effects of rapamycin with bortezomib on hepatocellular carcinoma cells and orthotopic tumor model. BMC Cancer. 2012;12:166. doi: 10.1186/1471-2407-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko BS, Chang TC, Chen CH, Liu CC, Kuo CC, Hsu C, Shen YC, Shen TL, Golubovskaya VM, Chang CC, et al. Bortezomib suppresses focal adhesion kinase expression via interrupting nuclear factor-kappa B. Life Sci. 2010;86:199–206. doi: 10.1016/j.lfs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Hui B, Shi YH, Ding ZB, Zhou J, Gu CY, Peng YF, Yang H, Liu WR, Shi GM, Fan J. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer. 2012;118:5560–5571. doi: 10.1002/cncr.27586. [DOI] [PubMed] [Google Scholar]
- 15.Chen KF, Liu CY, Lin YC, Yu HC, Liu TH, Hou DR, Chen PJ, Cheng AL. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene. 2010;29:6257–6266. doi: 10.1038/onc.2010.357. [DOI] [PubMed] [Google Scholar]
- 16.Chen KF, Yu HC, Liu TH, Lee SS, Chen PJ, Cheng AL. Synergistic interactions between sorafenib and bortezomib in hepatocellular carcinoma involve PP2A-dependent Akt inactivation. J Hepatol. 2010;52:88–95. doi: 10.1016/j.jhep.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL. Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2008;68:6698–6707. doi: 10.1158/0008-5472.CAN-08-0257. [DOI] [PubMed] [Google Scholar]
- 18.Yu HC, Hou DR, Liu CY, Lin CS, Shiau CW, Cheng AL, Chen KF. Cancerous inhibitor of protein phosphatase 2A mediates bortezomib-induced autophagy in hepatocellular carcinoma independent of proteasome. PLoS One. 2013;8:e55705. doi: 10.1371/journal.pone.0055705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spratlin JL, Pitts TM, Kulikowski GN, Morelli MP, Tentler JJ, Serkova NJ, Eckhardt SG. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011;31:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- 20.Baiz D, Pozzato G, Dapas B, Farra R, Scaggiante B, Grassi M, Uxa L, Giansante C, Zennaro C, Guarnieri G, et al. Bortezomib arrests the proliferation of hepatocellular carcinoma cells HepG2 and JHH6 by differentially affecting E2F1, p21 and p27 levels. Biochimie. 2009;91:373–382. doi: 10.1016/j.biochi.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 21.Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun HX, Xu Y, Yang XR, Wang WM, Bai H, Shi RY, Nayar SK, Devbhandari RP, He YZ, Zhu QF, et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/ E2F transcription factor 1-dependent apoptotic pathway. Hepatology. 2013;57:1088–1097. doi: 10.1002/hep.26188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YL, Uen YH, Li CF, Horng KC, Chen LR, Wu WR, Tseng HY, Huang HY, Wu LC, Shiue YL. The E2F transcription factor 1 transactives stathmin 1 in hepatocellular carcinoma. Ann Surg Oncol. 2013;20:4041–4054. doi: 10.1245/s10434-012-2519-8. [DOI] [PubMed] [Google Scholar]
- 24.Farra R, Dapas B, Pozzato G, Scaggiante B, Agostini F, Zennaro C, Grassi M, Rosso N, Giansante C, Fiotti N, et al. Effects of E2F1-cyclin E1-E2 circuit down regulation in hepatocellular carcinoma cells. Dig Liver Dis. 2011;43:1006–1014. doi: 10.1016/j.dld.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology. 2009;50:113–121. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- 26.Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS, Teng XM, Zhang X, Huang J, Han ZG. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res. 2010;70:782–791. doi: 10.1158/0008-5472.CAN-09-3082. [DOI] [PubMed] [Google Scholar]
- 27.Xiao F, Zhang W, Chen L, Chen F, Xie H, Xing C, Yu X, Ding S, Chen K, Guo H, et al. MicroRNA-503 inhibits the G1/S transition by downregulating cyclin D3 and E2F3 in hepatocellular carcinoma. J Transl Med. 2013;11:195. doi: 10.1186/1479-5876-11-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Y, Yim SH, Xu HD, Jung SH, Yang SY, Hu HJ, Jung CK, Chung YJ. A potential oncogenic role of the commonly observed E2F5 overexpression in hepatocellular carcinoma. World J Gastroenterol. 2011;17:470–477. doi: 10.3748/wjg.v17.i4.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grassi G, Scaggiante B, Farra R, Dapas B, Agostini F, Baiz D, Rosso N, Tiribelli C. The expression levels of the translational factors eEF1A 1/2 correlate with cell growth but not apoptosis in hepatocellular carcinoma cell lines with different differentiation grade. Biochimie. 2007;89:1544–1552. doi: 10.1016/j.biochi.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209:497–499. doi: 10.1126/science.6248960. [DOI] [PubMed] [Google Scholar]
- 31.Fujise K, Nagamori S, Hasumura S, Homma S, Sujino H, Matsuura T, Shimizu K, Niiya M, Kameda H, Fujita K. Integration of hepatitis B virus DNA into cells of six established human hepatocellular carcinoma cell lines. Hepatogastroenterology. 1990;37:457–460. [PubMed] [Google Scholar]
- 32.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 33.Palaiologou M, Koskinas J, Karanikolas M, Fatourou E, Tiniakos DG. E2F-1 is overexpressed and pro-apoptotic in human hepatocellular carcinoma. Virchows Arch. 2012;460:439–446. doi: 10.1007/s00428-012-1220-4. [DOI] [PubMed] [Google Scholar]