

Abstract
Our understanding of the microbial involvement in inflammatory bowel disease (IBD) pathogenesis has increased exponentially over the past decade. The development of newer molecular tools for the global assessment of the gut microbiome and the identification of nucleotide-binding oligomerization domain-containing protein 2 in 2001 and other susceptibility genes for Crohn’s disease in particular has led to better understanding of the aetiopathogenesis of IBD. The microbial studies have elaborated the normal composition of the gut microbiome and its perturbations in the setting of IBD. This altered microbiome or “dysbiosis” is a key player in the protracted course of inflammation in IBD. Numerous genome-wide association studies have identified further genes involved in gastrointestinal innate immunity (including polymorphisms in genes involved in autophagy: ATG16L1 and IGRM), which have helped elucidate the relationship of the local innate immunity with the adjacent luminal bacteria. These developments have also spurred the search for specific pathogens which may have a role in the metamorphosis of the gut microbiome from a symbiotic entity to a putative pathogenic one. Here we review advances in our understanding of microbial involvement in IBD pathogenesis over the past 10 years and offer insight into how this will shape our therapeutic management of the disease in the coming years.
Keywords: Inflammatory bowel disease, Crohn’s disease, Ulcerative colitis, Gut microbiota, Innate immune response, Probiotics, Prebiotics, Faecal transplant
Core tip: In the last decade there have been enormous strides in our understanding of the role of gut microbiota in the aetiopathogenesis of inflammatory bowel disease (IBD). Newer molecular and genetic diagnostic tools have elucidated distinct changes in the gut microbiota in IBD patients and clarified the deficiencies of innate immunity. A link between environmental factors like diet, host immunity and the gut microbiota has been established. This review aims to enumerate these diverse strands of converging research in the last decade to outline the exciting prospects of possible personalized therapeutic interventions for patients with IBD in the coming years.
INTRODUCTION
Inflammatory bowel disease (IBD) comprises two distinct conditions, ulcerative colitis (UC) and Crohn’s disease (CD) that are characterized by chronic relapsing inflammation of the gut in genetically susceptible individuals exposed to defined environmental risk factors[1,2]. IBD was historically considered to be a “Western” disease but in the last decade there has been a definite increase in its incidence and prevalence suggesting that it is progressively emerging as a global epidemic[3]. In the high prevalence regions the incidence of IBD has continued to rise in the past decade[4,5].
There has been a parallel rise in our understanding of the critical role of the gut microbiota in the aetiopathogenesis of IBD. This is aptly exemplified by entering the key words, “microbiota” or “microflora” and “inflammatory bowel disease” into the PubMed database. On restricting the search to the last 10 years, over 800 articles published on this subject can be retrieved as opposed to 100 articles in the decade preceding it. This radical explosion of interest has been primarily due to the advent of culture-independent techniques like next generation sequencing and metagenomics which has enabled the global assessment of the gut microbiota much more accurately and in a vastly more sophisticated manner[6,7]. The largest and perhaps the most ambitious initiative that has emerged in the last decade is the NIH sponsored Human Microbiome Project with a total budget of $115 million to study the changes of the human microbiome in health and disease[8]. It has recently led to the publication of 5177 microbial taxonomic profiles from a population of 242 healthy adults sampled at 15 or 18 body sites up to three times, with over 3.5 terabases of metagenomic sequence so far, which will serve as a comprehensive framework for future research in this field[9].
This expansion of knowledge in the last decade has also shifted the search from external environmental triggers to a trigger within the complex luminal microbiome or the so called “in-vironment” that we harvest within ourselves[10-12]. Prior to these radical developments research had focussed on unearthing a pathogen amidst the vast plethora of microbes in the gut lumen, which could be held responsible for initiating the inflammatory cascade that is typical of IBD[13]. This endeavour was akin to searching for the veritable “needle in the haystack”. The findings in the last decade has turned this whole concept on its head by revealing that the gut microbiome as a whole is altered in IBD, suggesting that perhaps the entire “haystack” is faulty. This concept of an altered gut microbiome or dysbiosis is possibly the most significant development in the field of IBD research in the past decade.
The other major shift in our knowledge of the aetiopathogenesis of inflammatory bowel disease has been from the host perspective. The dogma that CD and UC are typical autoimmune disorders was based on the characteristic histological appearance of these conditions and the response to immune-modulator drugs but the veil has lifted from this deep-embedded misconception[14,15]. Over the past decade, genome wide association studies and newer genetic technologies have elucidated distinct genetic defects in IBD patients. This has particular relevance with respect to host-microbial interaction at the luminal surface in the gut. A similar analysis on the PubMed database with the search items “genetics” and “inflammatory bowel disease” leads to a staggering yield of more than 5600 publications in the last decade as opposed to 2000 articles in the decade prior. It must be said that the avenue of research in this field was first opened up in 2001 when the first association of the nucleotide-binding oligomerization domain-containing protein 2 (NOD-2) gene mutation and susceptibility to Crohn’s disease was documented[16,17]. This has resulted in a drastic paradigm shift wherein IBD is no longer considered an autoimmune disease but may be an immunodeficient condition instead[15]. This putative genetic susceptibility leads to a complex interaction between the diverse gut microbiome and the local innate immune system and forms the current basis for the aetiopathogenesis of IBD (Figure 1).
Figure 1.
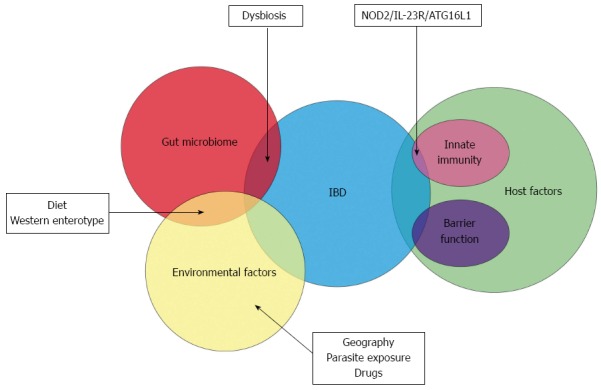
The Venn diagram depicts the overlapping role of the gut microbiome, host and environmental factors in the aetiopathogenesis of inflammatory bowel disease. Dysbiotic changes in the gut microbiome may be influenced by diet and other environmental factors and predispose to inflammatory bowel disease (IBD). A small proportion of IBD patients have demonstrable genetic susceptibility factors. NOD2: Nucleotide-binding oligomerization domain-containing protein 2; ATG16L1: Autophagy related protein 16-like 1; IL-23R: Interleukin 23 receptor.
DYSBIOSIS
The normal gut microbiome comprises 100 trillion diverse microbes, mostly bacteria, encompassing over 1100 prevalent species, with at least 160 species in each individual[18]. An exhaustive analysis of normal global gut bacterial communities suggests the possible existence of distinct enterotypes (Bacteroides, Prevotella or Ruminococcus) which are predominantly driven by dietary intake but independent of age or BMI[19,20]. Further analysis suggests that the Bacteroides enterotype is associated with a “western” protein rich diet as opposed to the Prevotella enterotype which was associated with a carbohydrate rich diet[21]. It remains to be seen whether this western enterotype turns out to be a distinct risk factor for developing IBD.
Dysbiosis or a definitive change of the normal gut microbiome with a breakdown of host- microbial mutualism is probably the defining event in the development of IBD. The shift from predominant “symbiont” microbes to potential harmful “pathobiont” microbes has now been well documented[22]. Some of these changes in the gut microbiome have been detected in the common subset of IBD patients but some have been clearly delineated either in CD or in UC patients. The most well defined change that has been noted in patients with IBD is the reduced abundance of the phyla Firmicutes[23-25]. Amongst the Firmicutes, the reduced presence of Faecalibacterium prausnitzii has been well documented in patients with CD as opposed to controls[23,26-30]. This has been countered in a paediatric cohort of patients with CD where there were increased levels of Faecalibacterium prausnitzii suggesting a more dynamic role for this bacterium with a putative protective effect at the point of onset of IBD[31]. In addition, there was a definite decrease in diversity of Firmicutes, with fewer of its constituent species detected in patients with IBD[23,32,33]. Unlike Firmicutes, there have been reports of increased number of bacteria from the phylum Bacteroidetes in patients with IBD[34-36]. Paradoxically, there have been some studies which have shown reduction in these bacterial species as well[23]. There is a suggestion that there may be spatial reorganization of the Bacteroides species in patients with IBD, with Bacteroides fragilis being responsible for a greater proportion of the biofilm mass in patients with IBD compared to controls, suggesting increased adherence[37]. Bacteria belonging to these two phyla make up for 90% of the phylogenetic categories in the normal microbiome and it is interesting to see the disparate ways in which they are altered in IBD.
Most of the known pathogenic bacteria in humans belong to the phylum Proteobacteria, which have been increasingly found to have a key role in IBD[38]. Microbial diversity analysis has shown a shift towards an increase in bacterial species belonging to this phylum, suggesting an aggressor role in the initiation of chronic inflammation in patients with IBD[39-42]. More specifically, increased concentrations of Escherichia coli including pathogenic variants have been documented in ileal CD[28,43].
This interesting shift within the gut microbiome with a decrease in obligate anaerobes of the phylum Firmicutes and an increase in facultative anaerobes of Proteobacteria has given rise to a putative “oxygen” hypothesis wherein disruption in anaerobiosis points to a role for oxygen in intestinal dysbiosis[44]. Similar functional disruptions associated with changes of the gut microbiome in patients with IBD may have more long reaching effects. Metagenomic analysis has revealed that the altered microbiome in IBD has 25% fewer genes and metaproteomic studies have shown a depletion of proteins and functional pathways[18,45]. The ileal CD patients were found to have alterations in bacterial carbohydrate metabolism, bacterial-host interactions, as well as human host-secreted enzymes[45]. Elucidation of the functional impact of the changes seen as a result of dysbiosis will help design remedial measures that will help in the treatment of IBD patients.
The immediate question which follows is how the host responds to dysbiosis. Host genetics factors, specifically those pertaining to the innate immunity arm, is expected to play a role in the aetiopathogenesis of IBD. The “chicken and the egg” question is what comes first. Are the changes in the gut microbiome a result of an aberrant immune response in a genetically susceptible individual or does the abnormality in the gut microbiota lead to an aberrant immune response in such an individual? Twin studies have shown that disease phenotype rather than host genotype plays a greater role in determining changes in gut microbiota[46]. However, studying the microbiota in subsets of patients with and without NOD2 and autophagy related protein 16-like 1 (ATG16L1) risk alleles showed that the affected genotypes were significantly associated with microbial compositional change but disease phenotype played a role as well[47]. The confounding factor is that these two alleles are associated with ileal CD and not colonic CD. It makes it difficult to attribute these genetic defects as a cause of dysbiosis but highlights the intricate role of innate immunity in IBD.
INNATE IMMUNITY AND IBD
The gastrointestinal microbiota is a major source of immune stimulation. The colonic epithelium lies in close proximity to a high density of diverse microbes leading to a continuous network of communication between host cells and microbes. This continual communication is essential for the maintenance of normal homeostasis though contribution to processes including supply of nutrients, xenobiotic metabolism and protection from pathogenic microorganisms, can have deleterious effects and contribute to intestinal inflammation[48,49]. In patients with IBD this delicate balance is disturbed as a result of host immune defects in microbial recognition or handling/clearance strategies[50]. Pattern recognition receptors (PRRs) are essential in distinguishing “friend from foe” in this very complex interaction and hold the key to understanding how genetic factors lead to an abnormal immune environment wherein normal commensal organisms can lead to pathological chronic inflammation. Ten years ago toll-like receptors (TLRs) and NOD2 were known to be involved in IBD pathogenesis although our understanding of their location, function and involvement was still very rudimentary. Evidence from IBD genetic studies had demonstrated that several innate immune genes had functionally relevant polymorphisms. Of those studied NOD2 genetic variants confer the greatest risk.
A decade ago the novel association between the recently characterised TLR4 Asp299Gly was described for both CD and UC[51]. This finding supported previous evidence of PRR genetic influence in IBD susceptibility which had shown that polymorphisms in NOD2 (Arg702Trp, Gly908Arg, and leu1007fsinsC) and the CD14-159C/T promoter polymorphism were associated with CD[16,17]. Since then, additional polymorphisms in TLRs have been identified including TLR1 R80T and TLR2 R753G which have been associated with pancolitis in UC patients[52]. The TLR9-1237T/C promoter polymorphism (TLR9-1237), which is associated with increased nuclear factor kappa B (NF-κB) binding affinity, has also been associated with CD[53,54].
The normal colonic epithelium constitutively expresses a variety of PRRs although expression levels are generally low with many receptors located basolaterally thus preventing interaction with luminal antigens[55]. Nevertheless, intestinal epithelial cells are responsive to TLR ligands and recognise/respond to commensal bacteria secreting antimicrobial proteins and cytokines which facilitate intercellular interactions[48]. Primary human intestinal epithelial cells express constitutive TLR3 and TLR5 and low levels of TLR2 and TLR4[56]. TLR2, 4 and 5 are expressed on the cell surface and recognise extracellular microbes. TLR3 detects viral particles and is located intracellularly on early endosomal vesicles. The critical role of TLR4 as a first line of defence against potential bacterial pathogens is now beyond doubt. Impairment of TLR4 function permits bacterial invasion and persistence, and leads to the characteristic inflammation of IBD. The importance of TLR5 in intestinal homeostasis has also been effectively demonstrated using microbiota transfer from knockout mice[57,58].
Distinct changes in TLR expression have been documented in IBD. TLR4 is found to be up regulated in both UC and CD, whereas the levels of TLR2 and TLR5 remain unchanged[56,59]. Altered TLR2 and TLR4 expression has been documented in the intestinal macrophages compared to peripheral monocytes, and a higher percentage of intestinal dendritic cells (DCs) have been shown to express TLR2 and TLR4 in IBD compared to control subjects[59]. Intestinal macrophage signalling through PRRs has also been shown to be affected by increased expression of suppressor of cytokine signalling 1 and sterile and Armadillo motif-containing protein[60,61]. Dysregulation of β-catenin and phosphotidylinositol-3-kinase pathways are also involved with alterations in these pathways involved in colitis susceptibility[62,63]. In IBD patients increased cytokine production is seen by lamina propria DCs and macrophages, consistent with dysregulated tolerance[64-66]. These changes can explain some of the abnormal response to the resident gut microbiota. However, it is difficult to elucidate whether the change in TLR expression initiates disease or is an epiphenomenon resulting from pro-inflammatory cytokine release. In many cases absence of epithelial cell-derived antimicrobial pathways increases susceptibility to intestinal inflammation, with IBD patients expressing lower levels of α-defensin compared to healthy individuals[67,68].
Animal models had played a significant role in driving forward our understanding of IBD pathogenesis, especially murine colitis models. TLR4 and MyD88 knockout mice have been shown to demonstrate distinctly less pathology following chemical induction of colitis with dextran-sodium sulphate although bacterial translocation to mesenteric lymph nodes was more commonly detected[69]. Impairment of TLR4 and TLR5 function has been shown to facilitate bacterial invasion and persistence (TLR4) and impact on intestinal homeostasis; development of metabolic syndrome (TLR5)[57,58].
Alterations in NOD2 function due to genetic polymorphisms have demonstrated an inability to respond to bacterial muramyl dipeptide (MDP) leading to ineffective downstream signalling of NF-κB[70]. NOD2, is expressed on several different cell types including myeloid-derived, epithelial and endothelial cells. As with impairment of TLR function, NOD2 deficiency increases translocation of enteric bacteria to the lamina propria, with alterations in cytokine expression following exposure of peripheral blood mononuclear cells to MDP also reported potentially explaining the alterations in cytokine profiles typically seen in CD[71,72]. Interestingly, NOD2 has more recently been shown to respond to viruses[73]. With increasing interest in non-bacterial microbes in IBD pathogenesis, namely viruses and fungi, this may prove to be an increasing area of consideration. A decrease in the protective, anti-inflammatory Th-2 cytokine IL-10 has been documented in NOD2 mutants further adding to our understanding of the functional abnormalities characteristic of CD[74].
Counter-intuitively, NOD2 can also contribute to down-regulation of inflammatory responses with chronic stimulation of NOD2 acting to tolerise cells against bacterial stimulation and ultimately down regulating other PRRs[75-77]. Hence, in CD patients with dysfunctional NOD2 this restraint is removed and the inflammatory response from other PRRs increases.
NOD2 is also implicated in mechanisms of microbial killing. Autophagy, an important mechanism of microbial cell clearance, is regulated through PRRs[78]. NOD2 interacts with ATG16L1[78,79]. Therefore dysregulation of NOD2 impacts not only on microbial recognition but also handling. Genetic variants in ATG16L1 and also a second autophagy gene, immunity-related GTPase family M have been associated with CD[80,81].
ROLE OF INDIVIDUAL PATHOGENS IN IBD
The rapid development of molecular techniques has also kindled hopes in the search for specific pathogenic agents initiating the inflammatory process of IBD. The pathophysiology of IBD does suggest that either primary or secondary pathogens play an important role in the cycle of inflammation. Many organisms have been proposed but those deemed to have been of the most interest over the last ten years are discussed below (Table 1).
Table 1.
Evidence for the role of specific major pathogens in the aetiopathogenesis of inflammatory bowel disease in the last decade n (%)
| Year | Pathogen | Disease | Sample type |
Detection rate |
Reference | ||
| CD | UC | Control | |||||
| 2003 | MAP | CD | Tissue | 34/37 (92) | 9/34 (26) | [85] | |
| 2003 | MAP | CD and UC | Tissue | 0/24 (0) | 1/28 (4) | 6/19 (32) | [94] |
| 2003 | Helicobacter | CD and UC | Tissue | 0/9 (0) | 0/11 (0) | 0/10 (0) | [109] |
| 2004 | MAP | CD and UC | Blood | 107/283 (37.8) | 50/144 (34.7) | 135/402 (33.6) | [92] |
| 2004 | MAP | CD and UC | Blood | 13/28 (46) | 4/9 (45) | 3/15 (20) | [86] |
| 2004 | H. pylori | UC | Tissue | 8/42 (19) | 7/74 (9.5) | [110] | |
| 2004 | Helicobacter | CD and UC | Tissue | 1/25 (4) | 5/33 (15.2) | 0/29 (0) | [111] |
| 2004 | Helicobacter | CD and UC | Tissue | 0/30 (0) | 0/26 (0) | 0/25 (0) | [112] |
| 2004 | EHH | CD and UC | Tissue | 3/25 (12) | 3/18 (17) | 1/23 (4) | [113] |
| H. pullorum | 2/25 (8) | 0/18 (0) | 1/23 (4) | ||||
| H. fennelliae | 1/25 (4) | 3/18 (17) | 0/23 (0) | ||||
| H. pylori | 8/25 (32) | 5/18 (28) | 14/23 (61) | ||||
| 2004 | Helicobacter | CD, UC and IC | Tissue | 0/11 (0) | 1/20 (5) | 0/37 (0) | [114] |
| 2004 | E. coli | CD and UC | Tissue | 11/14 (79) | 8/21 (38) | 10/24 (42) | [156] |
| AIEC | 10/14 (71) | 10/21 (48) | 7/24 (29) | ||||
| 2004 | AIEC | CD | Tissue | 7/63 (11.1) | 1/16 (6.3) | [154] | |
| 2004 | E. coli | CD | Tissue | 12/15 (80) | 1/10 (10) | [158] | |
| 2006 | E. coli | CD and UC | Tissue | 9/12 (75) | 7/7 (100) | 2/8 (25) | [159] |
| 2007 | AIEC | CD and UC | Tissue | 8/13 (61.5) | 11/19 (57.9) | 4/15 (26.7) | [160] |
| 2008 | Helicobacter | CD | Faeces | 17/29 (59) | 1/11 (9) | [115] | |
| EHH | 11/29 (38) | 1/11 (9) | |||||
| H. pylori | 6/29 (21) | 0/11 (0) | |||||
| H. trogontum | 4/29 (14) | 1/11 (9) | |||||
| H. canis | 5/29 (17) | 0/11 (0) | |||||
| H. bilis | 4/29 (14) | 0/11 (0) | |||||
| H. cinaedi | 1/29 (3) | 0/11 (0) | |||||
| 2009 | AIEC | CD | Tissue | 14/27 (51.9) | 4/24 (16.7) | [155] | |
| 2009 | Helicobacter | CD | Tissue | 32/73 (43.8) | 43/92 (46.7) | [116] | |
| EHH | 18/73 (24.7) | 16/92 (17.4) | |||||
| H. pylori | 29/73 (39.7) | 39/92 (42.4) | |||||
| H. pullorum | 8/73 (11) | 6/92 (6.5) | |||||
| H. canndensis | 10/73 (13.7) | 10/92 (10.9) | |||||
| 2009 | Campylobacter | CD | Tissue | 27/33 (82) | 12/52 (23) | [131] | |
| C. concisus | 17/33 (51) | 1/52 (2) | |||||
| C. showae | 3/33 (9) | 0/52 (0) | |||||
| C. hominis | 2/33 (6) | 2/52 (4) | |||||
| C. gracilis | 2/33 (6) | 0/52 (0) | |||||
| C. rectus | 1/33 (3) | 2/52 (4) | |||||
| C. jejuni | 1/33 (3) | 3/52 (6) | |||||
| C. ureolyticus | 1/33 (3) | 2/52 (4) | |||||
| 2010 | Helicobacter | CD | Tissue | 32/77 (41.6) | 23/102 (22.5) | [117] | |
| EHH | 18/77 (23.4) | 12/102 (11.8) | |||||
| H. pylori | 14/77 (18.2) | 11/102 (10.8) | |||||
| H. bilis | 1/77 (1.3) | 1/102 (1.0) | |||||
| H. canis | 2/77 (2.6) | 0/102 (0.0) | |||||
| H. hepaticus | 2/77 (2.6) | 2/102 (2.0) | |||||
| H. trogontum | 5/77 (6.5) | 4/102 (3.9) | |||||
| 2010 | Campylobacter | CD | Faeces | 39/54 (72) | 10/33 (10) | [132] | |
| C. concisus | 35/54 (65) | 11/33 (33) | |||||
| 2010 | C. concisus | CD and UC | Saliva | 13/13 (100) | 5/5 (100) | 57/59 (97) | [136] |
| 2011 | Helicobacter | UC | Tissue | 32/77 (42) | 11/59 (19) | [118] | |
| EHH | 30/77 (39) | 2/59 (3) | |||||
| H. pylori | 2/77 (3) | 9/59 (15) | |||||
| 2011 | C. concisus | CD, UC and IC | Tissue | 8/12 (66.7) | 3/8 (37.5) | 11/26 (42.3) | [133] |
| 2011 | Campylobacter | UC | Tissue | 51/69 (73.9) | 15/65 (23.1) | [135] | |
| C. concisus | 23/69 (33.3) | 7/65 (10.8) | |||||
| C. ureolyticus | 15/69 (21.7) | 2/65 (10.8) | |||||
| C. hominis | 14/69 (20.3) | 5/65 (7.7) | |||||
| C. curvus | 3/69 (4.3) | 4/65 (6.2) | |||||
| C. showae | 4/69 (5.8) | 0/65 (0) | |||||
| C. jejuni | 2/69 (2.9) | 0/65 (0) | |||||
| C. gracilis | 1/69 (1.4) | 0/65 (0) | |||||
| 2011 | Campylobacter | CD and UC | Tissue | 12/15 (80) | 11/13 (85) | 18/33 (48) | [134] |
| C. concisus | 10/15 (67) | 9/13 (69) | 12/33 (36) | ||||
| C. showae | 1/15 (7) | 2/13 (15) | 2/33 (6) | ||||
| C. hominis | 1/15 (7) | 1/13 (8) | 3/33 (9) | ||||
| C. ureolyticus | 2/15 (13) | 1/13 (8) | 2/33 (6) | ||||
| C. gracilis | 1/15 (7) | 1/13 (8) | 0/33 (0) | ||||
| C. rectus | 0/15 (0) | 1/13 (8) | 0/33 (0) | ||||
| C. jejuni | 1/15 (7) | 0/13 (0) | 0/33 (0) | ||||
| 2012 | AIEC | CD and UC | Tissue | 1/17 (5.9) | 1/10 (10) | 0/23 (0) | [166] |
| 2013 | Helicobacter | CD and UC | Tissue | 4/29 (13.8) | 1/13 (7.7) | 5/42 (11.9) | [119] |
| H. brantae | 1/59 (3.4) | 0/13 (0) | 0/42 (0) | ||||
| H. hepaticus | 1/59 (3.4) | 0/13 (0) | 0/42 (0) | ||||
| 2013 | Campylobacter | CD and UC | Tissue | 22/29 (75.9) | 9/13 (69) | 32/42 (76.2) | [119] |
| C. concisus | 13/29 (44.8) | 4/13 (30.8) | 16/42 (38.1) | ||||
| C. curvus | 2/29 (6.9) | 0/13 (0) | 3/42 (7.1) | ||||
| C. gracilis | 1/29 (3.4) | 0/13 (0) | 2/42 (4.8) | ||||
| C. hominis | 9/29 (31.0) | 5/13 (38.5) | 14/42 (33.3) | ||||
| C. rectus | 0/29 (0) | 0/13 (0) | 4/42 (9.5) | ||||
| C. showae | 9/29 (31.0) | 5/13 (38.5) | 9/42 (21.4) | ||||
| C. ureolyticus | 0/29 (0) | 0/13 (0) | 2/42 (4.8) | ||||
CD: Crohn’s disease; UC: Ulcerative colitis; IC: Indeterminate colitis; IBD: Inflammatory bowel disease; MAP: Mycobacterium avium subspecies paratuberculosis; EHH: Enterohepatic Helicobacter; E. coli: Escherichia coli; AIEC: Adherent-invasive E. coli.
Mycobacterium avium subspecies paratuberculosis
Mycobacterial infection has been postulated in the aetiology of Crohn’s disease since its first description in 1913. The association stems from the observed similarity between Crohn’s disease and the bovine condition Johne’s disease, a condition caused by Mycobacterium avium paratuberculosis (MAP) infection leading to granulomatous enterocolitis. There have been vast numbers of studies in this area but the role of MAP remains uncertain[50,82,83].
MAP can be widely isolated from meat, dairy products and water, indicating sources of infection and supporting its role[82,83]. However, a large study found a lack of epidemiological support for environmental exposure[84]. Over the last decade research into the prevalence of MAP in IBD patients has been inconclusive. A large number of researchers have successfully shown a higher prevalence of MAP in Crohn’s patients compared to controls but, it seems for each of these there has been an equivalent study yielding no association[85-97].
In support of its role the ability of MAP to invade gut epithelial cells, inducing tissue damage and inflammation, has been shown[98]. A dominant T-cell response to MAP has also been seen in CD patients and macrophages infected with viable MAP are associated with high production of tumour necrosis factor-alpha (TNF-α), a marker for CD[95,99,100]. Using mouse models, MAP has been found to induce full-thickness necrotizing colitis after subcutaneous and transluminal injection[101].
The discovered association between CD and the autophagy gene ATG16L1 lends further credence to the theory as it is known that autophagy is essential for inhibition of mycobacterium tuberculosis in infected macrophages[102,103]. Defective innate immune killing mechanisms in patients with NOD2 polymorphisms at first also seem to support the idea, and indeed it has been found that monocytes heterozygous for a NOD2 polymorphism are more permissive to the growth of MAP[104]. Beyond contemplation, however, evidence for this hypothesis is limited. MAP has been detected most commonly in colonic disease; this is in direct contrast with the prevalence of NOD2 mutation in ileal disease[105,106]. In fact a study directly looking at the relationship between NOD-2 and MAP serology found no association[107]. Combining this with the response of CD to immunosuppressant and anti-TNF therapy, known to cause MAP proliferation and a lack of success of anti-mycobacterial therapy, the role of MAP is clearly still in doubt[108].
Helicobacter
Helicobacters, as human gastrointestinal pathogens, have assumed great research interest since the discovery by Robin Warren and Barry Marshall of Helicobacter pylori as the infectious agent in gastric and duodenal ulceration. Also, similar to MAP, one of the main prompters of research into the role of Helicobacter in IBD has been their propensity to cause colitis in animal models like Cotton-top tamarin monkeys (Saguinus oedipus). Despite this, there has been a lack of success in the last decade establishing presence of Helicobacter in IBD patients. The findings of studies looking into the molecular evidence of Helicobacter presence are varied and studies aimed at culturing viable Helicobacter from IBD tissue have failed[109-121]. Interestingly the only seemingly universally accepted action of Helicobacter in IBD is the apparent protective effect of Helicobacter pylori which has convincingly been found to be negatively correlated with IBD[113,118-122]. This may conform to the “hygiene hypothesis” for the development of IBD[123].
The evidence for an association is much stronger with enterohepatic Helicobacter species. Non-pylori Helicobacter organisms have been shown to induce colitis in a number of rodent models; Helicobacter hepaticus and Helicobacter bilis (H. bilis) most prominently but, also Helicobacter trogontum, Helicobacter rodentium and Helicobacter typhlonius with cytokine patterns which were very similar to that of human IBD[124-128]. When studying the response to H. bilis, Jergens et al[126] showed that there was an IgG mediated response to the microbiota prior to the development of colitis, suggesting the ability of H. bilis to induce the hosts immune response to commensal bacteria, leading to the observed immune-mediated intestinal inflammation of IBD. In human subjects, enterohepatic Helicobacter species prevalence was significantly higher in colonic biopsy samples from patients with UC group compared to control subjects[118].
Campylobacter
Campylobacter is a relatively new and important player in IBD. Unlike the other IBD suspects, Campylobacter does not have a suitable animal disease model; instead interest stems from the recognition of Campylobacter jejuni (C. jejuni) as the leading cause of gastroenteritis worldwide[129].
The role of C. jejuni in human disease has been long recognised and its prevalence in IBD investigated[130]. The main advance in the last decade has been the recognition of the importance of non-jejuni Campylobacter as human pathogens. Zhang et al[131] found a higher prevalence of Campylobacter concisus (C. concisus) DNA and IgG levels in newly diagnosed paediatric patients with Crohn’s disease, even managing to culture C. concisus from a biopsy sample, indicating viability. Another study using faecal samples from newly diagnosed CD patients also found a significant association with C. concisus, 35 of 54 CD patients testing positive and only 11 of 33 healthy controls. This study also found that C. hominis was present in 13% of Crohn’s samples, Campylobacter ureolyticus in 9%, Campylobacter showae (C. showae) in 4%, Campylobacter gracilis (C. gracilis) in 2% and C. rectus in 2%. Interestingly C. gracilis, Campylobacter rectus and C. showae were only detected in patient samples[132]. Similar results have been obtained in a number of studies in adult patients[119,133-136]. Mahendran et al[134] also showed an increased prevalence in UC, a finding supported by Mukhopadhya et al[135] who found C. concisus DNA in biopsy samples in 23/69 (33.3%) of UC patients compared to 7/65 (10.8%) of controls. This study also found C. ureolyticus to be in higher prevalence in UC patients. The most recent study found that although Campylobacter appear to be surprisingly common, with positive PCR in 33/44 IBD patients and 32/42 controls, there was no association with IBD[119]. A dominant serological antibody response to C. concisus has been documented in IBD patients indicating the prevalence of infection[137,138]. Specifically CD patients have been shown to recognise flagellin B, ATP synthase F α subunit and outer membrane protein 18 of C. concisus[137].
The origins of Campylobacter have led to a few researchers looking into the risks of developing IBD after acute gastroenteritis. A long term study published in 2009 documents the risk of developing IBD after acute infection with Campylobacter (C. jejuni) or Salmonella[130]. The findings indicated a significant increased risk in the exposed group for subsequently developing IBD, which has been supported by similar studies[139-141].
The pathogenesis of C. jejuni had been fairly well established prior to the last decade. C. jejuni has been used to induce colitis in rodent models and previous exposure correlated with disease severity[142]. The ability of C. jejuni to attach and invade the gut epithelium is well documented[143]. The newest discovery has been that C. jejuni can promote translocation of commensal luminal bacteria. This is a natural process thought to be essential for immunological tolerance and mucosal surveillance in the GI tract. Up regulation could affect the normal mucosal response to the intestinal microbiota leading to the chronic immune-mediated intestinal inflammation of IBD[144]
A number of studies have demonstrated the ability of C. concisus to colonise and adhere to intestinal epithelial cells, causing cell damage and microvillus degradation[146]. Man et al[145] comprehensively described the method of C. concisus attachment and invasion. They showed C. concisus to attach to the intracellular junction, disrupting barrier function - increasing permeability by causing a loss of tight junction proteins and decreasing transepithelial electrical resistance and to invade by a process mediated by polar flagellum[146]. Other non-jejuni Campylobacters have also been shown to be invasive and induce pro-inflammatory cytokines as well as producing a number of virulence factors such as haemolysins, cytolethal distending toxin and zonula occludens toxin[129,146-150]. These mechanisms could have an important bearing when one considers a causative role for this group of pathogens in IBD.
Adherent and invasive Escherichia coli
A specific pathogenetic group of Escherichia coli (E. coli), adherent-invasive E. coli (AIEC) have recently been extensively implicated in human IBD and are currently one of the most exciting players in the pathogen story. This group are characterised by their ability to adhere and invade epithelial cells using actin microfilaments and microtubule recruitment. AIEC strains have been shown to be the cause of granulomatous colitis in boxer dogs and to induce granulomas, similar to early epithelioid granulomas, in vitro[151-153]. Similarly to the previously discussed bacteria, they have been documented to induce colitis in infected animals.
There is a growing body of evidence supporting the prevalence of AIEC in human disease. A number of studies initially showed a disproportionate increase in Enterobacteria as a whole[36,47]. When looking at AIEC organisms specifically, Darfeuille-Michaud et al[154] found them to be more prevalent in ileal Crohn’s lesion tissue (36.4%) then controls (6.2%). This study also found that AIEC seemed to be rarely found in colonic tissue with 3.7% detected from Crohn’s patients and 1.9% from controls, and none in UC specimens. This suggests a specific association of AIEC with ileal Crohn’s. The findings of this initial study have been backed up by many researchers obtaining similar results[28,155-161]. Additionally, antibodies to the E. coli membrane protein C and the CD associated bacterial sequence I2 have been shown to not only be more prevalent in CD but also to be associated with more severe disease, with small bowel involvement, faster disease progression and increased need for surgical intervention[157,162].
The mechanism by which AIEC might induce colitis has been fairly well established towards the end of the decade. AIEC have type one pili and flagella that can bind to host adhesion receptor carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6)[163,164]. CEACAM6 has been shown to be more highly expressed in ileal CD tissue, to be increased after-γ or TNF-α stimulation and to be upregulated by AIEC itself[163,164]. AIEC have also been shown to possess long polar fimbriae and so can cross the mucosal barrier to access lymphoid cells[165]. They can then invade macrophages without inducing cell death, allowing them to replicate and continuously activate immune cells, triggering TNF-alpha release and granuloma formation, which are hallmarks of Crohn’s disease. In fact the use of TNF-alpha antibodies has been shown to decrease the number of intramacrophagic bacteria, relating this to the success of anti-TNF therapy and further supporting the role of AIEC[166].
Other putative bacterial pathogens
In the last decade, there has been renewed focus in studying the role of various other bacterial strains in the aetiopathogenesis of IBD as well. The role of Fusobacterium was studied in mucosal biopsies of patients with IBD and was found to be significant compared to controls in a number of studies prior to this review period. More recently seropositivity to Fusobacterium varium (F. varium) infection was found to be higher in UC patients as opposed to controls with increased severity of disease in seropositive UC patients[167]. Further study has revealed the ability of F. varium to adhere to and invade colonic epithelial cells, increasing IL-8 and TNF-α secretion, providing a mechanism whereby F. varium infection may induce inflammation similar to that seen in IBD[168].
A similar association has also been found with Klebsiella infection, with Anti-Klebsiella antibodies found more commonly in IBD patients than in controls with the bacteria being implicated in disease relapses[169]. Klebsiella pneumoniae has recently been shown to increase the severity of colitis in mouse models, increasing COX-2, IL-1β, IL-6 and TNF-α expression and reducing tight junction associated proteins[170]. Colitis has been shown to be induced even in wild type mice highlighting the high pathogenic potential of this bacteria[171].
The role of Salmonella infection has been postulated from numerous studies that have documented the risk of developing IBD after acute Salmonella gastroenteritis[130,139-141]. When searching for a mechanism it has been found that a Salmonella virulence factor, the invasion-associated type Darfeuille-Michaud secretion system induces inflammation by activation of the NOD1/NOD2 signalling pathways[172]. This ties Salmonella nicely to the growing body of research into the genetics of IBD, supporting the role of the pathogen.
The potential role of Yersinia was proposed at the beginning of the last decade based upon the observed parallel increase in IBD and refrigeration, “the cold chain hypothesis”[173]. Similarly to Salmonella and Campylobacter there has been evidence that acute Y. enterocolitica infection increases the short and long term risk of developing IBD[174]. Despite some later successes in isolation Yersinia from IBD patients there has yet to be a compelling body of evidence coupling the prevalence of Yersinia with established IBD[175-177].
MODULATION OF THE GUT MICROBIOTA AS A TREATMENT OPTION IN IBD PATIENTS
The past decade has seen rapid and definitive strides in determining distinct changes in the gut microbiota in patients with IBD. The effects seem to be global, involving not only the physical composition of the principal components but also significantly altering their function. The role of individual pathogens in this complex milieu still needs to be elucidated. The proof of concept of “dysbiosis” as an important step towards developing IBD needs to be proven with therapeutic trials attempting to reverse the process.
Role of probiotics and prebiotics
Probiotics are beneficial microorganisms that, when ingested, may influence the gut microbiota composition, metabolic activity and immunomodulation to confer benefit to the host[178]. They can alter microbial diversity through competitive inhibition of other microbes, increase mucosal barrier function through the production of short chain fatty acids (SCFA) and interact with intestinal DC to stimulate an anti-inflammatory response[179-182]. These probiotic strains must be of human origin, be non-pathogenic and have the intrinsic ability to survive the gastrointestinal transit in order to confer maximal benefit[183]. The most common probiotics used in the treatment of IBD have been Lactobacillus sp, Bifidobacterium sp, Sacchromyces bouladrii, E. coli Nissle 1917 and the probiotic combination VSL#3[184-191]. The strongest indication for the use of probiotics in IBD has been in the treatment of pouchitis in the post-operative setting in UC patients[192]. E. coli Nissle 1917 and VSL#3 have been found to be effective in preventing relapse and inducing remission in this setting[193-198]. The data is not very robust with respect to the role of probiotics maintaining remission in UC and a recent Cochrane Database System Review has not recommended its use[197]. The current body of evidence does not show any demonstrable benefit in patients with CD[198,199].
Prebiotics are non-digestible oligosaccharides that are selectively fermented in the colon into SCFAs and can alter microbial composition and activity and confer benefit to the host[200]. Examples include inulin, fructooligosaccharide (FOS), galactooligosaccharides and lactulose[201-206]. Prebiotics can selectively stimulate the growth of certain probiotics such as Lactobacillus and Bifidobacterium, decrease intraluminal pH and increase the production of SCFA, such as acetate and butyrate, which play an important role in epithelial and DC function[207,208]. SCFAs have also been found to have an anti-inflammatory effect[209]. In an open labelled trial FOSuse decreased the disease activity index in patients with active CD and resulted in increased faecal Bifidobacterium, but this benefit was not demonstrated in a subsequent randomized placebo controlled trial[205,206]. Another prebiotic inulin showed some promise in a randomized controlled trial in patients with UC and was also found to decrease inflammation in patients with pouchitis[202,203]. A couple of studies have also found a potential role of germinated barley foodstuff in maintaining remission in patients with active UC[210,211]. Lastly, some benefit was also accrued with the use of Ispaghula husk in patients with UC[212]. The important studies and their brief outcomes are summarized in Table 2.
Table 2.
Probiotics and prebiotics in inflammatory bowel disease
| Active component | Study | Design | n | Duration | Intervention | Result | Reference |
| Lactobacillus | CD remission | RCT | 98 | 6 mo | Lactobacillus johnsonii LA1 4 × 109 cfu/d | No difference | [184] |
| IBD | 40 | 1 mo | Lactobacillus rhamnosus GR-1 and L. reuteri RC-14 supplemented yogurt | Anti-inflammatory effects | [185] | ||
| Bifidobacterium | Active UC | RCT | 20 | 12 wk | Bifido-fermented milk [B. breve, B. bifidum and acidophilus] (1 × 1010) or placebo | Decreased clinical activity (P < 0.05) decreased endoscopic/histological scores (P < 0.01) | [186] |
| Active UC | Open label | 12 | 4 wk | BGS 4.5 g/d | Decrease in clinical activity index (P < 0.01) and endoscopic scores (P < 0.05) | [187] | |
| C57BL/6 mice | Experimental | 16 | 3 d | B. bifidum S17 | Decrease in microscopic inflammation and reduction in inflammatory cytokines | [188] | |
| E. coli Nissle 1917 | UC remission | 327 | 12 mo | 200 mg E. coli Nissle 1917 or 1500 mg mesalazine/d | E. coli Nissle 1917 was equivalent to mesalazine in maintaining remission | [189] | |
| VSL#3 | UC remission | Open label | 34 | 6 wk | VSL#3, 3.6 × 1012, bacteria/d | ITT analysis demonstrated remission in 18/34 and response in 8/34 | [190] |
| Active UC | RCT | 29 | 12 mo | VSL#3 450-1800 billion bacteria/d | Remission was achieved in 13/14 VSL#3 and 4/15 placebo (P < 0.001) | [191] | |
| Relapses within 1 yr of followup occurred in 3/14 VSL#3 and 11/15 placebo | |||||||
| Endoscopic and histological score were significantly lower in VSL#3 vs placebo (P < 0.05) | |||||||
| Inulin | Active UC | RCT | 19 | 2 wk | 3 g/d mesalazine and either 12 g/d oligofructose-enriched inulin or placebo | Dyspeptic symptoms scale decreased significantly and an early reduction of calprotectin was observed in oligofructose-enriched inulin group | [202] |
| Pouchitis | RCT | 20 | 3 wk | 24 g/d inulin or placebo | Reduction in inflammation, increase butyrate conc and decreased inflammation associated factors | [203] | |
| Inulin and FOS | HLA-B27 rat model IBD | 12 wk | 8 g/kg body weight inulin or FOS | FOS increased Bifidobacterium spp. FOS and inulin reduced Clostridium cluster XI and C. difficile toxin gene expression correlating with a reduction of chronic intestinal inflammation | [204] | ||
| FOS | Active CD | RCT | 103 | 4 wk | 15 g/d FOS or placebo | No clinical benefit, despite impacting on DC function | [205] |
| Active CD | Open label | 10 | 3 wk | 15 g/d | Significant reduction in Harvey Bradshaw index (P < 0.01) significant increase in faecal bifidobacteria conc. (P < 0.001) and modifies DC function | [206] |
CD: Crohn’s disease; UC: Ulcerative colitis; IBD: Inflammatory bowel disease; RCT: Randomized control trial; FOS: Fructooligosaccharides; E. coli: Escherichia coli.
Antibiotics
A couple of recent meta-analyses on antibiotics in IBD found that the use of antibiotics improved clinical outcomes of patients with IBD[213,214]. There is evidence that metronidazole and ciprofloxacin are useful in the treatment of CD and pouchitits[215,216]. Support for the use of antibiotics as the primary treatment in UC is less convincing, however, there are some studies which suggest that rifaximin and ciprofloxacin could be useful as an adjunctive treatment for UC[213]. The mechanisms through which antibiotics are thought to benefit patients with CD are through the inhibition of pathogenic bacteria or through reducing overall bacterial numbers. The main issues with antibiotic treatment include lack of understanding of which bacteria may be involved in the initiation of inflammation, lack of specificity and the potential for antibiotic resistance. There have been several trials in the past studying the specific role of anti-mycobacterials in the treatment of CD and this has been summarized in a European consensus document which has deemed the futility of such treatment[217].
Faecal transplantation
Faecal transplantation or faecal microbial therapy (FMT) as it is more commonly known is a technique in which stool is taken from a healthy surrogate and inserted into an unhealthy person, with curative intent[218]. The origins of faecal transplantation as a method of treating enteric pathology can be traced back for more than two millennia, when it was used as a traditional Chinese medicine to treat diarrhoea[219].
In recent times, FMT is perhaps best known for its potential role in treating Clostridium difficile (C. difficile) infectious diarrhoea[220]. After donor-faeces infusion in a group of patients infected with C. difficile, there was an alteration in the gut microflora with an increased faecal bacterial diversity, similar to that in healthy donors, with an increase in Bacteroidetes species and Clostridium clusters and a decrease in Proteobacteria species. This therapeutic benefit by FMT as documented in this trial, would theoretically have a beneficial effect in patients with IBD as well. The use of this form of intervention is still restricted to few exploratory trials. Donor faecal enemas were given to a group of ten children over five days with moderate to severe UC and resulted in 78% clinical response after a week and 67% with sustained response after a month in the nine children who could tolerate the treatment[221]. This was similarly documented in a subset of six adult UC patients who were treated over a period of 5 d. Complete reversal of symptoms was achieved in all patients by 4 mo, by which time all other UC medications had been ceased and at 1 to 13 years post FMT and without any UC medication, there was no clinical, colonoscopic, or histologic evidence of UC in any patient[222]. However, a single infusion in six adult patients with severe UC did not have a similar beneficial effect and the faecal microbiota changed to the donor phenotype in only 50% of those treated, suggesting that as opposed to treatment of C. difficile a prolonged treatment is indicated in IBD[223]. Although the data described above is certainly promising, there is clearly a need to move on from individual case reports and conduct more large scale randomised control trials before any benefit of FMT can be claimed with any certainty. Some concerns have also been raised regarding safety and side effects, with some IBD patients suffering mild side effects following FMT, and the obvious issues surrounding potential transmission of host infectious disease[224]. The efficacies of different administration techniques and dosing regimens for FMT also need to be refined and investigated. Literature to date describes a range of methods including colonoscopy, duodenal or gastric tubes and self-administered enema yet to date there is no clear evidence to support one method over any other. There is no doubt that manipulation of the gut microbiota could have enormous therapeutic potential and FMT will play an important role in its future.
CONCLUSION
The understanding of the aetiopathogenesis of IBD has undergone radical shifts in the past decade with the advent of modern molecular techniques that can characterize the gut microbiome more accurately and host genomic analysis that can explore the vast genetic universe of IBD. At the heart of the inflammatory process in IBD is “dysbiosis” of the gut microbiome, which may be driven by host genetics and environmental factors like diet. The next decade will help unravel the intricacies of the host immune defences that determine this intriguing host-microbiome ecology. The relationship of the genotype of the host and the extent to which it determines the composition of the microbiome needs to be elucidated. It will open the doors to more “personalized” therapeutic interventions, which would encompass the host genotype and serotype, the disease phenotype, the gene expression profiles of the immune cells and the microbiome composition to decide the best strategy for treating patients with IBD. This will usher in a paradigm shift in patient management with a move away from standard generic therapy to a scientific, tailored approach based on the needs of individual patients.
Footnotes
P- Reviewers: Hokama A, Lakatos PL, Pellicano R S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
- 1.Schirbel A, Fiocchi C. Inflammatory bowel disease: Established and evolving considerations on its etiopathogenesis and therapy. J Dig Dis. 2010;11:266–276. doi: 10.1111/j.1751-2980.2010.00449.x. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 3.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42; quiz e30. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Henderson P, Hansen R, Cameron FL, Gerasimidis K, Rogers P, Bisset WM, Reynish EL, Drummond HE, Anderson NH, Van Limbergen J, et al. Rising incidence of pediatric inflammatory bowel disease in Scotland. Inflamm Bowel Dis. 2012;18:999–1005. doi: 10.1002/ibd.21797. [DOI] [PubMed] [Google Scholar]
- 5.Kappelman MD, Moore KR, Allen JK, Cook SF. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Dig Dis Sci. 2013;58:519–525. doi: 10.1007/s10620-012-2371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489:250–256. doi: 10.1038/nature11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zoetendal EG, Rajilic-Stojanovic M, de Vos WM. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut. 2008;57:1605–1615. doi: 10.1136/gut.2007.133603. [DOI] [PubMed] [Google Scholar]
- 8.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danese S, Sans M, Fiocchi C. Inflammatory bowel disease: the role of environmental factors. Autoimmun Rev. 2004;3:394–400. doi: 10.1016/j.autrev.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9:599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 12.Fiocchi C. Genes and ‘in-vironment’: how will our concepts on the pathophysiology of inflammatory bowel disease develop in the future? Dig Dis. 2012;30 Suppl 3:2–11. doi: 10.1159/000342585. [DOI] [PubMed] [Google Scholar]
- 13.Prantera C, Scribano ML. Crohn’s disease: the case for bacteria. Ital J Gastroenterol Hepatol. 1999;31:244–246. [PubMed] [Google Scholar]
- 14.Ehrhardt RO. New insights into the immunopathology of chronic inflammatory bowel disease. Semin Gastrointest Dis. 1996;7:144–150. [PubMed] [Google Scholar]
- 15.Vinh DC, Behr MA. Crohn’s as an immune deficiency: from apparent paradox to evolving paradigm. Expert Rev Clin Immunol. 2013;9:17–30. doi: 10.1586/eci.12.87. [DOI] [PubMed] [Google Scholar]
- 16.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 17.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 18.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur N, Chen CC, Luther J, Kao JY. Intestinal dysbiosis in inflammatory bowel disease. Gut Microbes. 2011;2:211–216. doi: 10.4161/gmic.2.4.17863. [DOI] [PubMed] [Google Scholar]
- 23.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Doré J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:106–111. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- 25.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 27.Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 28.Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, Tysk C, Jansson JK. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 29.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, Thomas M, Wells JM, Langella P. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16:255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, Mukhopadhya I, Bisset WM, Barclay AR, Bishop J, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am J Gastroenterol. 2012;107:1913–1922. doi: 10.1038/ajg.2012.335. [DOI] [PubMed] [Google Scholar]
- 32.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neut C, Bulois P, Desreumaux P, Membré JM, Lederman E, Gambiez L, Cortot A, Quandalle P, van Kruiningen H, Colombel JF. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am J Gastroenterol. 2002;97:939–946. doi: 10.1111/j.1572-0241.2002.05613.x. [DOI] [PubMed] [Google Scholar]
- 35.Andoh A, Kuzuoka H, Tsujikawa T, Nakamura S, Hirai F, Suzuki Y, Matsui T, Fujiyama Y, Matsumoto T. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J Gastroenterol. 2012;47:1298–1307. doi: 10.1007/s00535-012-0605-0. [DOI] [PubMed] [Google Scholar]
- 36.Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43:3380–3389. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012;9:219–230. doi: 10.1038/nrgastro.2012.14. [DOI] [PubMed] [Google Scholar]
- 39.Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1:403–418. doi: 10.1038/ismej.2007.52. [DOI] [PubMed] [Google Scholar]
- 40.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–4141. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Doré J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 44.Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 2013;7:1256–1261. doi: 10.1038/ismej.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 47.Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140:1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hansen R, Thomson JM, El-Omar EM, Hold GL. The role of infection in the aetiology of inflammatory bowel disease. J Gastroenterol. 2010;45:266–276. doi: 10.1007/s00535-009-0191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Devière J, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pierik M, Joossens S, Van Steen K, Van Schuerbeek N, Vlietinck R, Rutgeerts P, Vermeire S. Toll-like receptor-1, -2, and -6 polymorphisms influence disease extension in inflammatory bowel diseases. Inflamm Bowel Dis. 2006;12:1–8. doi: 10.1097/01.mib.0000195389.11645.ab. [DOI] [PubMed] [Google Scholar]
- 53.Ng MT, Van’t Hof R, Crockett JC, Hope ME, Berry S, Thomson J, McLean MH, McColl KE, El-Omar EM, Hold GL. Increase in NF-kappaB binding affinity of the variant C allele of the toll-like receptor 9 -1237T/C polymorphism is associated with Helicobacter pylori-induced gastric disease. Infect Immun. 2010;78:1345–1352. doi: 10.1128/IAI.01226-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto-Furusho JK, Podolsky DK. Innate immunity in inflammatory bowel disease. World J Gastroenterol. 2007;13:5577–5580. doi: 10.3748/wjg.v13.i42.5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhee SH, Im E, Riegler M, Kokkotou E, O’brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci USA. 2005;102:13610–13615. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hausmann M, Kiessling S, Mestermann S, Webb G, Spöttl T, Andus T, Schölmerich J, Herfarth H, Ray K, Falk W, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 60.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005;115:66–75. doi: 10.1172/JCI19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smythies LE, Shen R, Bimczok D, Novak L, Clements RH, Eckhoff DE, Bouchard P, George MD, Hu WK, Dandekar S, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem. 2010;285:19593–19604. doi: 10.1074/jbc.M109.069955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329:849–853. doi: 10.1126/science.1188510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamada N, Hisamatsu T, Okamoto S, Sato T, Matsuoka K, Arai K, Nakai T, Hasegawa A, Inoue N, Watanabe N, et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol. 2005;175:6900–6908. doi: 10.4049/jimmunol.175.10.6900. [DOI] [PubMed] [Google Scholar]
- 66.Weber B, Saurer L, Mueller C. Intestinal macrophages: differentiation and involvement in intestinal immunopathologies. Semin Immunopathol. 2009;31:171–184. doi: 10.1007/s00281-009-0156-5. [DOI] [PubMed] [Google Scholar]
- 67.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut. 2008;57:903–910. doi: 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- 69.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–G1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 70.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 71.Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, Naber TH, Drenth JP, Girardin SE, Kullberg BJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 72.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 73.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noguchi E, Homma Y, Kang X, Netea MG, Ma X. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol. 2009;10:471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci USA. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hedl M, Abraham C. Secretory mediators regulate Nod2-induced tolerance in human macrophages. Gastroenterology. 2011;140:231–241. doi: 10.1053/j.gastro.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 79.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 80.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 81.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greenstein RJ. Is Crohn’s disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis, and Johne’s disease. Lancet Infect Dis. 2003;3:507–514. doi: 10.1016/s1473-3099(03)00724-2. [DOI] [PubMed] [Google Scholar]
- 83.Sartor RB. Does Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease? Gut. 2005;54:896–898. doi: 10.1136/gut.2004.055889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abubakar I, Myhill DJ, Hart AR, Lake IR, Harvey I, Rhodes JM, Robinson R, Lobo AJ, Probert CS, Hunter PR. A case-control study of drinking water and dairy products in Crohn’s Disease--further investigation of the possible role of Mycobacterium avium paratuberculosis. Am J Epidemiol. 2007;165:776–783. doi: 10.1093/aje/kwk067. [DOI] [PubMed] [Google Scholar]
- 85.Bull TJ, McMinn EJ, Sidi-Boumedine K, Skull A, Durkin D, Neild P, Rhodes G, Pickup R, Hermon-Taylor J. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s disease. J Clin Microbiol. 2003;41:2915–2923. doi: 10.1128/JCM.41.7.2915-2923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Naser SA, Ghobrial G, Romero C, Valentine JF. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet. 2004;364:1039–1044. doi: 10.1016/S0140-6736(04)17058-X. [DOI] [PubMed] [Google Scholar]
- 87.Romero C, Hamdi A, Valentine JF, Naser SA. Evaluation of surgical tissue from patients with Crohn’s disease for the presence of Mycobacterium avium subspecies paratuberculosis DNA by in situ hybridization and nested polymerase chain reaction. Inflamm Bowel Dis. 2005;11:116–125. doi: 10.1097/00054725-200502000-00004. [DOI] [PubMed] [Google Scholar]
- 88.Sechi LA, Scanu AM, Molicotti P, Cannas S, Mura M, Dettori G, Fadda G, Zanetti S. Detection and Isolation of Mycobacterium avium subspecies paratuberculosis from intestinal mucosal biopsies of patients with and without Crohn’s disease in Sardinia. Am J Gastroenterol. 2005;100:1529–1536. doi: 10.1111/j.1572-0241.2005.41415.x. [DOI] [PubMed] [Google Scholar]
- 89.Kirkwood CD, Wagner J, Boniface K, Vaughan J, Michalski WP, Catto-Smith AG, Cameron DJ, Bishop RF. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn’s disease. Inflamm Bowel Dis. 2009;15:1643–1655. doi: 10.1002/ibd.20967. [DOI] [PubMed] [Google Scholar]
- 90.Lee A, Griffiths TA, Parab RS, King RK, Dubinsky MC, Urbanski SJ, Wrobel I, Rioux KP. Association of Mycobacterium avium subspecies paratuberculosis with Crohn Disease in pediatric patients. J Pediatr Gastroenterol Nutr. 2011;52:170–174. doi: 10.1097/MPG.0b013e3181ef37ba. [DOI] [PubMed] [Google Scholar]
- 91.Autschbach F, Eisold S, Hinz U, Zinser S, Linnebacher M, Giese T, Löffler T, Büchler MW, Schmidt J. High prevalence of Mycobacterium avium subspecies paratuberculosis IS900 DNA in gut tissues from individuals with Crohn’s disease. Gut. 2005;54:944–949. doi: 10.1136/gut.2004.045526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Juste RA, Elguezabal N, Garrido JM, Pavon A, Geijo MV, Sevilla I, Cabriada JL, Tejada A, García-Campos F, Casado R, et al. On the prevalence of M. avium subspecies paratuberculosis DNA in the blood of healthy individuals and patients with inflammatory bowel disease. PLoS One. 2008;3:e2537. doi: 10.1371/journal.pone.0002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bernstein CN, Blanchard JF, Rawsthorne P, Collins MT. Population-based case control study of seroprevalence of Mycobacterium paratuberculosis in patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2004;42:1129–1135. doi: 10.1128/JCM.42.3.1129-1135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bernstein CN, Nayar G, Hamel A, Blanchard JF. Study of animal-borne infections in the mucosas of patients with inflammatory bowel disease and population-based controls. J Clin Microbiol. 2003;41:4986–4990. doi: 10.1128/JCM.41.11.4986-4990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clancy R, Ren Z, Turton J, Pang G, Wettstein A. Molecular evidence for Mycobacterium avium subspecies paratuberculosis (MAP) in Crohn’s disease correlates with enhanced TNF-alpha secretion. Dig Liver Dis. 2007;39:445–451. doi: 10.1016/j.dld.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 96.Parrish NM, Radcliff RP, Brey BJ, Anderson JL, Clark DL, Koziczkowski JJ, Ko CG, Goldberg ND, Brinker DA, Carlson RA, et al. Absence of mycobacterium avium subsp. paratuberculosis in Crohn’s patients. Inflamm Bowel Dis. 2009;15:558–565. doi: 10.1002/ibd.20799. [DOI] [PubMed] [Google Scholar]
- 97.Sasikala M, Reddy DN, Pratap N, Sharma SK, Balkumar PR, Sekaran A, Banerjee R, Reddy DB. Absence of Mycobacterium avium ss paratuberculosis-specific IS900 sequence in intestinal biopsy tissues of Indian patients with Crohn’s disease. Indian J Gastroenterol. 2009;28:169–174. doi: 10.1007/s12664-009-0068-2. [DOI] [PubMed] [Google Scholar]
- 98.Golan L, Livneh-Kol A, Gonen E, Yagel S, Rosenshine I, Shpigel NY. Mycobacterium avium paratuberculosis invades human small-intestinal goblet cells and elicits inflammation. J Infect Dis. 2009;199:350–354. doi: 10.1086/596033. [DOI] [PubMed] [Google Scholar]
- 99.Nakase H, Tamaki H, Matsuura M, Chiba T, Okazaki K. Involvement of mycobacterium avium subspecies paratuberculosis in TNF-α production from macrophage: possible link between MAP and immune response in Crohn’s disease. Inflamm Bowel Dis. 2011;17:E140–E142. doi: 10.1002/ibd.21750. [DOI] [PubMed] [Google Scholar]
- 100.Olsen I, Tollefsen S, Aagaard C, Reitan LJ, Bannantine JP, Andersen P, Sollid LM, Lundin KE. Isolation of Mycobacterium avium subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of Crohn’s disease patients. PLoS One. 2009;4:e5641. doi: 10.1371/journal.pone.0005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Momotani E, Ozaki H, Hori M, Yamamoto S, Kuribayashi T, Eda S, Ikegami M. Mycobacterium avium subsp. paratuberculosis lipophilic antigen causes Crohn’s disease-type necrotizing colitis in Mice. Springerplus. 2012;1:47. doi: 10.1186/2193-1801-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 103.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glubb DM, Gearry RB, Barclay ML, Roberts RL, Pearson J, Keenan JI, McKenzie J, Bentley RW. NOD2 and ATG16L1 polymorphisms affect monocyte responses in Crohn’s disease. World J Gastroenterol. 2011;17:2829–2837. doi: 10.3748/wjg.v17.i23.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsianos EV, Katsanos KH, Tsianos VE. Role of genetics in the diagnosis and prognosis of Crohn’s disease. World J Gastroenterol. 2011;17:5246–5259. doi: 10.3748/wjg.v17.i48.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cantó E, Ricart E, Busquets D, Monfort D, García-Planella E, González D, Balanzó J, Rodriguez-Sanchez JL, Vidal S. Influence of a nucleotide oligomerization domain 1 (NOD1) polymorphism and NOD2 mutant alleles on Crohn’s disease phenotype. World J Gastroenterol. 2007;13:5446–5453. doi: 10.3748/wjg.v13.i41.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bernstein CN, Wang MH, Sargent M, Brant SR, Collins MT. Testing the interaction between NOD-2 status and serological response to Mycobacterium paratuberculosis in cases of inflammatory bowel disease. J Clin Microbiol. 2007;45:968–971. doi: 10.1128/JCM.02062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Selby W, Pavli P, Crotty B, Florin T, Radford-Smith G, Gibson P, Mitchell B, Connell W, Read R, Merrett M, et al. Two-year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn’s disease. Gastroenterology. 2007;132:2313–2319. doi: 10.1053/j.gastro.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 109.Bell SJ, Chisholm SA, Owen RJ, Borriello SP, Kamm MA. Evaluation of Helicobacter species in inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18:481–486. doi: 10.1046/j.1365-2036.2003.01703.x. [DOI] [PubMed] [Google Scholar]
- 110.Oliveira AG, das Graças Pimenta Sanna M, Rocha GA, Rocha AM, Santos A, Dani R, Marinho FP, Moreira LS, de Lourdes Abreu Ferrari M, Moura SB, et al. Helicobacter species in the intestinal mucosa of patients with ulcerative colitis. J Clin Microbiol. 2004;42:384–386. doi: 10.1128/JCM.42.1.384-386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Streutker CJ, Bernstein CN, Chan VL, Riddell RH, Croitoru K. Detection of species-specific helicobacter ribosomal DNA in intestinal biopsy samples from a population-based cohort of patients with ulcerative colitis. J Clin Microbiol. 2004;42:660–664. doi: 10.1128/JCM.42.2.660-664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huijsdens XW, Linskens RK, Koppes J, Tang YL, Meuwissen SG, Vandenbroucke-Grauls CM, Savelkoul PH. Detection of Helicobacter species DNA by quantitative PCR in the gastrointestinal tract of healthy individuals and of patients with inflammatory bowel disease. FEMS Immunol Med Microbiol. 2004;41:79–84. doi: 10.1016/j.femsim.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 113.Bohr UR, Glasbrenner B, Primus A, Zagoura A, Wex T, Malfertheiner P. Identification of enterohepatic Helicobacter species in patients suffering from inflammatory bowel disease. J Clin Microbiol. 2004;42:2766–2768. doi: 10.1128/JCM.42.6.2766-2768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Basset C, Holton J, Bazeos A, Vaira D, Bloom S. Are Helicobacter species and enterotoxigenic Bacteroides fragilis involved in inflammatory bowel disease? Dig Dis Sci. 2004;49:1425–1432. doi: 10.1023/b:ddas.0000042241.13489.88. [DOI] [PubMed] [Google Scholar]
- 115.Man SM, Zhang L, Day AS, Leach S, Mitchell H. Detection of enterohepatic and gastric helicobacter species in fecal specimens of children with Crohn’s disease. Helicobacter. 2008;13:234–238. doi: 10.1111/j.1523-5378.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 116.Laharie D, Asencio C, Asselineau J, Bulois P, Bourreille A, Moreau J, Bonjean P, Lamarque D, Pariente A, Soulé JC, et al. Association between entero-hepatic Helicobacter species and Crohn’s disease: a prospective cross-sectional study. Aliment Pharmacol Ther. 2009;30:283–293. doi: 10.1111/j.1365-2036.2009.04034.x. [DOI] [PubMed] [Google Scholar]
- 117.Kaakoush NO, Holmes J, Octavia S, Man SM, Zhang L, Castaño-Rodríguez N, Day AS, Leach ST, Lemberg DA, Dutt S, et al. Detection of Helicobacteraceae in intestinal biopsies of children with Crohn’s disease. Helicobacter. 2010;15:549–557. doi: 10.1111/j.1523-5378.2010.00792.x. [DOI] [PubMed] [Google Scholar]
- 118.Thomson JM, Hansen R, Berry SH, Hope ME, Murray GI, Mukhopadhya I, McLean MH, Shen Z, Fox JG, El-Omar E, et al. Enterohepatic helicobacter in ulcerative colitis: potential pathogenic entities? PLoS One. 2011;6:e17184. doi: 10.1371/journal.pone.0017184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hansen R, Berry SH, Mukhopadhya I, Thomson JM, Saunders KA, Nicholl CE, Bisset WM, Loganathan S, Mahdi G, Kastner-Cole D, et al. The microaerophilic microbiota of de-novo paediatric inflammatory bowel disease: the BISCUIT study. PLoS One. 2013;8:e58825. doi: 10.1371/journal.pone.0058825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang S, Zhong B, Chao K, Xiao Y, Cui Y, Gao X, Chen B, He Y, Hu P, Chen M, et al. Role of Helicobacter species in Chinese patients with inflammatory bowel disease. J Clin Microbiol. 2011;49:1987–1989. doi: 10.1128/JCM.02630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Keenan JI, Mitchell HM, Day AS. Interactions between gastric and enteric infections: clues to the pathogenesis of inflammatory bowel disease? N Z Med J. 2011;124:62–67. [PubMed] [Google Scholar]
- 122.Sonnenberg A, Genta RM. Low prevalence of Helicobacter pylori infection among patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2012;35:469–476. doi: 10.1111/j.1365-2036.2011.04969.x. [DOI] [PubMed] [Google Scholar]
- 123.Lidar M, Lipschitz N, Langevitz P, Barzilai O, Ram M, Porat-Katz BS, Pagnoux C, Guilpain P, Sinico RA, Radice A, et al. Infectious serologies and autoantibodies in Wegener’s granulomatosis and other vasculitides: novel associations disclosed using the Rad BioPlex 2200. Ann N Y Acad Sci. 2009;1173:649–657. doi: 10.1111/j.1749-6632.2009.04641.x. [DOI] [PubMed] [Google Scholar]
- 124.Zhang L, Danon SJ, Grehan M, Chan V, Lee A, Mitchell H. Natural colonization with Helicobacter species and the development of inflammatory bowel disease in interleukin-10-deficient mice. Helicobacter. 2005;10:223–230. doi: 10.1111/j.1523-5378.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 125.Liu Z, Ramer-Tait AE, Henderson AL, Demirkale CY, Nettleton D, Wang C, Hostetter JM, Jergens AE, Wannemuehler MJ. Helicobacter bilis colonization enhances susceptibility to Typhlocolitis following an inflammatory trigger. Dig Dis Sci. 2011;56:2838–2848. doi: 10.1007/s10620-011-1701-3. [DOI] [PubMed] [Google Scholar]
- 126.Jergens AE, Wilson-Welder JH, Dorn A, Henderson A, Liu Z, Evans RB, Hostetter J, Wannemuehler MJ. Helicobacter bilis triggers persistent immune reactivity to antigens derived from the commensal bacteria in gnotobiotic C3H/HeN mice. Gut. 2007;56:934–940. doi: 10.1136/gut.2006.099242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chichlowski M, Sharp JM, Vanderford DA, Myles MH, Hale LP. Helicobacter typhlonius and Helicobacter rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL10-deficient mice. Comp Med. 2008;58:534–541. [PMC free article] [PubMed] [Google Scholar]
- 128.Whary MT, Danon SJ, Feng Y, Ge Z, Sundina N, Ng V, Taylor NS, Rogers AB, Fox JG. Rapid onset of ulcerative typhlocolitis in B6.129P2-IL10tm1Cgn (IL-10-/-) mice infected with Helicobacter trogontum is associated with decreased colonization by altered Schaedler’s flora. Infect Immun. 2006;74:6615–6623. doi: 10.1128/IAI.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Man SM. The clinical importance of emerging Campylobacter species. Nat Rev Gastroenterol Hepatol. 2011;8:669–685. doi: 10.1038/nrgastro.2011.191. [DOI] [PubMed] [Google Scholar]
- 130.Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology. 2009;137:495–501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 131.Zhang L, Man SM, Day AS, Leach ST, Lemberg DA, Dutt S, Stormon M, Otley A, O’Loughlin EV, Magoffin A, et al. Detection and isolation of Campylobacter species other than C. jejuni from children with Crohn’s disease. J Clin Microbiol. 2009;47:453–455. doi: 10.1128/JCM.01949-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Man SM, Zhang L, Day AS, Leach ST, Lemberg DA, Mitchell H. Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm Bowel Dis. 2010;16:1008–1016. doi: 10.1002/ibd.21157. [DOI] [PubMed] [Google Scholar]
- 133.Mukhopadhya I, Hansen R, Nicholl CE, Alhaidan YA, Thomson JM, Berry SH, Pattinson C, Stead DA, Russell RK, El-Omar EM, et al. A comprehensive evaluation of colonic mucosal isolates of Sutterella wadsworthensis from inflammatory bowel disease. PLoS One. 2011;6:e27076. doi: 10.1371/journal.pone.0027076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mahendran V, Riordan SM, Grimm MC, Tran TA, Major J, Kaakoush NO, Mitchell H, Zhang L. Prevalence of Campylobacter species in adult Crohn’s disease and the preferential colonization sites of Campylobacter species in the human intestine. PLoS One. 2011;6:e25417. doi: 10.1371/journal.pone.0025417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mukhopadhya I, Thomson JM, Hansen R, Berry SH, El-Omar EM, Hold GL. Detection of Campylobacter concisus and other Campylobacter species in colonic biopsies from adults with ulcerative colitis. PLoS One. 2011;6:e21490. doi: 10.1371/journal.pone.0021490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang L, Budiman V, Day AS, Mitchell H, Lemberg DA, Riordan SM, Grimm M, Leach ST, Ismail Y. Isolation and detection of Campylobacter concisus from saliva of healthy individuals and patients with inflammatory bowel disease. J Clin Microbiol. 2010;48:2965–2967. doi: 10.1128/JCM.02391-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kovach Z, Kaakoush NO, Lamb S, Zhang L, Raftery MJ, Mitchell H. Immunoreactive proteins of Campylobacter concisus, an emergent intestinal pathogen. FEMS Immunol Med Microbiol. 2011;63:387–396. doi: 10.1111/j.1574-695X.2011.00864.x. [DOI] [PubMed] [Google Scholar]
- 138.Sørensen NB, Nielsen HL, Varming K, Nielsen H. Neutrophil activation by Campylobacter concisus. Gut Pathog. 2013;5:17. doi: 10.1186/1757-4749-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jess T, Simonsen J, Nielsen NM, Jørgensen KT, Bager P, Ethelberg S, Frisch M. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut. 2011;60:318–324. doi: 10.1136/gut.2010.223396. [DOI] [PubMed] [Google Scholar]
- 140.García Rodríguez LA, Ruigómez A, Panés J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology. 2006;130:1588–1594. doi: 10.1053/j.gastro.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 141.Ternhag A, Törner A, Svensson A, Ekdahl K, Giesecke J. Short- and long-term effects of bacterial gastrointestinal infections. Emerg Infect Dis. 2008;14:143–148. doi: 10.3201/eid1401.070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.O’Hara JR, Feener TD, Fischer CD, Buret AG. Campylobacter jejuni disrupts protective Toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect Immun. 2012;80:1563–1571. doi: 10.1128/IAI.06066-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zheng J, Meng J, Zhao S, Singh R, Song W. Adherence to and invasion of human intestinal epithelial cells by Campylobacter jejuni and Campylobacter coli isolates from retail meat products. J Food Prot. 2006;69:768–774. doi: 10.4315/0362-028x-69.4.768. [DOI] [PubMed] [Google Scholar]
- 144.Kalischuk LD, Inglis GD, Buret AG. Campylobacter jejuni induces transcellular translocation of commensal bacteria via lipid rafts. Gut Pathog. 2009;1:2. doi: 10.1186/1757-4749-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kaakoush NO, Deshpande NP, Wilkins MR, Tan CG, Burgos-Portugal JA, Raftery MJ, Day AS, Lemberg DA, Mitchell H. The pathogenic potential of Campylobacter concisus strains associated with chronic intestinal diseases. PLoS One. 2011;6:e29045. doi: 10.1371/journal.pone.0029045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Man SM, Kaakoush NO, Leach ST, Nahidi L, Lu HK, Norman J, Day AS, Zhang L, Mitchell HM. Host attachment, invasion, and stimulation of proinflammatory cytokines by Campylobacter concisus and other non-Campylobacter jejuni Campylobacter species. J Infect Dis. 2010;202:1855–1865. doi: 10.1086/657316. [DOI] [PubMed] [Google Scholar]
- 147.Burgos-Portugal JA, Kaakoush NO, Raftery MJ, Mitchell HM. Pathogenic potential of Campylobacter ureolyticus. Infect Immun. 2012;80:883–890. doi: 10.1128/IAI.06031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kaakoush NO, Man SM, Lamb S, Raftery MJ, Wilkins MR, Kovach Z, Mitchell H. The secretome of Campylobacter concisus. FEBS J. 2010;277:1606–1617. doi: 10.1111/j.1742-4658.2010.07587.x. [DOI] [PubMed] [Google Scholar]
- 149.Engberg J, Bang DD, Aabenhus R, Aarestrup FM, Fussing V, Gerner-Smidt P. Campylobacter concisus: an evaluation of certain phenotypic and genotypic characteristics. Clin Microbiol Infect. 2005;11:288–295. doi: 10.1111/j.1469-0691.2005.01111.x. [DOI] [PubMed] [Google Scholar]
- 150.Istivan TS, Smith SC, Fry BN, Coloe PJ. Characterization of Campylobacter concisus hemolysins. FEMS Immunol Med Microbiol. 2008;54:224–235. doi: 10.1111/j.1574-695X.2008.00467.x. [DOI] [PubMed] [Google Scholar]
- 151.Meconi S, Vercellone A, Levillain F, Payré B, Al Saati T, Capilla F, Desreumaux P, Darfeuille-Michaud A, Altare F. Adherent-invasive Escherichia coli isolated from Crohn’s disease patients induce granulomas in vitro. Cell Microbiol. 2007;9:1252–1261. doi: 10.1111/j.1462-5822.2006.00868.x. [DOI] [PubMed] [Google Scholar]
- 152.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun. 2006;74:4778–4792. doi: 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun. 2006;74:4778–4792. doi: 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 155.Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15:872–882. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 156.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 157.Papp M, Altorjay I, Norman GL, Shums Z, Palatka K, Vitalis Z, Foldi I, Lakos G, Tumpek J, Udvardy ML, et al. Seroreactivity to microbial components in Crohn’s disease is associated with ileal involvement, noninflammatory disease behavior and NOD2/CARD15 genotype, but not with risk for surgery in a Hungarian cohort of IBD patients. Inflamm Bowel Dis. 2007;13:984–992. doi: 10.1002/ibd.20146. [DOI] [PubMed] [Google Scholar]
- 158.Ryan P, Kelly RG, Lee G, Collins JK, O’Sullivan GC, O’Connell J, Shanahan F. Bacterial DNA within granulomas of patients with Crohn’s disease--detection by laser capture microdissection and PCR. Am J Gastroenterol. 2004;99:1539–1543. doi: 10.1111/j.1572-0241.2004.40103.x. [DOI] [PubMed] [Google Scholar]
- 159.Conte MP, Schippa S, Zamboni I, Penta M, Chiarini F, Seganti L, Osborn J, Falconieri P, Borrelli O, Cucchiara S. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut. 2006;55:1760–1767. doi: 10.1136/gut.2005.078824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kotlowski R, Bernstein CN, Sepehri S, Krause DO. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut. 2007;56:669–675. doi: 10.1136/gut.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Negroni A, Costanzo M, Vitali R, Superti F, Bertuccini L, Tinari A, Minelli F, Di Nardo G, Nuti F, Pierdomenico M, et al. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:913–924. doi: 10.1002/ibd.21899. [DOI] [PubMed] [Google Scholar]
- 162.Mow WS, Vasiliauskas EA, Lin YC, Fleshner PR, Papadakis KA, Taylor KD, Landers CJ, Abreu-Martin MT, Rotter JI, Yang H, et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology. 2004;126:414–424. doi: 10.1053/j.gastro.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 163.Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007;117:1566–1574. doi: 10.1172/JCI30504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol Microbiol. 2003;48:781–794. doi: 10.1046/j.1365-2958.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 165.Chassaing B, Rolhion N, de Vallée A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Söderholm JD, Hugot JP, Colombel JF, et al. Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest. 2011;121:966–975. doi: 10.1172/JCI44632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bringer MA, Billard E, Glasser AL, Colombel JF, Darfeuille-Michaud A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-α secretion. Lab Invest. 2012;92:411–419. doi: 10.1038/labinvest.2011.156. [DOI] [PubMed] [Google Scholar]
- 167.Minami M, Ando T, Okamoto A, Sasaki N, Ohkura T, Torii K, Hasegawa T, Ohta M, Goto H. Seroprevalence of Fusobacterium varium in ulcerative colitis patients in Japan. FEMS Immunol Med Microbiol. 2009;56:67–72. doi: 10.1111/j.1574-695X.2009.00550.x. [DOI] [PubMed] [Google Scholar]
- 168.Ohkusa T, Okayasu I, Ogihara T, Morita K, Ogawa M, Sato N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut. 2003;52:79–83. doi: 10.1136/gut.52.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rashid T, Wilson C, Ebringer A. The link between ankylosing spondylitis, Crohn’s disease, Klebsiella, and starch consumption. Clin Dev Immunol. 2013;2013:872632. doi: 10.1155/2013/872632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee IA, Kim DH. Klebsiella pneumoniae increases the risk of inflammation and colitis in a murine model of intestinal bowel disease. Scand J Gastroenterol. 2011;46:684–693. doi: 10.3109/00365521.2011.560678. [DOI] [PubMed] [Google Scholar]
- 171.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Keestra AM, Winter MG, Klein-Douwel D, Xavier MN, Winter SE, Kim A, Tsolis RM, Bäumler AJ. A Salmonella virulence factor activates the NOD1/NOD2 signaling pathway. MBio. 2011;2:pii: e00266–11. doi: 10.1128/mBio.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hugot JP, Alberti C, Berrebi D, Bingen E, Cézard JP. Crohn’s disease: the cold chain hypothesis. Lancet. 2003;362:2012–2015. doi: 10.1016/S0140-6736(03)15024-6. [DOI] [PubMed] [Google Scholar]
- 174.Saebo A, Vik E, Lange OJ, Matuszkiewicz L. Inflammatory bowel disease associated with Yersinia enterocolitica O: 3 infection. Eur J Intern Med. 2005;16:176–182. doi: 10.1016/j.ejim.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 175.Leu SB, Shulman SC, Steelman CK, Lamps LW, Bulut OP, Abramowsky CR, Gold BD, Szlam S, Stockwell C, Havens J, et al. Pathogenic yersinia DNA in intestinal specimens of pediatric patients with Crohn’s disease. Fetal Pediatr Pathol. 2013;32:367–370. doi: 10.3109/15513815.2013.768744. [DOI] [PubMed] [Google Scholar]
- 176.Knösel T, Schewe C, Petersen N, Dietel M, Petersen I. Prevalence of infectious pathogens in Crohn’s disease. Pathol Res Pract. 2009;205:223–230. doi: 10.1016/j.prp.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 177.Tamboli CP, Good MR, Reynolds EM, Sharma P, Mitros FA. Anti-Yersinia antibodies are not associated with microscopic colitis in an American case-control study. Scand J Gastroenterol. 2011;46:1442–1448. doi: 10.3109/00365521.2011.627450. [DOI] [PubMed] [Google Scholar]
- 178.Veerappan GR, Betteridge J, Young PE. Probiotics for the treatment of inflammatory bowel disease. Curr Gastroenterol Rep. 2012;14:324–333. doi: 10.1007/s11894-012-0265-5. [DOI] [PubMed] [Google Scholar]
- 179.Sengupta R, Altermann E, Anderson RC, McNabb WC, Moughan PJ, Roy NC. The role of cell surface architecture of lactobacilli in host-microbe interactions in the gastrointestinal tract. Mediators Inflamm. 2013;2013:237921. doi: 10.1155/2013/237921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Greer RL, Morgun A, Shulzhenko N. Bridging immunity and lipid metabolism by gut microbiota. J Allergy Clin Immunol. 2013;132:253–262; quiz 263. doi: 10.1016/j.jaci.2013.06.025. [DOI] [PubMed] [Google Scholar]
- 181.Mann ER, Landy JD, Bernardo D, Peake ST, Hart AL, Al-Hassi HO, Knight SC. Intestinal dendritic cells: their role in intestinal inflammation, manipulation by the gut microbiota and differences between mice and men. Immunol Lett. 2013;150:30–40. doi: 10.1016/j.imlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 182.Ng SC, Benjamin JL, McCarthy NE, Hedin CR, Koutsoumpas A, Plamondon S, Price CL, Hart AL, Kamm MA, Forbes A, et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in Crohn’s disease. Inflamm Bowel Dis. 2011;17:2027–2037. doi: 10.1002/ibd.21590. [DOI] [PubMed] [Google Scholar]
- 183.Hardy H, Harris J, Lyon E, Beal J, Foey AD. Probiotics, prebiotics and immunomodulation of gut mucosal defences: homeostasis and immunopathology. Nutrients. 2013;5:1869–1912. doi: 10.3390/nu5061869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Marteau P, Lémann M, Seksik P, Laharie D, Colombel JF, Bouhnik Y, Cadiot G, Soulé JC, Bourreille A, Metman E, et al. Ineffectiveness of Lactobacillus johnsonii LA1 for prophylaxis of postoperative recurrence in Crohn’s disease: a randomised, double blind, placebo controlled GETAID trial. Gut. 2006;55:842–847. doi: 10.1136/gut.2005.076604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lorea Baroja M, Kirjavainen PV, Hekmat S, Reid G. Anti-inflammatory effects of probiotic yogurt in inflammatory bowel disease patients. Clin Exp Immunol. 2007;149:470–479. doi: 10.1111/j.1365-2249.2007.03434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kato K, Mizuno S, Umesaki Y, Ishii Y, Sugitani M, Imaoka A, Otsuka M, Hasunuma O, Kurihara R, Iwasaki A, et al. Randomized placebo-controlled trial assessing the effect of bifidobacteria-fermented milk on active ulcerative colitis. Aliment Pharmacol Ther. 2004;20:1133–1141. doi: 10.1111/j.1365-2036.2004.02268.x. [DOI] [PubMed] [Google Scholar]
- 187.Suzuki A, Mitsuyama K, Koga H, Tomiyasu N, Masuda J, Takaki K, Tsuruta O, Toyonaga A, Sata M. Bifidogenic growth stimulator for the treatment of active ulcerative colitis: a pilot study. Nutrition. 2006;22:76–81. doi: 10.1016/j.nut.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 188.Philippe D, Heupel E, Blum-Sperisen S, Riedel CU. Treatment with Bifidobacterium bifidum 17 partially protects mice from Th1-driven inflammation in a chemically induced model of colitis. Int J Food Microbiol. 2011;149:45–49. doi: 10.1016/j.ijfoodmicro.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 189.Kruis W, Fric P, Pokrotnieks J, Lukás M, Fixa B, Kascák M, Kamm MA, Weismueller J, Beglinger C, Stolte M, et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004;53:1617–1623. doi: 10.1136/gut.2003.037747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bibiloni R, Fedorak RN, Tannock GW, Madsen KL, Gionchetti P, Campieri M, De Simone C, Sartor RB. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am J Gastroenterol. 2005;100:1539–1546. doi: 10.1111/j.1572-0241.2005.41794.x. [DOI] [PubMed] [Google Scholar]
- 191.Miele E, Pascarella F, Giannetti E, Quaglietta L, Baldassano RN, Staiano A. Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am J Gastroenterol. 2009;104:437–443. doi: 10.1038/ajg.2008.118. [DOI] [PubMed] [Google Scholar]
- 192.Ewaschuk JB, Dieleman LA. Probiotics and prebiotics in chronic inflammatory bowel diseases. World J Gastroenterol. 2006;12:5941–5950. doi: 10.3748/wjg.v12.i37.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ewaschuk JB, Tejpar QZ, Soo I, Madsen K, Fedorak RN. The role of antibiotic and probiotic therapies in current and future management of inflammatory bowel disease. Curr Gastroenterol Rep. 2006;8:486–498. doi: 10.1007/s11894-006-0039-z. [DOI] [PubMed] [Google Scholar]
- 194.Mimura T, Rizzello F, Helwig U, Poggioli G, Schreiber S, Talbot IC, Nicholls RJ, Gionchetti P, Campieri M, Kamm MA. Once daily high dose probiotic therapy (VSL#3) for maintaining remission in recurrent or refractory pouchitis. Gut. 2004;53:108–114. doi: 10.1136/gut.53.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gionchetti P, Rizzello F, Helwig U, Venturi A, Lammers KM, Brigidi P, Vitali B, Poggioli G, Miglioli M, Campieri M. Prophylaxis of pouchitis onset with probiotic therapy: a double-blind, placebo-controlled trial. Gastroenterology. 2003;124:1202–1209. doi: 10.1016/s0016-5085(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 196.Schultz M. Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1012–1018. doi: 10.1002/ibd.20377. [DOI] [PubMed] [Google Scholar]
- 197.Mallon P, McKay D, Kirk S, Gardiner K. Probiotics for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2007;(4):CD005573. doi: 10.1002/14651858.CD005573.pub2. [DOI] [PubMed] [Google Scholar]
- 198.Butterworth AD, Thomas AG, Akobeng AK. Probiotics for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2008;(3):CD006634. doi: 10.1002/14651858.CD006634.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rolfe VE, Fortun PJ, Hawkey CJ, Bath-Hextall F. Probiotics for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2006;(4):CD004826. doi: 10.1002/14651858.CD004826.pub2. [DOI] [PubMed] [Google Scholar]
- 200.Langlands SJ, Hopkins MJ, Coleman N, Cummings JH. Prebiotic carbohydrates modify the mucosa associated microflora of the human large bowel. Gut. 2004;53:1610–1616. doi: 10.1136/gut.2003.037580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Roberfroid M. Prebiotics: the concept revisited. J Nutr. 2007;137:830S–837S. doi: 10.1093/jn/137.3.830S. [DOI] [PubMed] [Google Scholar]
- 202.Casellas F, Borruel N, Torrejón A, Varela E, Antolin M, Guarner F, Malagelada JR. Oral oligofructose-enriched inulin supplementation in acute ulcerative colitis is well tolerated and associated with lowered faecal calprotectin. Aliment Pharmacol Ther. 2007;25:1061–1067. doi: 10.1111/j.1365-2036.2007.03288.x. [DOI] [PubMed] [Google Scholar]
- 203.Welters CF, Heineman E, Thunnissen FB, van den Bogaard AE, Soeters PB, Baeten CG. Effect of dietary inulin supplementation on inflammation of pouch mucosa in patients with an ileal pouch-anal anastomosis. Dis Colon Rectum. 2002;45:621–627. doi: 10.1007/s10350-004-6257-2. [DOI] [PubMed] [Google Scholar]
- 204.Koleva PT, Valcheva RS, Sun X, Gänzle MG, Dieleman LA. Inulin and fructo-oligosaccharides have divergent effects on colitis and commensal microbiota in HLA-B27 transgenic rats. Br J Nutr. 2012;108:1633–1643. doi: 10.1017/S0007114511007203. [DOI] [PubMed] [Google Scholar]
- 205.Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL, Kamm MA, Sanderson JD, Knight SC, Forbes A, et al. Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn’s disease. Gut. 2011;60:923–929. doi: 10.1136/gut.2010.232025. [DOI] [PubMed] [Google Scholar]
- 206.Lindsay JO, Whelan K, Stagg AJ, Gobin P, Al-Hassi HO, Rayment N, Kamm MA, Knight SC, Forbes A. Clinical, microbiological, and immunological effects of fructo-oligosaccharide in patients with Crohn’s disease. Gut. 2006;55:348–355. doi: 10.1136/gut.2005.074971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Damaskos D, Kolios G. Probiotics and prebiotics in inflammatory bowel disease: microflora ‘on the scope’. Br J Clin Pharmacol. 2008;65:453–467. doi: 10.1111/j.1365-2125.2008.03096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hedin C, Whelan K, Lindsay JO. Evidence for the use of probiotics and prebiotics in inflammatory bowel disease: a review of clinical trials. Proc Nutr Soc. 2007;66:307–315. doi: 10.1017/S0029665107005563. [DOI] [PubMed] [Google Scholar]
- 209.Viladomiu M, Hontecillas R, Yuan L, Lu P, Bassaganya-Riera J. Nutritional protective mechanisms against gut inflammation. J Nutr Biochem. 2013;24:929–939. doi: 10.1016/j.jnutbio.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kanauchi O, Suga T, Tochihara M, Hibi T, Naganuma M, Homma T, Asakura H, Nakano H, Takahama K, Fujiyama Y, et al. Treatment of ulcerative colitis by feeding with germinated barley foodstuff: first report of a multicenter open control trial. J Gastroenterol. 2002;37 Suppl 14:67–72. doi: 10.1007/BF03326417. [DOI] [PubMed] [Google Scholar]
- 211.Bamba T, Kanauchi O, Andoh A, Fujiyama Y. A new prebiotic from germinated barley for nutraceutical treatment of ulcerative colitis. J Gastroenterol Hepatol. 2002;17:818–824. doi: 10.1046/j.1440-1746.2002.02709.x. [DOI] [PubMed] [Google Scholar]
- 212.Hallert C, Kaldma M, Petersson BG. Ispaghula husk may relieve gastrointestinal symptoms in ulcerative colitis in remission. Scand J Gastroenterol. 1991;26:747–750. doi: 10.3109/00365529108998594. [DOI] [PubMed] [Google Scholar]
- 213.Wang SL, Wang ZR, Yang CQ. Meta-analysis of broad-spectrum antibiotic therapy in patients with active inflammatory bowel disease. Exp Ther Med. 2012;4:1051–1056. doi: 10.3892/etm.2012.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, Talley NJ, Moayyedi P. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–673. doi: 10.1038/ajg.2011.72. [DOI] [PubMed] [Google Scholar]
- 215.Shen B. Acute and chronic pouchitis--pathogenesis, diagnosis and treatment. Nat Rev Gastroenterol Hepatol. 2012;9:323–333. doi: 10.1038/nrgastro.2012.58. [DOI] [PubMed] [Google Scholar]
- 216.Kale-Pradhan PB, Zhao JJ, Palmer JR, Wilhelm SM. The role of antimicrobials in Crohn’s disease. Expert Rev Gastroenterol Hepatol. 2013;7:281–288. doi: 10.1586/egh.13.6. [DOI] [PubMed] [Google Scholar]
- 217.Travis SP, Stange EF, Lémann M, Oresland T, Chowers Y, Forbes A, D’Haens G, Kitis G, Cortot A, Prantera C, et al. European evidence based consensus on the diagnosis and management of Crohn’s disease: current management. Gut. 2006;55 Suppl 1:i16–i35. doi: 10.1136/gut.2005.081950b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bakken JS, Borody T, Brandt LJ, Brill JV, Demarco DC, Franzos MA, Kelly C, Khoruts A, Louie T, Martinelli LP, et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol. 2011;9:1044–1049. doi: 10.1016/j.cgh.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhang F, Luo W, Shi Y, Fan Z, Ji G. Should we standardize the 1,700-year-old fecal microbiota transplantation? Am J Gastroenterol. 2012;107:1755; author reply p.1755-p.1756. doi: 10.1038/ajg.2012.251. [DOI] [PubMed] [Google Scholar]
- 220.van Nood E, Dijkgraaf MG, Keller JJ. Duodenal infusion of feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:2145. doi: 10.1056/NEJMc1303919. [DOI] [PubMed] [Google Scholar]
- 221.Kunde S, Pham A, Bonczyk S, Crumb T, Duba M, Conrad H, Cloney D, Kugathasan S. Safety, tolerability, and clinical response after fecal transplantation in children and young adults with ulcerative colitis. J Pediatr Gastroenterol Nutr. 2013;56:597–601. doi: 10.1097/MPG.0b013e318292fa0d. [DOI] [PubMed] [Google Scholar]
- 222.Borody TJ, Warren EF, Leis S, Surace R, Ashman O. Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol. 2003;37:42–47. doi: 10.1097/00004836-200307000-00012. [DOI] [PubMed] [Google Scholar]
- 223.Kump PK, Gröchenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM, Deutschmann A, Wenzl HH, Petritsch W, Krejs GJ, et al. Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis. 2013;19:2155–2165. doi: 10.1097/MIB.0b013e31829ea325. [DOI] [PubMed] [Google Scholar]
- 224.Vrieze A, de Groot PF, Kootte RS, Knaapen M, van Nood E, Nieuwdorp M. Fecal transplant: a safe and sustainable clinical therapy for restoring intestinal microbial balance in human disease? Best Pract Res Clin Gastroenterol. 2013;27:127–137. doi: 10.1016/j.bpg.2013.03.003. [DOI] [PubMed] [Google Scholar]