

Abstract
Chronic abdominal pain accompanying intestinal inflammation emerges from the hyperresponsiveness of neuronal, immune and endocrine signaling pathways within the intestines, the peripheral and the central nervous system. In this article we review how the sensory nerve information from the healthy and the hypersensitive bowel is encoded and conveyed to the brain. The gut milieu is continuously monitored by intrinsic enteric afferents, and an extrinsic nervous network comprising vagal, pelvic and splanchnic afferents. The extrinsic afferents convey gut stimuli to second order neurons within the superficial spinal cord layers. These neurons cross the white commissure and ascend in the anterolateral quadrant and in the ipsilateral dorsal column of the dorsal horn to higher brain centers, mostly subserving regulatory functions. Within the supraspinal regions and the brainstem, pathways descend to modulate the sensory input. Because of this multiple level control, only a small proportion of gut signals actually reaches the level of consciousness to induce sensation or pain. In inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS) patients, however, long-term neuroplastic changes have occurred in the brain-gut axis which results in chronic abdominal pain. This sensitization may be driven on the one hand by peripheral mechanisms within the intestinal wall which encompasses an interplay between immunocytes, enterochromaffin cells, resident macrophages, neurons and smooth muscles. On the other hand, neuronal synaptic changes along with increased neurotransmitter release in the spinal cord and brain leads to a state of central wind-up. Also life factors such as but not limited to inflammation and stress contribute to hypersensitivity. All together, the degree to which each of these mechanisms contribute to hypersensitivity in IBD and IBS might be disease- and even patient-dependent. Mapping of sensitization throughout animal and human studies may significantly improve our understanding of sensitization in IBD and IBS. On the long run, this knowledge can be put forward in potential therapeutic targets for abdominal pain in these conditions.
Keywords: Afferent nerves, Chronic pain, Inflammatory bowel disease, Irritable bowel syndrome, Sensitisation, Sensory nerves, Visceral hypersensitivity
Core tip: This review reports on the neuroanatomy of gastrointestinal pain disorders. The encoding and conveying of information from the gastrointestinal wall to the central nervous system is described with emphasis on the peripheral level, the spinal cord and the higher brain centers. Besides the basic principles of visceral hypersensitivity are reviewed taking into consideration peripheral sensitization (and the main mediators involved) and central wind-up phenomena in order to better understand the mechanisms involved in chronic abdominal pain in inflammatory bowel disease and irritable bowel syndrome patients.
INTRODUCTION
Chronic abdominal pain frequently originates from a local inflammatory reaction in the lower gastrointestinal (GI) tract. Indeed, 50%-70% of patients with inflammatory bowel disease (IBD), encompassing ulcerative colitis (UC) and Crohn’s disease (CD), present with abdominal discomfort or pain[1,2]. The bulk of IBD research merely focuses on treatment algorithms for the achievement and the maintenance of endoscopic remission of mucosal lesions but this approach insufficiently resolves concomitant pain[3,4]. A retrospective 5 year follow-up study in CD patients revealed a high prevalence of chronic narcotic users which exemplifies the analgesic shortcoming[5]. Farrokhyar and colleagues reported symptoms compliant to Rome II diagnostic criteria for irritable bowel syndrome (IBS) in approximately one-third of patients with inactive CD[4,6]. IBS entails a heterogeneous group of functional lower GI disorders characterized by abdominal pain and altered bowel habits[7]. While routine diagnostic tests exclude alarming features for organic pathology, some though not all investigators discovered subclinical evidence of a low-grade ileitis and colitis in several IBS patients such as mildly elevated levels of fecal calprotectin[8,9], an increase in proinflammatory cytokines including IL-6, IL-8, tumor necrosis factor (TNF)-α and IL-1β[10], colonic lymphocytosis and mastocytosis[11]. Aside from concerns of some regarding these findings[12,13], observations linking symptoms to a bout of enteritis hypothesize that a previous transient inflammation contributes to IBS etiology[14]. IBD on the other hand manifests as a chronic uncontrolled immunologic reaction with recurrent flares of inflammation[15]. Hence, there are indications supporting and opposing overlap between IBD and IBS. A more acknowledged statement, regardless of the inflammatory origin, is that the pathology has imprinted changes in the gut-brain neuronal connection and so manifests in a diffuse and poorly localized chronic pain in the abdomen. The aforementioned features characterize pain in IBS and IBD patients and are partially attributed to the complex sensory innervation pattern of the pelvis. The colorectum is extensively innervated, albeit only a low density of extrinsic afferents provides the substantial link with the central nervous system (CNS) for perception. The extrinsic colon afferents branch to other organs (e.g., colorectum, bladder, reproductive organs) within the abdominal cavity and organize into web-like plexuses scattered through the abdomen. Due to this anatomical organization signals of multiple pelvic organs, rather than restricted to the colon, converge to a relatively extensive number of spinal cord segments[16]. Within the spine other body signals may join and together the information travels via supraspinal levels to the higher brain centers. Importantly it is known that somatic input outnumbers the visceral input and that the visceral input is entering the spinal cord at different segments. This ambiguous signal transduction from the gut via the three-neuron chain to the brain explains why the experienced pain has a poor topological relation and cannot always be pinpointed. In IBD and IBS, each level of control is susceptible to neuromodulatory changes or sensitization. Therefore, we review how stimuli in the healthy and the hypersensitive bowel are detected, encoded, and conveyed to the brain to be either unconsciously, consciously or painfully perceived. For reasons of clarity, we will start with a description of the anatomical structures involved.
PERIPHERAL AFFERENT PATHWAYS SUPPLYING THE GASTROINTESTINAL TRACT
Intrinsic innervation: the enteric nervous system
The intestinal wall of the esophagus, the stomach, the small intestine and the colon house a dense neuronal network (about 108 neurons), the enteric nervous system (ENS), also referred to as the “little brain-of-the-gut’’. This intrinsic network comprises enteric nerve cell bodies of sensory, inter- and motor neurons grouped into ganglia and interconnected by bundles of nerve processes forming plexuses of which the best characterised are the myenteric plexus (Auerbach’s plexus) and the submucosal plexus (Meissner’s plexus)[17-19]. The ENS controls motility, mucosal secretion and absorption, mucosal growth, local blood flow and the immune function in the gut[18]. The connective link between the CNS and the ENS is bidirectional: the brain influences the function of the ENS and vice versa. When the brain encounters stressful life events, the lower gut gets overstimulated resulting in diarrhea. When the lower gut responds to food poisoning with powerful propulsive colon contractions, the body experiences aversion towards the ingested meal and abdominal cramping pain. With referral to the latter, high-amplitude propagating contractions in the ileocecum and sigmoid colon of IBS patients in response to eating correlate to abdominal pain[20]. Support to this hypothesis comes from reports on antispasmodics giving short-term pain relief in at least a subset of diarrhea-predominant IBS patients[21]. Likewise, antispasmodic agents may be effective in IBD, especially in those patients who are in remission and have mild to moderate chronic pain[22]. Besides their role in ileocolonic dysmotility, intrinsic enteric afferents containing serotonin, substance P, CGRP can initiate or intensify neurogenic inflammation upon release and thereby sensitise adjacent extrinsic gut nerves. The relevance of the enteric nervous system to pain primarily lies within the excitation of these extrinsic afferents by neuropeptides.
Extrinsic sensory innervation of the gastrointestinal tract
The extrinsic primary afferents of the GI tract provide the anatomical connection with the CNS and so a basis for both nonpainful (e.g., satiety, passage of gas, etc.) and painful (e.g., inflammation, ischemia, extensive distension) gut sensations. The GI tract receives a dual innervation with complementary roles in gut signaling: a splanchnic and a vagal plus pelvic afferent population. These afferents run alongside the efferent orthosympathetic (splanchnic nerves) and parasympathetic nervous system (vagal/pelvic nerves) respectively, but are never referred to as such[23]. It is assumed that the vagal/pelvic nerves subserve homeostatic functions, whereas the splanchnic innervation principally conveys nociception. This simple dichotomy of function, however, appears far more complex than formerly assumed.
Vagal innervation: The vagal nerve is the largest sensory pathway in the body with up to 80% of the fibers being afferents. The vagal nerve branches to the entire gut, except the transverse and distal portion of the colon. The vagal cell bodies reside in the ganglion nodosum and the central nerve endings terminate in the nucleus of the solitary tract in the dorsal medulla. Vagal afferents mainly regulate feeding behavior by upper gut reflexes (e.g., gastric accommodation, gastric emptying, gastric/pancreatic secretion, emesis) and the perception of hunger, fullness, satisfaction, bloating and nausea. Three types of vagal fibers were characterized in the mouse by an in vitro vagus-gastro-esophageal set-up: mechanoreceptors, tension receptors and specific chemoreceptors activated by bile. Vagal mechanical afferent endings within the muscle layers are classified into two types: intramuscular arrays and intraganglionic laminar endings. Intramuscular arrays run parallel in either the longitudinal or circular muscle layers and have been suggested to respond to muscle stretch. Intraganglionic laminar endings branch extensively in connective tissue surrounding myenteric ganglia and convey info about distension and muscle contraction[24]. The tension-sensitive afferents respond maximally at distensions within the physiological range and are activated by normal peristaltic contractions. This implies that the vagal afferents take care of physiological perception of mechanical stimuli[25,26], whereas pain evoked by distension of the upper GI tract is probably mediated via the splanchnic afferents. However, vagal afferents have been shown to be implicated in the pain reactions evoked by gastric acid challenge[27]. The activity of lower gut vagal afferents modulates spinal transmission. The latter is supported by the observation of increased pain responses to colorectal distension after subdiaphragmatic vagotomy in rats[28,29]. In humans, lower thresholds for the perception of pain have been described in patients who had previously undergone vagotomy in the course of a Billroth I gastrectomy compared with pain thresholds in healthy controls[30]. The exact mechanism is still controversial, but likely involves specific relay nuclei such as the nucleus raphe magnus and ventral locus coeruleus. On the one hand vagal afferents have been shown to facilitate nociceptive transmission[25], whereas on the other hand the vagal nerve appears to participate in an antinociceptive descending pathway mediated by nanomolecules such as but not limited to opioids[28,31]. This discrepancy may be explained by differences in stimulation parameters: low intensity stimulation of vagal afferents facilitates, while high intensity stimulation inhibits nociception[31]. IBD patients may benefit from chronic vagal stimulation since the vagal nerve stimulation exerts anti-inflammatory effects. Previous studies in a sepsis model showed that the vagal nerve regulates the cholinergic tonus so that the immune response of macrophages and immunocytes is dampened[32].
Pelvic innervation: The pelvic nerves mainly innervate pelvic structures: the colorectum, the bladder and the reproductive organs. Pelvic afferents represent 30%-50% of the total number of neurons and converge visceral information onto spinal neurons in the lumbosacral segments L6-S2 of the spinal cord in mice and rats[33]. Their cell bodies are located in the lumbosacral dorsal root ganglia (DRG). The pelvic nerve contains serosal, mucosal, muscular (e.g., intraganglionic laminar ending and intramuscular arrays), muscular/mucosal afferents and is specialized to detect circular stretch, the primary stimulus generated by low-intensity colorectal distension or stool passage[34]. Pelvic afferents transmit similar modalities of information as the vagal afferent system i.e., physiological sensation (e.g., urgency, desire to defecate, etc.). In addition, animal data support the idea that they form the afferent branch of extrinsic gut reflexes such as the cologastric inhibitory reflex[35,36]. They are a subject of interest in pain research as a bilateral pelvic nerve section almost entirely abolished pain-related behavior to noxious colorectal distension in rats[23,34]. Following TNBS colitis, however, pain responses partially recovered[37]. Hence, the pelvic nerve is involved in normal physiology and acute pain, rather than in inflammatory pain which is more specifically mediated by splanchnic afferents. However, animal experiments in rat have shown pelvic fiber sensitization when a chemical irritant is applied to colonic tissue, posing a role in nociception of the pelvic nerve under inflammatory circumstances[36].
Splanchnic innervations: The splanchnic nerves innervate the entire GI tract and are the functional counterpart of the vagal plus pelvic nerves. Visceral afferents located in splanchnic nerves project to the spinal cord. Unlike the pelvic afferents, visceral information from the colorectum carried by the splanchnic nerves project onto thoracolumbar segments T10 - L2 in mice and rats[33]. Their cell bodies are located in the thoracolumbar DRG near the spinal cord, with peripheral projections ending at various levels within the gut wall. These splanchnic afferent fibers course through the prevertebral ganglia (celiac, superior and inferior mesenteric ganglion) where they may form ‘‘en passant’’ synapses with efferent sympathic neurons. From animal data splanchnic afferents are thought to constitute the main nociceptive pathway from the gut as they signal different modalities of mechanosensory information[34,38]. In the upper GI tract splanchnic afferents appear to mediate gastric mechano-nociception, but not gastric chemo-nociception which is mediated by vagal sensory neurons. This is supported by the finding that in rodents pain-related responses to gastric distension are blocked by splanchnicectomy, but not by vagotomy[39]. In the lower GI tract, splanchnic afferents convey signals of abdominal discomfort. Indeed, the majority of splanchnic afferents of the colon encountered in mice are located in the serosa (36%) and mesenteric (50%) membranes, often associated with mesenteric blood vessels. Likewise, the mucosal spinal afferents are often found near submucosal blood vessels where they form varicose branching axons. The bare endings of the submucosa and mesenteric afferents are likely to respond to distortion of the gut during stretch or contraction, especially at levels that give rise to pain[40,41]. Hence, they are tuned to encode stimuli into the noxious range[34]. In the rat, both anatomical and functional evidence points to a specific role of splanchnic afferents in pain during colitis[37,42].
Very recently Brookes and co-authors suggest a novel classification into five “structurally distinct” types of sensory endings within the gut wall forming the anatomical extrinsic sensory pathways described by these authors as the vagal pathway, the thoracolumbar spinal pathway projecting via the splanchnic nerves and the lumbosacral spinal pathway projecting via the pelvic and rectal nerves[23].
CENTRAL PATHWAYS CONVEYING VISCERAL SIGNALS FROM THE LOWER GI TRACT
Extrinsic primary afferents innervating the lower GI tract primarily synapse with second-order neurons in the dorsal horn of the thoracolumbar and lumbosacral spinal cord segment. Fibers terminate predominantly in the superficial laminae I and II, but also reach deeper layers such as the laminae V and X of the gray matter. Ascending pathways project to higher brain centers where pathways origin and descend to fine-tune the sensory input[43,44].
Ascending pathways
Central ascending pathways involved in bowel sensations include both pathways ascending in the anterolateral quadrant (ALQ) of the white commissure and the dorsal column of the dorsal horn (Figure 1). Pathways ascending in the ALQ transmit noxious cutaneous stimuli and also carry nociceptive information of visceral origin. This idea is largely based on anterolateral cordotomies performed in the 20th century to relieve pain due to damage to the spinal cord by disease or trauma[45]. The pathways in the ALQ are the spinoreticular, spinomesencephalic, spinohypothalamic and spinothalamic tracts[46]. The former three tracts mainly subserve regulatory functions below the level of consciousness. The spinoreticular tract projects to the dorsal reticular nucleus in the brainstem, which is involved in the affective-motivational properties (emotional component of pain) of visceral stimulation. The spinomesencephalic tract conveys information from the spinal cord to the periaqueductal gray (PAG) and other midbrain regions. The spinohypothalamic tract conducts sensory information from the spinal cord directly to the hypothalamus. The hypothalamus together with other parts of the limbic system (amygdala, medial thalamus, ACC), locus coeruleus and PAG regulate arousal and emotional, autonomic and behavioral responses. The spinothalamic tract mediates the sensations of pain, cold and heat, and also contributes to touch sensation. Projections of the spinothalamic tract have been traced to the thalamus in humans and in laboratory animals. The thalamus is a major relay station where multiple somatic and visceral inputs converge. Before the information is conveyed via the third order neurons to the cortex, the thalamus will process the nociceptive information. Human observations coupled with an extensive repertoire of experimental data suggest that particularly the posterolateral nucleus of the thalamus is involved in the processing of visceral information, including both innocuous and noxious visceral inputs. The thalamus, relays to cortical regions such as the pregenual anterior cingulate cortex (pACC), mid cingulate cortex, the insula and the somatosensory cortex. Notably, visceral sensation is primarily represented in the secondary somatosensory cortex[47]. In these cortical regions the nociceptive signals are processed, integrated and eventually perceived as ‘‘painful’’. Brain images provided by H(2)(15)O micro positron-emission tomography (PET) scanning performed during colorectal distension in rats suggest that the cerebellum is also involved in visceral nociception[48], which is supported by findings in healthy humans documenting cerebellar activation in response to painful visceral stimuli such as distension of the colorectum[49]. A number of recent studies has pointed to a specific role of the dorsal funiculus [dorsal column (DC) in animals] in viscerosensory transmission and visceral nociception. Experimental data from different groups have identified the DC as being more important in visceral nociceptive transmission than the spinothalamic, spinohypothalamic, spinomesencephalic and spinoreticular tracts[50,51]. The bulk of evidence rests on the great effectiveness of limited midline myelotomy in reducing intractable pelvic cancer-related pain in humans and on a number of experimental observations in animals[52]. The DC contains collateral branches of primary afferent fibers that ascend from the dorsal root entry level to the medulla. In addition, it contains the ascending axons of tract cells of the dorsal horn. These tract cells form the postsynaptic DC pathway, which along with primary afferent axons, travel in the DC and synapses in the DC nuclei. The postsynaptic DC cells in rats and monkeys were shown to receive inputs from the colon, the ureter, the pancreas and epigastric structures[53]. A DC lesion does not reduce pain caused by noxious cutaneous stimuli in humans[54], which argues for a selective role of the DC pathway in visceral pain such as pain evoked by enteritis.
Figure 1.
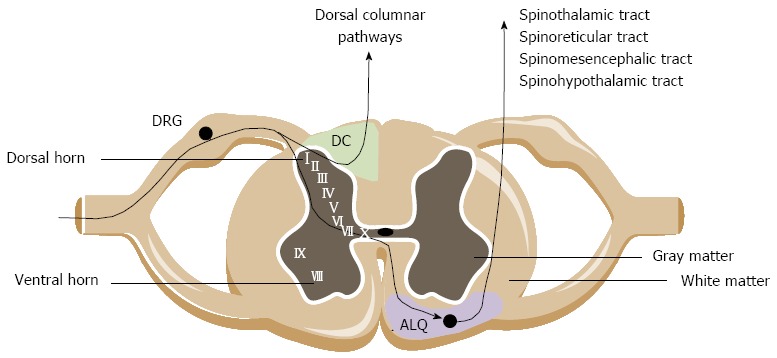
Cross section of the spinal cord. The central branches of the visceral afferents innervating the lower gastrointestinal tract travel via the dorsal root ganglia (DRG) and project onto the second order neurons in laminae I, II, V, X of the spinal gray matter. Ascending pathways arise in the anterolateral quadrant (ALQ; purple zone) and the dorsal column (DC; green zone) region in the spinal cord and project to higher brain centers (, medulla, thalamus).
Descending pathways
It is well recognized that spinal nociceptive transmission is modulated by descending pathways from various supraspinal structures, including the nucleus raphe magnus, the periventricular gray of the hypothalamus and the midbrain PAG. At cortical level, the ACC is the most important source of descending modulatory pathways, projecting to the amygdala and the PAG which is probably the key pain modulatory region. Descending modulation of spinal nociceptive processing can be either inhibitory or facilitatory. In the late 1960s it was shown that focal electrical stimulation in the midbrain PAG of the rat permitted abdominal surgery in the absence of general anesthesia due to the pain suppressive effects of stimulation of this specific region[55]. The PAG - rostral ventromedial medulla (RVM) - dorsal horn circuitry is the best characterized nociceptive modulatory pathway through which pain is endogenously inhibited. Endogenous opioids are key mediators in the descending pain inhibitory pathways. Specifically the pACC is assumed to send inhibitory signals to pontomedullary networks since it contains a high content of opioids. Additionally, monoaminergic neurotransmitters such as noradrenaline, serotonin and dopamine positively or negatively modulate pain signaling with remarkably opposing effects, depending on the extent of transmitter release, the receptor type, receptor affinity and the location in the spinal cord the descending pathways project towards[47,56,57]. Further, it is shown that the excitability of spinal dorsal horn neurons to peripheral sensory stimulation are enhanced or increased by stimulation of the RVM including the reticular formation of the serotonergic nucleus raphe magnus[58-60]. These findings support a role of the RVM and raphe magnus in a facilitatory descending pathway.
PRINCIPLES OF VISCERAL HYPERSENSITIVITY
Visceral hypersensitivity refers to an increased perception of stimuli arising from the viscera. Specific terms are used to describe the hypersensitivity: allodynia and hyperalgesia. The perception of pain in response to stimuli that are normally not perceived as painful is referred to as allodynia. The term allodynia strictly does not apply to visceral pain since the visceral organs are normally almost insensate but the concept of visceral allodynia is useful to understand sensitization in a variety of gut disorders. An increase in pain perception to stimuli that are normally perceived as painful is referred to as hyperalgesia[61]. Concerning IBD and IBS, we focus this review on colorectal hypersensitivity. A hypersensitive colorectum is considered the hallmark feature of all IBS subtypes[62,63] as altered rectal perception is documented in 61% of IBS patients meeting Rome II criteria[64]. It is currently the most widely accepted mechanism for abdominal pain. Some investigators have even suggested that this physiological hallmark is useful in clinical diagnosis[65]. Based on the current scientific evidence, the mechanisms of visceral hypersensitivity have been formulated in a number of hypotheses. These include (1) the sensitization of peripheral visceral afferent neurons; (2) the sensitization of spinal cord dorsal horn neurons; (3) the altered descending excitatory and inhibitory influences to the spinal cord nociceptive neurons; and (4) the misinterpretation of innocuous sensation as noxious due to cognitive and emotional biasing (e.g., hypervigilance, pain catastrophizing)[47,66]. The degree to which each of these mechanisms generate visceral hypersensitivity and therefore pain symptoms is still unclear. However, it is assumed that these mechanisms are rather complementary than mutually exclusive.
Peripheral sensitization
The gut is not only provided with an extensive neuronal network, it also houses highly specialized immunocytes and epithelial cells equipped with the machinery to participate in sensitization in the event of a potential threat[67]. In IBD and some IBS subsets, inflammation likely triggers the peripheral sensitization. Enterochromaffin cells (ECC) and mast cells function as intermediaries between the ‘‘inflammatory soup’’ (e.g., tissue acidosis, cytokines, arachidonic acid metabolites) and the neuroenteric system (Figure 2). ECC are interposed between epithelial cells of the GI mucosa where they act as sensors or ‘‘taste bottoms’’ of the intraluminal milieu. EEC contain large numbers of electron-dense secretory granules with a range of peptides such as but not limited to serotonin, cholecystokinin and secretin positioned towards their basement membrane. In response to luminal nutrients, toxins and mechanical stimulation the ECC release their content into the gut wall which influences the neuromuscular apparatus. Serotonin release for instance is well known to activate vagal afferent endings in the upper GI tract serving as an emetic trigger[68]. A proportion of postinfectious irritable bowel syndrome (PI-IBS) patients have ECC hyperplasia and multivariate analysis has shown that ECC count is an important predictor of developing PI-IBS (relative risk 3.8)[4,69]. Also the endocrine cell population in patients with CD ileitis showed an increase in ECC number, both at affected and nonaffected sites of the ileum. In a study on colonic tissue, the ECC area was likewise significantly increased in active CD and UC[47]. The same was found in colorectal tissue from UC patients in remission. Recently, a nematode-infected (Trichuris muris) immunodeficient mice model revealed an interaction between CD4+ T cells and ECC. The infection evoked Th2 response lead to ECC hyperplasia via the presence of IL-13 receptors on ECC, resulting in an increase in serotonin production[70]. The 5-HT receptor subtypes that are involved in visceral hypersensitivity are 5-HT3, 5-HT4 and 5-HT2B. 5-HT3 antagonists (alosetron and cilansetron) prevent the activation of 5-HT3 receptors on extrinsic afferent neurons and decrease hyperalgesia and abdominal pain in IBS patients[71]. More recently, evidence emerged that 5-HT4 receptor-mediated mechanisms regulate visceral sensitivity as tegaserod, a partial 5-HT4 agonist, normalized postinflammatory hypersensitive colon in the rat[72]. In a recent patient study, tegaserod significantly reduced the inhibitory effects of colorectal distension on the RIII reflex in 12 of 15 patients[73]. Finally a role for 5-HT2B has been stated, but needs further verification. Serotonergic mechanisms are likely implicated in PI-IBS patients based on an increased number of ECC[74-76], an increased mast cell population[77], an increased postprandial serotonin release[78]. The metabolism of 5-HT might also be disrupted in both IBS and IBD. In this regard, it has been suggested that decreased serotonin-selective reuptake transporter (SERT) expression in IBD and IBS patients is associated with GI dysfunction in these disorders[79-81]. SERT, which is expressed on enterocytes, terminates the actions of serotonin by removing it from the interstitial space. The role of SERT in GI pathology is further supported by the observation that colonic sensitivity to CRD was attenuated in mice after long-term treatment with paroxetine, a SERT inhibitor[82]. Polymorphisms of the serotonin re-uptake transporter gene may also play a role in disturbance of gut function. IBS patients with deletion/deletion genotype of SERT polymorphism more often experience abdominal pain compared to those expressing other SERT polymorphisms[83].
Figure 2.
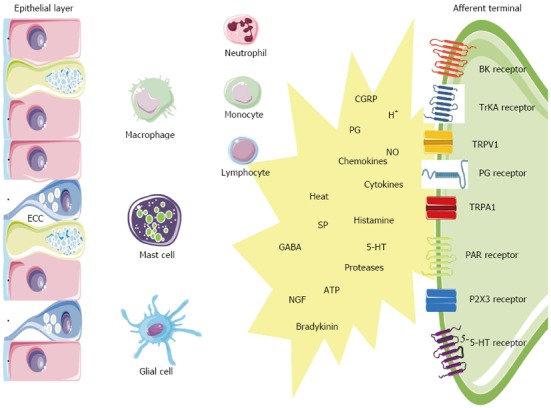
Scheme is oversimplified and limited to the cell types and mediators discussed in this review and represents a subset of cells and inflammatory mediators responsible for activation of gut sensory afferents after an initial inflammatory response. 5-HT: 5-hydroxytryptamine; BK: Bradykinin; CGRP: Calcitonin-gene-related peptide; ECC: Enterochromaffin cell; GABA: Gamma-amino butyric acid; NGF: Nerve growth factor; NO: Nitric oxide; PAR: Proteinase-activated receptor; PG: Prostaglandin; SP: Substance P; TrKA: Tyrosine receptor kinase A; TRPA1: Transient receptor potential ankyrin-1; TRPV1: Transient receptor potential vanilloid-1; P2X3: Purinergic P2X3 receptor.
Mast cells are bone-marrow derived cells that circulate in the bloodstream as immature progenitors and maturate and reside within the mucosal and connective tissues (Figure 2). Mast cells possess a plethora of mediators that can be rapidly released out of preformed granules like histamine, serotonin, serine proteases (e.g., tryptase), proteoglycans or that can be de novo synthetized such as prostaglandins (e.g., PGE2, PGD2), leukotrienes (e.g., LTC4, LTD4), platelet activating factor (PAF), and cytokines (e.g., TNFα, IL-6)[84,85]. In mice, serotonin is also present in mucosal mast cells in the lamina propria and some studies have suggested that human mast cells may also contain serotonin especially in conditions associated with mastocytosis[86]. Within the GI wall the close proximity between mast cells and neurons is intriguing and a bidirectional interaction between them is generally accepted[67,87,88]. Recently, it was shown that the number of mast cells close to afferent fibers was significantly increased in rats with DSS colitis, and that nerve fibers reacted stronger to compound 48/80-evoked mast cell degranulation[89]. Visceral hypersensitivity, evoked by the chemical irritant TNBS but also by chronic juvenile stress from maternal separation, can be treated with the mast cell stabilizer ketotifen and is abolished in mast cell deficient rats[90,91]. In patients, ketotifen increases the threshold for discomfort in patients with IBS with visceral hypersensitivity, reduces IBS symptoms and improves health-related quality of life regardless from the poor correlation with mast cell activation in biopsies[92]. There is considerable clinical evidence for mast cell involvement in human IBD. In colorectal mucosa from patients with CD and UC, the amount of mast cell tryptase was significantly increased, as was the number of mast cells in the lamina propria and submucosa[93]. The same observations were made in the afflicted ileum of CD patients[4,94]. The secretion profile of mast cells derived from UC patients was also shifted, releasing greater amounts of histamine, PGs and leukotrienes[95,96]. Moreover, rates of tryptase secretion were increased in both inflamed and noninflamed tissue from UC patients indicating that mast cell activation/proliferation can be altered remote from the site of active inflammation[4,97]. Also in IBS patients sufficient data support a role for mast cells. Mast cell infiltration was associated with symptoms of bloating in IBS patients[98]. In addition, activated mast cells in close proximity to colonic nerves were significantly correlated with the severity and frequency of abdominal pain or discomfort[99]. The experimental observation that mast cell mediators, released from the colonic mucosal biopsies from IBS patients but not of healthy patients, excite rat nociceptive visceral afferents further provide evidence of a pivotal role of mast cells in IBS hypersensitivity and have been confirmed by several other groups in different countries[100,101].
Protease-activated receptors (PARs) are a family of 4 receptor types activated by serine proteases such as thrombin and tryptase (Figure 2). PARs have a widespread distribution throughout the GI tract enabling the involvement in all the aspects of gut physiology, including inflammation and nociception. PAR1, 2 and 4 have been implicated in the modulation of nociceptive mechanisms, since they are expressed by nociceptive DRG neurons containing CGRP and SP. However, the function of these receptors in nociceptive signaling might be opposite. PAR2 was found to be activated by trypsin and the mast cell mediator tryptase, whereas PAR1 and 4 are activated by thrombin[102]. In rodents, PAR1 and PAR4 exert antinociceptive signals whereas PAR2 is clearly a pronociceptive agent regarding both mast cell- and formalin-induced hyperalgesia in mice[103]. A role for PARs in inflammatory disorders is evidenced by several studies: the expression of PAR1 and PAR2 is upregulated in tissues from CD or UC patients[104,105], the levels of the PAR2 agonists trypsin and mast cell tryptase are elevated in mouse colon[106], elevated colonic luminal serine protease activity has been observed in IBS-D patients[107]. The generation of pain symptoms has been suggested by the observation that mice injected with mediators released from colonic biopsies of IBS patients, exhibit enhanced nociceptive responses to CRD, whereas transgenic mice without PAR2 failed to show such mechanical hyperalgesia[107,108]. From these findings, it would appear that PAR2 antagonists and PAR1 and PAR4 agonists have potential in the control of visceral pain and hyperalgesia symptoms in both IBD and IBS. In mice, PAR2-mediated mechanical hyperalgesia requires sensitization of the ion channel transient receptor potential vanilloid 4 (TRPV4), since deletion of TRPV4 prevented PAR2 agonist-induced mechanical hyperalgesia and sensitization[109,110]. Accordingly, mast cell tryptase-induced PAR2 activation is proposed as a mechanism for TRPA1 sensitization as it was shown that PAR2-induced hyperalgesia was absent in TRPA1 knockout mice[111].
Nerve growth factor (NGF) is synthetized by epithelial cells and mast cells when triggered by IL-1β and TNFα (Figure 2)[112]. NGF influences development and function of afferents by binding to its high affinity TrKA receptor. Indeed, NGF can modulate the expression of membrane bound receptors such as TRPV1 and TRPA1 localized at peripheral afferents. The described NGF-mediated mechanism could regulate inflammatory hyperalgesia seen in IBD, as hypersensitivity in rats with inflamed colon can be reversed by anti-NGF antibody treatment[113]. NGF has been implicated in several chronic inflammatory processes. In CD, NGF mRNA is increased in 60% and TrkA mRNA in 54% in UC, NGF mRNA expression was enhanced in 58% (2.4-fold; P < 0.01) and TrkA mRNA expression in 50% of the patients. Enhanced expression of NGF and TrkA in both neural and non-neural structures suggests activation of this neuroimmune pathway in chronic inflammation in CD and UC[114].
A population of cells that is recently taken into account in the modulation of neuroimmune interactions are the peripheral glial cells. These cells are capable of modulating enteric neurotransmission, modulate inflammation and control intestinal barrier function. They are capable of these interactions as they contain precursors for neurotransmitters such as GABA and NO; they express receptors for purines and they are able to produce cytokines (IL-1β, IL-6, TNFα), NGF and neuropeptides (NKA and SP) after activation[115]. There is recent evidence for a paracrine purinergic neuro-glial communication and also after injection of endotoxins in mice glial cells are activated[116,117]. Changes in enteric glial cells have been described in IBD[118]. Recently, the role of glial cells has been investigated in rectal biopsies of UC patients; the expression of S100, a marker for enteric glial cells, was associated with an increase of inducible nitric oxide synthase expression[119]. Inflammation increases the synthesis of PGs through upregulation of cyclooxygenase-2 (COX-2). For instance, in patients with active CD and UC a six- to eightfold increase in COX-2 mRNA was demonstrated in the bowel wall[120]. Although suppression of PG production in the gut by COX inhibitors carries the risk of severe GI mucosal damage, blockade of PG receptors expressed by sensory neurons appears to be an alternative way of preventing the proalgesic action of PGs. PGE2 and PGI2 have been proven to be key mediators of inflammatory hyperalgesia. Primary afferent neurons express PG receptors of the EP1, EP2, EP3, EP4 and IP type. PG receptors are also found at the central synapse in the spinal cord. For instance, PGE2 is recognized as playing a prominent role in the CNS as well as peripheral tissues[121].
Perhaps the most intriguing players in peripheral hyperalgesia and pain are the transient receptor potential (TRP) ion channels of the vanilloid type 1 (TRPV1), the vanilloid type 4 (TRPV4), the ankyrin type 1 (TRPA1) and the melastatin type 8 (TRPM8) (Figure 2). These TRPs are expressed on gut afferents including those that conduct noxious stimuli to the spinal cord and co-express with the above listed G protein-coupled receptors (e.g., EP1, 5HT3, BK1, PAR2) and growth factor receptors (e.g., TrKA receptors). Their close proximity allows the TRPs to couple their activity to mutual downstream pathways which enables them to integrate a diversity of stimuli present in the inflammatory milieu[122]. TRPV1, the best characterized TRP, acts as an immediate sensory alarm in response to mechanical stretch or distension, mild acidification (pH < 5.9), noxious heat (> 42 °C) and spice ingredients such as capsaicin[123,124]. Also endovanilloids such as anandamide, unsatured N-acyldopamines and lipoxygenases of arachidonic acid are known to directly activate TRPV1. Many proalgesic factors are associated with TRPV1-induced hyperalgesia (e.g., bradykinin, 5-HT, NGF, PAR2, endogenous metabolites)[125]. Experimental animal models have shown that TRPV-/- mice have decreased pain-related responses to colorectal distension, whereas capsaicin application will increase pain to colorectal distension in animals. These data in animals suggest the involvement of TRPV1 signalling pathways in colonic pain. Of clinical relevance is that TPRV1 expression is increased in UC and CD[126]. TRPV1 is upregulated not only in inflammation but also in the absence of overt inflammation as is typical of functional GI disorders. This is true for patients with IBS in which increased density of TRPV1 in the rectosigmoid correlated with pain severity[127]. A similar correlation between pain intensity and number of mucosal TRPV1-positive nerve fibers is found in patients with quiescent IBD who continue to complain of abdominal pain[126,127]. TRPA1 is a receptor characterized by a long ankyrin repeat at the N-terminal site that serves as a binding site for a range of environmental irritants, oxidants and spices such as mustard oil, wasabi and horseradish[128]. Recent findings have also found that TRPA1 is involved in cold transduction and mechanosensation[129,130]. Moreover, TRPA1 contributes to inflammatory hyperalgesia via PAR2 activation[111]. Clinically, an autosomal dominant mutation in the fourth transmembrane of TRPA1 was described in one family that underlies a familial episodic pain syndrome[131]. Further, TRPA1 is a candidate mechanosensor for mechanical hyperalgesia in colitis and overactive bladder[130,132]. TRPA1 is almost exclusively present in a TPRV1-positive population of sensory nociceptors and does not co-express with TRPV1 in other tissues. In this regard, a study quantifying TRPA1-positive neurons in trigeminal ganglia has demonstrated that TRPA1 is expressed in anout 55% of TRPV1-positive neurons while NGF treatment of trigeminal ganglia increases TRPA1 expression to anout 80% of TRPV1-positive cells. Several lines of evidence has shown that TRPV1 exerts a modulatory role on TRPA1 channels[133]. With concern to hypersensitivity of the colon, we recently have shown that TRPV1 and TRPA1 synergistically decrease visceromotor responses in rats with TNBS colitis but not in control rats[134].
Central sensitization
Apart from sensitization in the periphery, the gut impulses are modulated or amplified in the spinal cord and higher brain centers; a process referred to as central sensitization. The co-morbidity of IBS with disorders such as but not limited to depression, anxiety, and painful bladder syndrome or of IBD with interstitial cystitis may originate from central sensitization[16,135,136]. The leading hypothesis to explain these co-occurrences is a viscerovisceral and a viscerosomatic cross-sensitization, with somatic and visceral afferents converging onto the same second order neuron in the spinal cord or third order neuron in the supraspinal centers and an overlap within yet undefined brain fields[137]. Clinical evidence for a role of CNS sensitization in visceral pain comes from functional resonance magnetic imaging (fMRI) and PET studies on referred pain to adjacent structures or at remote distance from the (actual) injured organ[138]. More direct evidence for enhanced spinal processing in IBS patients has been confirmed through analysis of rectal distensions on the RIII reflex, a nociceptive withdrawal reflex used as an objective tool to investigate pain processing at the spinal and supraspinal level. Whereas slow ramp rectal distension induced inhibition of this reflex in healthy volunteers, it facilitated the reflex in IBS[139]. Proof of altered brain activity has been shown with brain imaging studies and the potential of this research should be further explored[140]. The current know-how on brain imaging can be extensively consulted in review articles by Van Oudenhove et al[141], Smith et al[142] and Mayer et al[57,140].
Sensitized ascending and descending pathways: Upon repetitive stimulation by extrinsic primary afferent neurons, intracellular signaling cascades are activated within the spinal dorsal horn neurons. This leads to amplified responses to both innocuous and noxious input due to two major mechanisms: the facilitation of excitatory synaptic responses (so-called wind-up) and the downregulation of descending inhibitory influences[47,143]. The main mediator of wind-up is the neurotransmitter glutamate. When the presynaptic release of glutamate is triggered, glutamate acts on the ligand-gated ion channels NMDA (N-methyl-D-aspartate) receptors, kainate, AMPA (α-amino-5-hydroxy-3-methyl-4-isoxazole propionic acid) and metabotropic glutamate receptors (mGLUR) expressed by the dorsal horn neurons. In addition to this direct effect, hyperstimulation of spinal neurons phosphorylates NMDA receptors which further increases NMDA receptor responsiveness to glutamate and increases synaptic strength[144]. AMPA receptor trafficking from the intracellular stores to the synaptic plasma membrane has also shown to augment glutamate responsiveness in a mice model of visceral nociception induced by intracolonic capsaicin[145]. The potential therapeutic effect of glutamate removal has also been investigated in experimental animal models. In this regard, it has been shown that ceftriaxone attenuates visceral hypersensitivity to CRD in rats with DSS and TNBS colitis. This effect was mediated via overexpression of spinal glutamate transporter-1 which increased removal of extracellular glutamate[146]. Other important mediators of central sensitization include substance P (SP), PGE2 and brain-derived neurotropic factor which respectively target spinal neurokinin-1 receptor expression, PGE2 receptors and tyrosine kinase B receptors[147]. For example, PGE2 suppresses glycinergic transmission via activation PGE2 receptors of the EP2 subtype and subsequent PKA-dependent blockade of glycine receptors containing the α3 subunit (GlyRα3)[148]. The result of this blockade is the discontinuance of dorsal horn nociceptive neurons from their inhibitory control by glycinergic neurons. This PGE2-evoked mechanism facilitates nociceptive input from the spinal cord. Similarly, a loss of GABAergic synaptic inhibition also increases nociceptive signaling[149]. COX-2, the enzyme that forms PGE2 is markedly upregulated in the spinal cord during acute and chronic peripheral inflammation. In the spinal cord, basal release of PGE2 is increased after peripheral inflammation[150]. Apart from neuron-neuron interactions, also glial cell-nerve interactions modulate signaling at the neuronal synapse, although this research is still in its infancy. Spinal glial cell activation is believed to be important in facilitation of nociceptive signals in various pain conditions. Under physiological conditions, glial cells are quiescent. However, during inflammation glial cells produce a variety of nociceptive agents such as TNFα, IL-1 and NO[151]. Most information has been obtained from experimental animal models of injury[152]. For instance, it has been shown that neonatal colonic irritation-induced visceral hypersensitivity in rats is accompanied by an increased expression of OX42, indicating glial cell proliferation. Visceral hypersensitivity was blocked with minocycline, an inhibitor of glial cell activation[153]. Recently, morphological remodeling of colonic afferent central nerve terminals was proposed in a mice model of hypersensitivity after TNBS inflammation. However, overall the ‘‘sprouting’’ theory of central afferent colonic nerve endings as a mechanism of central sensitization remains controversial[154]. Studies using functional brain imaging techniques have shown inflammation-induced modulation of activity in brain regions involved in visceral sensation, such as the ACC of the limbic system.
Electrophysiological studies in laboratory animals have shown that ACC sensitization occurs in viscerally hypersensitive rats[155]. It was revealed that for instance IBS was associated with decreased gray matter density in various brain areas, including medial and ventrolateral prefrontal cortex, posterior parietal cortex, ventral striatum, thalamus, and PAG. Further, IBS patients show brain responses consistent with hyperresponsiveness to gut distension in terms of vigilance, arousal and perhaps sensory sensitization[156]. Taken together, emerging evidence of structural brain changes in IBS is intriguing, but should be interpreted with great caution until more knowledge about the nature and implications of the observed alterations becomes available[63,157].
Accumulating evidence also suggests that descending facilitatory influences may contribute to the development and maintenance of hyperalgesia and thus contribute to chronic pain states. In this regard, a role for the RVM in the maintenance of hyperalgesic states following peripheral tissue injury activated by NMDA receptors, neurotensin receptors and NO is established[58]. Impaired ability to activate the descending pain inhibitory system has been hypothesized in IBS[57]. Aside from IBS patients, patients with active UC have been reported with reduced threshold to perception and reduced maximal tolerance to anorectal balloon distension[158]. CD children and adolescents suffering from abdominal pain despite remission had a lower rectal sensory pain threshold compared to healthy patients in a study conducted by Faure and co-workers[159]. Paradoxically, in other studies conducted in chronic quiescent intestinal inflammatory states such as CD or UC, patients experience attenuated rectal perception and increased threshold for discomfort. UC patients with mild mucosal inflammation of the rectum had lower thresholds for discomfort during rectosigmoid distension compared to healthy patients[2]. A central descending inhibitory mechanism of sensory pathways in chronic inflammatory states, which would not be active in IBS patients, might be responsible for this seemingly discrepancy. This concept is further supported by a study showing that colonic inflammation is not necessarily associated with increased afferent input to the brain and that, in response to colorectal distension, inhibition of limbic/paralimbic circuits was observed in UC and control patients, but not in IBS patients[57]. Strong inhibitory mechanisms counteracting inflammation-induced hypersensitivity can be activated in chronic inflammatory pathologies, but seem to be deficient in patients with IBS-associated visceral hypersensitivity[160].
A recent meta-analysis of published studies on brain responses to rectal distension have shown differences between IBS patients and healthy controls[161]. Recently, Larsson and co-workers have shown that hypersensitive patients with IBS had greater activation of the insula and reduced deactivation in the pregenual ACC during noxious rectal distension compared to healthy patients and normosensitive IBS patients[162].
FUTURE DIRECTIONS
New therapeutic strategies may arise from the progressing identification of molecular prognostic markers and characterization of the molecular basis of IBD and IBS. Recently, dysfunction of microRNAs (miRNAs), which are non-coding RNA molecules that regulate gene expression, was postulated to play a role in IBD and IBS. Wu et al[163] showed that miRNAs regulate colonic epithelial cell-derived chemokine expression and that colonic tissues from patients with ulcerative colitis have altered miRNA expression patterns. An increased expression of miR-29a was also observed in blood microvesicles, small bowel and colon tissues of IBS patients with increased intestinal membrane permeability[164]. Recent evidence suggests that miR-29 expression is upregulated in human dendritic cells in response to NOD2 signals with concomitant downregulation of interleukin-23[165]. Interestingly, dendritic cells with NOD2 polymorphisms from Crohn’s disease patients fail to induce miR-29 upon pattern recognition receptor stimulation[165]. Moreover, experimental colitis in miR-29-deficient mice is more severe and associated with significantly enhanced levels of IL-23 and T helper 17 in the intestinal mucosa[165]. In respect to visceral pain, it was recently suggested that epigenetic central mechanisms are involved in the regulation of stress-induced visceral hypersensitivity in rats[166]. Overall, these results suggest that modulation of genetic and epigenetic regulatory mechanisms and profiling of miRNAs may represent promising strategies for the treatment of pain associated with IBD and IBS.
CONCLUSION
Chronic abdominal pain in IBD and IBS requires notion of how the lower gut becomes highly sensitive to any kind of stimulus. Noninvasive markers, including PET and fMRI, combined with pharmacology are used to assess hypersensitivity in these pathologies. In support, functional anatomical and physiological studies in rodents are being conducted[167]. Together these approaches discovered a significant amount of the neuroanatomical substrates and molecules in gut hypersensitivity, yet the degree to which each of these mechanisms contribute to hypersensitivity remains unknown. In both IBD and IBS, the complex interplay of sensitization occurs at different sites of action among the brain-gut axis and can be broadly categorized: sensitization of visceral afferents, sensitization of spinal cord ascending afferents, altered descending excitatory and inhibitory influences to the spinal cord nociceptive neurons and misinterpretation of non-noxious sensation as noxious due to cognitive and emotional biasing (hypervigilance)[47].
IBS and IBD have many of the mechanisms and molecules in visceral peripheral and CNS sensitization in common. Currently, no unambiguous neuronal marker exists that discriminates IBD and IBS. However, an inactive descending inhibitory control is hypothesized in IBS, but not in IBD. A recent study has suggested differences in coping behavior between IBD and IBS[168]. These differences among IBD and IBS certainly merit more appraisal, however, should be interpreted with caution. There are proportionally less studies on sensitivity in IBD patients than in IBS patients, therefore making head-to-head comparisons difficult. The shortage of studies on sensitivity in IBD patients may be attributed to the risk of jeopardizing remission by the barostat-induced distensions performed for sensitivity measurements. Nevertheless in both cases neuroplastic changes are quite common and the observed differences may not per se reflect disorder-specific changes, but may be attributed to affective disturbances, negative emotions in anticipation of/during visceral stimulation, and altered pain-related expectations and learning processes[63]. Expectation of pain may explain up to 50% of the variation of pain ratings[169]. Indeed, abdominal pain is not linearly related to peripheral sensory input. A considerable proportion (about one third) of IBS patients have a normal rectal perception, and a proportion of both UC and CD patients had increased thresholds for perception and discomfort[2]. Due to the multi-factorial complexity of sensitization in IBD and IBS, there is presently a rather limited success of available therapeutic approaches for IBS and the functional IBS-like symptoms in IBD. Technological progress that allows mapping of sensitization may be interesting to screen patients. In patients with peripheral sensitization mechanisms, the combination of anti-inflammatory properties and analgesic properties within one drug seems a promising route for translational research. In patients with CNS disorders, approaches such as cognitive therapy or anti-depressive agents that decrease anxiety or hypervigilance might be beneficial.
In summary, the mechanisms of hypersensitivity in response to bowel inflammation are complex and need to be further unraveled. Determining the level of sensitization is crucial for the assessment of disease activity and for tailoring therapy. Several of these already defined mechanisms can be coined up as potential targets for the development of therapeutic options for inflammatory GI pain.
Footnotes
P- Reviewer: Wiley JW S- Editor: Zhai HH L- Editor: A E- Editor: Wang CH
References
- 1.Wagtmans MJ, Verspaget HW, Lamers CB, van Hogezand RA. Crohn’s disease in the elderly: a comparison with young adults. J Clin Gastroenterol. 1998;27:129–133. doi: 10.1097/00004836-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Chang L, Munakata J, Mayer EA, Schmulson MJ, Johnson TD, Bernstein CN, Saba L, Naliboff B, Anton PA, Matin K. Perceptual responses in patients with inflammatory and functional bowel disease. Gut. 2000;47:497–505. doi: 10.1136/gut.47.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geboes K, Collins S. Structural abnormalities of the nervous system in Crohn’s disease and ulcerative colitis. Neurogastroenterol Motil. 1998;10:189–202. doi: 10.1046/j.1365-2982.1998.00102.x. [DOI] [PubMed] [Google Scholar]
- 4.De Schepper HU, De Man JG, Moreels TG, Pelckmans PA, De Winter BY. Review article: gastrointestinal sensory and motor disturbances in inflammatory bowel disease - clinical relevance and pathophysiological mechanisms. Aliment Pharmacol Ther. 2008;27:621–637. doi: 10.1111/j.1365-2036.2008.03624.x. [DOI] [PubMed] [Google Scholar]
- 5.Cross RK, Wilson KT, Binion DG. Narcotic use in patients with Crohn’s disease. Am J Gastroenterol. 2005;100:2225–2229. doi: 10.1111/j.1572-0241.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- 6.Farrokhyar F, Marshall JK, Easterbrook B, Irvine EJ. Functional gastrointestinal disorders and mood disorders in patients with inactive inflammatory bowel disease: prevalence and impact on health. Inflamm Bowel Dis. 2006;12:38–46. doi: 10.1097/01.mib.0000195391.49762.89. [DOI] [PubMed] [Google Scholar]
- 7.Camilleri M. Management of the irritable bowel syndrome. Gastroenterology. 2001;120:652–668. doi: 10.1053/gast.2001.21908. [DOI] [PubMed] [Google Scholar]
- 8.Tibble JA, Sigthorsson G, Foster R, Forgacs I, Bjarnason I. Use of surrogate markers of inflammation and Rome criteria to distinguish organic from nonorganic intestinal disease. Gastroenterology. 2002;123:450–460. doi: 10.1053/gast.2002.34755. [DOI] [PubMed] [Google Scholar]
- 9.Grad C, David L, Portincasa P, Dumitraşcu DL. Diagnostic value of calprotectin in irritable bowel syndrome and in inflammatory bowel disease. Rom J Intern Med. 2012;50:3–6. [PubMed] [Google Scholar]
- 10.Dinan TG, Quigley EM, Ahmed SM, Scully P, O’Brien S, O’Mahony L, O’Mahony S, Shanahan F, Keeling PW. Hypothalamic-pituitary-gut axis dysregulation in irritable bowel syndrome: plasma cytokines as a potential biomarker? Gastroenterology. 2006;130:304–311. doi: 10.1053/j.gastro.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 11.Chadwick VS, Chen W, Shu D, Paulus B, Bethwaite P, Tie A, Wilson I. Activation of the mucosal immune system in irritable bowel syndrome. Gastroenterology. 2002;122:1778–1783. doi: 10.1053/gast.2002.33579. [DOI] [PubMed] [Google Scholar]
- 12.Spiller RC. Potential biomarkers. Gastroenterol Clin North Am. 2011;40:121–139. doi: 10.1016/j.gtc.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Swan C, Duroudier NP, Campbell E, Zaitoun A, Hastings M, Dukes GE, Cox J, Kelly FM, Wilde J, Lennon MG, et al. Identifying and testing candidate genetic polymorphisms in the irritable bowel syndrome (IBS): association with TNFSF15 and TNFα. Gut. 2013;62:985–994. doi: 10.1136/gutjnl-2011-301213. [DOI] [PubMed] [Google Scholar]
- 14.Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136:1979–1988. doi: 10.1053/j.gastro.2009.02.074. [DOI] [PubMed] [Google Scholar]
- 15.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 16.Malykhina AP. Neural mechanisms of pelvic organ cross-sensitization. Neuroscience. 2007;149:660–672. doi: 10.1016/j.neuroscience.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 17.Timmermans JP, Scheuermann DW, Stach W, Adriaensen D, De Groodt-Lasseel MH. Functional morphology of the enteric nervous system with special reference to large mammals. Eur J Morphol. 1992;30:113–122. [PubMed] [Google Scholar]
- 18.Costa M, Brookes SJ, Hennig GW. Anatomy and physiology of the enteric nervous system. Gut. 2000;47 Suppl 4:iv15–iv19; discussion iv26. doi: 10.1136/gut.47.suppl_4.iv15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furness JB. The organisation of the autonomic nervous system: peripheral connections. Auton Neurosci. 2006;130:1–5. doi: 10.1016/j.autneu.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Chey WY, Jin HO, Lee MH, Sun SW, Lee KY. Colonic motility abnormality in patients with irritable bowel syndrome exhibiting abdominal pain and diarrhea. Am J Gastroenterol. 2001;96:1499–1506. doi: 10.1111/j.1572-0241.2001.03804.x. [DOI] [PubMed] [Google Scholar]
- 21.Ford AC, Talley NJ, Spiegel BM, Foxx-Orenstein AE, Schiller L, Quigley EM, Moayyedi P. Effect of fibre, antispasmodics, and peppermint oil in the treatment of irritable bowel syndrome: systematic review and meta-analysis. BMJ. 2008;337:a2313. doi: 10.1136/bmj.a2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makharia GK. Understanding and treating abdominal pain and spasms in organic gastrointestinal diseases: inflammatory bowel disease and biliary diseases. J Clin Gastroenterol. 2011;45 Suppl:S89–S93. doi: 10.1097/MCG.0b013e31821fbd82. [DOI] [PubMed] [Google Scholar]
- 23.Blackshaw LA, Brookes SJ, Grundy D, Schemann M. Sensory transmission in the gastrointestinal tract. Neurogastroenterol Motil. 2007;19:1–19. doi: 10.1111/j.1365-2982.2006.00871.x. [DOI] [PubMed] [Google Scholar]
- 24.Powley TL, Wang XY, Fox EA, Phillips RJ, Liu LW, Huizinga JD. Ultrastructural evidence for communication between intramuscular vagal mechanoreceptors and interstitial cells of Cajal in the rat fundus. Neurogastroenterol Motil. 2008;20:69–79. doi: 10.1111/j.1365-2982.2007.00990.x. [DOI] [PubMed] [Google Scholar]
- 25.Grundy D. Neuroanatomy of visceral nociception: vagal and splanchnic afferent. Gut. 2002;51 Suppl 1:i2–i5. doi: 10.1136/gut.51.suppl_1.i2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Page AJ, Martin CM, Blackshaw LA. Vagal mechanoreceptors and chemoreceptors in mouse stomach and esophagus. J Neurophysiol. 2002;87:2095–2103. doi: 10.1152/jn.00785.2001. [DOI] [PubMed] [Google Scholar]
- 27.Holzer P. Afferent signalling of gastric acid challenge. J Physiol Pharmacol. 2003;54 Suppl 4:43–53. [PubMed] [Google Scholar]
- 28.Gschossmann JM, Mayer EA, Miller JC, Raybould HE. Subdiaphragmatic vagal afferent innervation in activation of an opioidergic antinociceptive system in response to colorectal distension in rats. Neurogastroenterol Motil. 2002;14:403–408. doi: 10.1046/j.1365-2982.2002.00345.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen SL, Wu XY, Cao ZJ, Fan J, Wang M, Owyang C, Li Y. Subdiaphragmatic vagal afferent nerves modulate visceral pain. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1441–G1449. doi: 10.1152/ajpgi.00588.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holtmann G, Goebell H, Jockenhoevel F, Talley NJ. Altered vagal and intestinal mechanosensory function in chronic unexplained dyspepsia. Gut. 1998;42:501–506. doi: 10.1136/gut.42.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Randich A, Gebhart GF. Vagal afferent modulation of nociception. Brain Res Brain Res Rev. 1992;17:77–99. doi: 10.1016/0165-0173(92)90009-b. [DOI] [PubMed] [Google Scholar]
- 32.Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. 2013;62:1214–1222. doi: 10.1136/gutjnl-2012-302550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christianson JA, Traub RJ, Davis BM. Differences in spinal distribution and neurochemical phenotype of colonic afferents in mouse and rat. J Comp Neurol. 2006;494:246–259. doi: 10.1002/cne.20816. [DOI] [PubMed] [Google Scholar]
- 34.Brierley SM, Jones RC, Gebhart GF, Blackshaw LA. Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology. 2004;127:166–178. doi: 10.1053/j.gastro.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Gué M, Junien JL, Buéno L. The kappa agonist fedotozine modulates colonic distention-induced inhibition of gastric motility and emptying in dogs. Gastroenterology. 1994;107:1327–1334. doi: 10.1016/0016-5085(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 36.De Schepper HU, De Man JG, Van Nassauw L, Timmermans JP, Herman AG, Pelckmans PA, De Winter BY. Acute distal colitis impairs gastric emptying in rats via an extrinsic neuronal reflex pathway involving the pelvic nerve. Gut. 2007;56:195–202. doi: 10.1136/gut.2006.104745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Traub RJ. Evidence for thoracolumbar spinal cord processing of inflammatory, but not acute colonic pain. Neuroreport. 2000;11:2113–2116. doi: 10.1097/00001756-200007140-00011. [DOI] [PubMed] [Google Scholar]
- 38.Grundy D. Speculations on the structure/function relationship for vagal and splanchnic afferent endings supplying the gastrointestinal tract. J Auton Nerv Syst. 1988;22:175–180. doi: 10.1016/0165-1838(88)90104-x. [DOI] [PubMed] [Google Scholar]
- 39.Lamb K, Kang YM, Gebhart GF, Bielefeldt K. Gastric inflammation triggers hypersensitivity to acid in awake rats. Gastroenterology. 2003;125:1410–1418. doi: 10.1016/j.gastro.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Lynn PA, Blackshaw LA. In vitro recordings of afferent fibres with receptive fields in the serosa, muscle and mucosa of rat colon. J Physiol. 1999;518(Pt 1):271–282. doi: 10.1111/j.1469-7793.1999.0271r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song X, Chen BN, Zagorodnyuk VP, Lynn PA, Blackshaw LA, Grundy D, Brunsden AM, Costa M, Brookes SJ. Identification of medium/high-threshold extrinsic mechanosensitive afferent nerves to the gastrointestinal tract. Gastroenterology. 2009;137:274–284, 284.e1. doi: 10.1053/j.gastro.2009.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Traub RJ, Murphy A. Colonic inflammation induces fos expression in the thoracolumbar spinal cord increasing activity in the spinoparabrachial pathway. Pain. 2002;95:93–102. doi: 10.1016/s0304-3959(01)00381-5. [DOI] [PubMed] [Google Scholar]
- 43.Ness TJ, Metcalf AM, Gebhart GF. A psychophysiological study in humans using phasic colonic distension as a noxious visceral stimulus. Pain. 1990;43:377–386. doi: 10.1016/0304-3959(90)90035-C. [DOI] [PubMed] [Google Scholar]
- 44.Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci USA. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gybels JM, Sweet WH. Neurosurgical treatment of persistent pain. Physiological and pathological mechanisms of human pain. Pain Headache. 1989;11:1–402. [PubMed] [Google Scholar]
- 46.Pasricha PJ, Willis WD, Gebhart GF. Chronic Abdominal and Visceral Pain: Theory and Practice. 1 st ed. New York: Informa; 2006. [Google Scholar]
- 47.Anand P, Aziz Q, Willert R, van Oudenhove L. Peripheral and central mechanisms of visceral sensitization in man. Neurogastroenterol Motil. 2007;19:29–46. doi: 10.1111/j.1365-2982.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 48.Wouters MM, Van Wanrooy S, Casteels C, Nemethova A, de Vries A, Van Oudenhove L, Van den Wijngaard RM, Van Laere K, Boeckxstaens G. Altered brain activation to colorectal distention in visceral hypersensitive maternal-separated rats. Neurogastroenterol Motil. 2012;24:678–685, e297. doi: 10.1111/j.1365-2982.2012.01919.x. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberger C, Thürling M, Forsting M, Elsenbruch S, Timmann D, Gizewski ER. Contributions of the cerebellum to disturbed central processing of visceral stimuli in irritable bowel syndrome. Cerebellum. 2013;12:194–198. doi: 10.1007/s12311-012-0413-3. [DOI] [PubMed] [Google Scholar]
- 50.Al-Chaer ED, Feng Y, Willis WD. A role for the dorsal column in nociceptive visceral input into the thalamus of primates. J Neurophysiol. 1998;79:3143–3150. doi: 10.1152/jn.1998.79.6.3143. [DOI] [PubMed] [Google Scholar]
- 51.Ness TJ. Evidence for ascending visceral nociceptive information in the dorsal midline and lateral spinal cord. Pain. 2000;87:83–88. doi: 10.1016/S0304-3959(00)00272-4. [DOI] [PubMed] [Google Scholar]
- 52.Palecek J, Paleckova V, Willis WD. The roles of pathways in the spinal cord lateral and dorsal funiculi in signaling nociceptive somatic and visceral stimuli in rats. Pain. 2002;96:297–307. doi: 10.1016/S0304-3959(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 53.Palecek J. The role of dorsal columns pathway in visceral pain. Physiol Res. 2004;53 Suppl 1:S125–S130. [PubMed] [Google Scholar]
- 54.Danziger N, Rémy P, Pidoux B, Dormont D, Samson Y, Fournier E, Wall PD, Willer JC. A clinical and neurophysiological study of a patient with an extensive transection of the spinal cord sparing only a part of one anterolateral quadrant. Brain. 1996;119(Pt 6):1835–1848. doi: 10.1093/brain/119.6.1835. [DOI] [PubMed] [Google Scholar]
- 55.Reynolds DV. Surgery in the rat during electrical analgesia induced by focal brain stimulation. Science. 1969;164:444–445. doi: 10.1126/science.164.3878.444. [DOI] [PubMed] [Google Scholar]
- 56.Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27:729–737. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Mayer EA, Berman S, Suyenobu B, Labus J, Mandelkern MA, Naliboff BD, Chang L. Differences in brain responses to visceral pain between patients with irritable bowel syndrome and ulcerative colitis. Pain. 2005;115:398–409. doi: 10.1016/j.pain.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 58.Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VP. Extrinsic primary afferent signalling in the gut. Nat Rev Gastroenterol Hepatol. 2013;10:286–296. doi: 10.1038/nrgastro.2013.29. [DOI] [PubMed] [Google Scholar]
- 59.Farmer AD, Aziz Q. Visceral pain hypersensitivity in functional gastrointestinal disorders. Br Med Bull. 2009;91:123–136. doi: 10.1093/bmb/ldp026. [DOI] [PubMed] [Google Scholar]
- 60.Straub RH, Wiest R, Strauch UG, Härle P, Schölmerich J. The role of the sympathetic nervous system in intestinal inflammation. Gut. 2006;55:1640–1649. doi: 10.1136/gut.2006.091322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merskey H, Bogduk N. Classification of Chronic Pain. In: Harold Merskey, Nikolai Bogduk., editors. Part III: Pain Terms, A Current List with Definitions and Notes on Usage. Seattle: IASP Press; 1994. pp. 209–214. [Google Scholar]
- 62.Mertz HR. Irritable bowel syndrome. N Engl J Med. 2003;349:2136–2146. doi: 10.1056/NEJMra035579. [DOI] [PubMed] [Google Scholar]
- 63.Elsenbruch S. Abdominal pain in Irritable Bowel Syndrome: a review of putative psychological, neural and neuro-immune mechanisms. Brain Behav Immun. 2011;25:386–394. doi: 10.1016/j.bbi.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 64.Posserud I, Syrous A, Lindström L, Tack J, Abrahamsson H, Simrén M. Altered rectal perception in irritable bowel syndrome is associated with symptom severity. Gastroenterology. 2007;133:1113–1123. doi: 10.1053/j.gastro.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 65.Bouin M, Plourde V, Boivin M, Riberdy M, Lupien F, Laganière M, Verrier P, Poitras P. Rectal distention testing in patients with irritable bowel syndrome: sensitivity, specificity, and predictive values of pain sensory thresholds. Gastroenterology. 2002;122:1771–1777. doi: 10.1053/gast.2002.33601. [DOI] [PubMed] [Google Scholar]
- 66.Hazlett-Stevens H, Craske MG, Mayer EA, Chang L, Naliboff BD. Prevalence of irritable bowel syndrome among university students: the roles of worry, neuroticism, anxiety sensitivity and visceral anxiety. J Psychosom Res. 2003;55:501–505. doi: 10.1016/s0022-3999(03)00019-9. [DOI] [PubMed] [Google Scholar]
- 67.De Winter BY, De Man JG. Interplay between inflammation, immune system and neuronal pathways: effect on gastrointestinal motility. World J Gastroenterol. 2010;16:5523–5535. doi: 10.3748/wjg.v16.i44.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bueno L, de Ponti F, Fried M, Kullak-Ublick GA, Kwiatek MA, Pohl D, Quigley EM, Tack J, Talley NJ. Serotonergic and non-serotonergic targets in the pharmacotherapy of visceral hypersensitivity. Neurogastroenterol Motil. 2007;19:89–119. doi: 10.1111/j.1365-2982.2006.00876.x. [DOI] [PubMed] [Google Scholar]
- 69.Dunlop SP, Jenkins D, Neal KR, Naesdal J, Borgaonker M, Collins SM, Spiller RC. Randomized, double-blind, placebo-controlled trial of prednisolone in post-infectious irritable bowel syndrome. Aliment Pharmacol Ther. 2003;18:77–84. doi: 10.1046/j.1365-2036.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- 70.Wang H, Steeds J, Motomura Y, Deng Y, Verma-Gandhu M, El-Sharkawy RT, McLaughlin JT, Grencis RK, Khan WI. CD4+ T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut. 2007;56:949–957. doi: 10.1136/gut.2006.103226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gershon MD. Nerves, reflexes, and the enteric nervous system: pathogenesis of the irritable bowel syndrome. J Clin Gastroenterol. 2005;39:S184–S193. doi: 10.1097/01.mcg.0000156403.37240.30. [DOI] [PubMed] [Google Scholar]
- 72.Greenwood-Van Meerveld B, Venkova K, Hicks G, Dennis E, Crowell MD. Activation of peripheral 5-HT receptors attenuates colonic sensitivity to intraluminal distension. Neurogastroenterol Motil. 2006;18:76–86. doi: 10.1111/j.1365-2982.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- 73.Coffin B, Farmachidi JP, Rueegg P, Bastie A, Bouhassira D. Tegaserod, a 5-HT4 receptor partial agonist, decreases sensitivity to rectal distension in healthy subjects. Aliment Pharmacol Ther. 2003;17:577–585. doi: 10.1046/j.1365-2036.2003.01449.x. [DOI] [PubMed] [Google Scholar]
- 74.Spiller RC, Jenkins D, Thornley JP, Hebden JM, Wright T, Skinner M, Neal KR. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. 2000;47:804–811. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dunlop SP, Jenkins D, Neal KR, Spiller RC. Relative importance of enterochromaffin cell hyperplasia, anxiety, and depression in postinfectious IBS. Gastroenterology. 2003;125:1651–1659. doi: 10.1053/j.gastro.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 76.Kim HS, Lim JH, Park H, Lee SI. Increased immunoendocrine cells in intestinal mucosa of postinfectious irritable bowel syndrome patients 3 years after acute Shigella infection--an observation in a small case control study. Yonsei Med J. 2010;51:45–51. doi: 10.3349/ymj.2010.51.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang LH, Fang XC, Pan GZ. Bacillary dysentery as a causative factor of irritable bowel syndrome and its pathogenesis. Gut. 2004;53:1096–1101. doi: 10.1136/gut.2003.021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thabane M, Marshall JK. Post-infectious irritable bowel syndrome. World J Gastroenterol. 2009;15:3591–3596. doi: 10.3748/wjg.15.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coates MD, Mahoney CR, Linden DR, Sampson JE, Chen J, Blaszyk H, Crowell MD, Sharkey KA, Gershon MD, Mawe GM, et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology. 2004;126:1657–1664. doi: 10.1053/j.gastro.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 80.Gershon MD. Review article: serotonin receptors and transporters -- roles in normal and abnormal gastrointestinal motility. Aliment Pharmacol Ther. 2004;20 Suppl 7:3–14. doi: 10.1111/j.1365-2036.2004.02180.x. [DOI] [PubMed] [Google Scholar]
- 81.Camilleri M, Andrews CN, Bharucha AE, Carlson PJ, Ferber I, Stephens D, Smyrk TC, Urrutia R, Aerssens J, Thielemans L, et al. Alterations in expression of p11 and SERT in mucosal biopsy specimens of patients with irritable bowel syndrome. Gastroenterology. 2007;132:17–25. doi: 10.1053/j.gastro.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coates MD, Johnson AC, Greenwood-Van Meerveld B, Mawe GM. Effects of serotonin transporter inhibition on gastrointestinal motility and colonic sensitivity in the mouse. Neurogastroenterol Motil. 2006;18:464–471. doi: 10.1111/j.1365-2982.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 83.Kumar S, Ranjan P, Mittal B, Ghoshal UC. Serotonin transporter gene (SLC6A4) polymorphism in patients with irritable bowel syndrome and healthy controls. J Gastrointestin Liver Dis. 2012;21:31–38. [PubMed] [Google Scholar]
- 84.Rijnierse A, Nijkamp FP, Kraneveld AD. Mast cells and nerves tickle in the tummy: implications for inflammatory bowel disease and irritable bowel syndrome. Pharmacol Ther. 2007;116:207–235. doi: 10.1016/j.pharmthera.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 85.De Winter BY, van den Wijngaard RM, de Jonge WJ. Intestinal mast cells in gut inflammation and motility disturbances. Biochim Biophys Acta. 2012;1822:66–73. doi: 10.1016/j.bbadis.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 86.Grundy D. 5-HT system in the gut: roles in the regulation of visceral sensitivity and motor functions. Eur Rev Med Pharmacol Sci. 2008;12 Suppl 1:63–67. [PubMed] [Google Scholar]
- 87.Stead RH, Dixon MF, Bramwell NH, Riddell RH, Bienenstock J. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology. 1989;97:575–585. doi: 10.1016/0016-5085(89)90627-6. [DOI] [PubMed] [Google Scholar]
- 88.Van Nassauw L, Adriaensen D, Timmermans JP. The bidirectional communication between neurons and mast cells within the gastrointestinal tract. Auton Neurosci. 2007;133:91–103. doi: 10.1016/j.autneu.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 89.Coldwell JR, Phillis BD, Sutherland K, Howarth GS, Blackshaw LA. Increased responsiveness of rat colonic splanchnic afferents to 5-HT after inflammation and recovery. J Physiol. 2007;579:203–213. doi: 10.1113/jphysiol.2006.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ohashi K, Sato Y, Kawai M, Kurebayashi Y. Abolishment of TNBS-induced visceral hypersensitivity in mast cell deficient rats. Life Sci. 2008;82:419–423. doi: 10.1016/j.lfs.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 91.Deiteren A, De Man JG, Moreels TG, Pelckmans PA, De Winter BY. Mast cells are involved in post-inflammatory visceral hypersensitivity and exert their effects partially through H1 receptors. Neurogastroenterol Motil. 2012;24:152 (abstract). [Google Scholar]
- 92.Klooker TK, Braak B, Koopman KE, Welting O, Wouters MM, van der Heide S, Schemann M, Bischoff SC, van den Wijngaard RM, Boeckxstaens GE. The mast cell stabiliser ketotifen decreases visceral hypersensitivity and improves intestinal symptoms in patients with irritable bowel syndrome. Gut. 2010;59:1213–1221. doi: 10.1136/gut.2010.213108. [DOI] [PubMed] [Google Scholar]
- 93.Raithel M, Winterkamp S, Pacurar A, Ulrich P, Hochberger J, Hahn EG. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:174–179. doi: 10.1080/003655201750065933. [DOI] [PubMed] [Google Scholar]
- 94.Wang XY, Zarate N, Soderholm JD, Bourgeois JM, Liu LW, Huizinga JD. Ultrastructural injury to interstitial cells of Cajal and communication with mast cells in Crohn’s disease. Neurogastroenterol Motil. 2007;19:349–364. doi: 10.1111/j.1365-2982.2006.00894.x. [DOI] [PubMed] [Google Scholar]
- 95.Barbara G, Stanghellini V, De Giorgio R, Corinaldesi R. Functional gastrointestinal disorders and mast cells: implications for therapy. Neurogastroenterol Motil. 2006;18:6–17. doi: 10.1111/j.1365-2982.2005.00685.x. [DOI] [PubMed] [Google Scholar]
- 96.Fox CC, Lazenby AJ, Moore WC, Yardley JH, Bayless TM, Lichtenstein LM. Enhancement of human intestinal mast cell mediator release in active ulcerative colitis. Gastroenterology. 1990;99:119–124. doi: 10.1016/0016-5085(90)91238-2. [DOI] [PubMed] [Google Scholar]
- 97.Sasaki Y, Tanaka M, Kudo H. Differentiation between ulcerative colitis and Crohn’s disease by a quantitative immunohistochemical evaluation of T lymphocytes, neutrophils, histiocytes and mast cells. Pathol Int. 2002;52:277–285. doi: 10.1046/j.1440-1827.2002.01354.x. [DOI] [PubMed] [Google Scholar]
- 98.Cremon C, Gargano L, Morselli-Labate AM, Santini D, Cogliandro RF, De Giorgio R, Stanghellini V, Corinaldesi R, Barbara G. Mucosal immune activation in irritable bowel syndrome: gender-dependence and association with digestive symptoms. Am J Gastroenterol. 2009;104:392–400. doi: 10.1038/ajg.2008.94. [DOI] [PubMed] [Google Scholar]
- 99.Barbara G, Stanghellini V, De Giorgio R, Cremon C, Cottrell GS, Santini D, Pasquinelli G, Morselli-Labate AM, Grady EF, Bunnett NW, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 100.Barbara G, Wang B, Stanghellini V, de Giorgio R, Cremon C, Di Nardo G, Trevisani M, Campi B, Geppetti P, Tonini M, et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology. 2007;132:26–37. doi: 10.1053/j.gastro.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 101.Hughes PA, Brierley SM, Martin CM, Liebregts T, Persson J, Adam B, Holtmann G, Blackshaw LA. TRPV1-expressing sensory fibres and IBS: links with immune function. Gut. 2009;58:465–466. doi: 10.1136/gut.2008.161760. [DOI] [PubMed] [Google Scholar]
- 102.Stenton GR, Nohara O, Déry RE, Vliagoftis H, Gilchrist M, Johri A, Wallace JL, Hollenberg MD, Moqbel R, Befus AD. Proteinase-activated receptor (PAR)-1 and -2 agonists induce mediator release from mast cells by pathways distinct from PAR-1 and PAR-2. J Pharmacol Exp Ther. 2002;302:466–474. doi: 10.1124/jpet.302.2.466. [DOI] [PubMed] [Google Scholar]
- 103.Vergnolle N. Protease-activated receptors as drug targets in inflammation and pain. Pharmacol Ther. 2009;123:292–309. doi: 10.1016/j.pharmthera.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 104.Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis. 2003;9:224–229. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 105.Vergnolle N. Clinical relevance of proteinase activated receptors (pars) in the gut. Gut. 2005;54:867–874. doi: 10.1136/gut.2004.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, Steinhoff M, Barbara G, Beck P, Bunnett NW, et al. Role for protease activity in visceral pain in irritable bowel syndrome. J Clin Invest. 2007;117:636–647. doi: 10.1172/JCI29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bradesi S, Herman J, Mayer EA. Visceral analgesics: drugs with a great potential in functional disorders? Curr Opin Pharmacol. 2008;8:697–703. doi: 10.1016/j.coph.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cenac N, Altier C, Motta JP, d’Aldebert E, Galeano S, Zamponi GW, Vergnolle N. Potentiation of TRPV4 signalling by histamine and serotonin: an important mechanism for visceral hypersensitivity. Gut. 2010;59:481–488. doi: 10.1136/gut.2009.192567. [DOI] [PubMed] [Google Scholar]
- 111.Cattaruzza F, Spreadbury I, Miranda-Morales M, Grady EF, Vanner S, Bunnett NW. Transient receptor potential ankyrin-1 has a major role in mediating visceral pain in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G81–G91. doi: 10.1152/ajpgi.00221.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharkey KA, Mawe GM. Neuroimmune and epithelial interactions in intestinal inflammation. Curr Opin Pharmacol. 2002;2:669–677. doi: 10.1016/s1471-4892(02)00215-1. [DOI] [PubMed] [Google Scholar]
- 113.Delafoy L, Raymond F, Doherty AM, Eschalier A, Diop L. Role of nerve growth factor in the trinitrobenzene sulfonic acid-induced colonic hypersensitivity. Pain. 2003;105:489–497. doi: 10.1016/S0304-3959(03)00266-5. [DOI] [PubMed] [Google Scholar]
- 114.di Mola FF, Friess H, Zhu ZW, Koliopanos A, Bley T, Di Sebastiano P, Innocenti P, Zimmermann A, Büchler MW. Nerve growth factor and Trk high affinity receptor (TrkA) gene expression in inflammatory bowel disease. Gut. 2000;46:670–679. doi: 10.1136/gut.46.5.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rühl A. Glial cells in the gut. Neurogastroenterol Motil. 2005;17:777–790. doi: 10.1111/j.1365-2982.2005.00687.x. [DOI] [PubMed] [Google Scholar]
- 116.Vanden Berghe P, Tack J, Boesmans W. Highlighting synaptic communication in the enteric nervous system. Gastroenterology. 2008;135:20–23. doi: 10.1053/j.gastro.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 117.De Filippis D, Esposito G, Cirillo C, Cipriano M, De Winter BY, Scuderi C, Sarnelli G, Cuomo R, Steardo L, De Man JG, et al. Cannabidiol reduces intestinal inflammation through the control of neuroimmune axis. PLoS One. 2011;6:e28159. doi: 10.1371/journal.pone.0028159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neunlist M, Van Landeghem L, Bourreille A, Savidge T. Neuro-glial crosstalk in inflammatory bowel disease. J Intern Med. 2008;263:577–583. doi: 10.1111/j.1365-2796.2008.01963.x. [DOI] [PubMed] [Google Scholar]
- 119.Cirillo C, Sarnelli G, Esposito G, Grosso M, Petruzzelli R, Izzo P, Calì G, D’Armiento FP, Rocco A, Nardone G, et al. Increased mucosal nitric oxide production in ulcerative colitis is mediated in part by the enteroglial-derived S100B protein. Neurogastroenterol Motil. 2009;21:1209–e112. doi: 10.1111/j.1365-2982.2009.01346.x. [DOI] [PubMed] [Google Scholar]
- 120.Roberts PJ, Morgan K, Miller R, Hunter JO, Middleton SJ. Neuronal COX-2 expression in human myenteric plexus in active inflammatory bowel disease. Gut. 2001;48:468–472. doi: 10.1136/gut.48.4.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 122.Holzer P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol Ther. 2011;131:142–170. doi: 10.1016/j.pharmthera.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brierley SM, Jones RC, Xu L, Gebhart GF, Blackshaw LA. Activation of splanchnic and pelvic colonic afferents by bradykinin in mice. Neurogastroenterol Motil. 2005;17:854–862. doi: 10.1111/j.1365-2982.2005.00710.x. [DOI] [PubMed] [Google Scholar]
- 124.Jones RC, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–10989. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blackshaw LA, Brierley SM, Hughes PA. TRP channels: new targets for visceral pain. Gut. 2010;59:126–135. doi: 10.1136/gut.2009.179523. [DOI] [PubMed] [Google Scholar]
- 126.Yiangou Y, Facer P, Dyer NH, Chan CL, Knowles C, Williams NS, Anand P. Vanilloid receptor 1 immunoreactivity in inflamed human bowel. Lancet. 2001;357:1338–1339. doi: 10.1016/s0140-6736(00)04503-7. [DOI] [PubMed] [Google Scholar]
- 127.Akbar A, Yiangou Y, Facer P, Walters JR, Anand P, Ghosh S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923–929. doi: 10.1136/gut.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Högestätt ED, Julius D, Jordt SE, Zygmunt PM. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Namer B, Kleggetveit IP, Handwerker H, Schmelz M, Jorum E. Role of TRPM8 and TRPA1 for cold allodynia in patients with cold injury. Pain. 2008;139:63–72. doi: 10.1016/j.pain.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 130.Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O’Donnell TA, Cooper NJ, Harrington AM, Adam B, Liebregts T, et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology. 2009;137:2084–2095.e3. doi: 10.1053/j.gastro.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, Woods CG, Jones NG, Paterson KJ, Fricker FR, et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Andrade EL, Ferreira J, André E, Calixto JB. Contractile mechanisms coupled to TRPA1 receptor activation in rat urinary bladder. Biochem Pharmacol. 2006;72:104–114. doi: 10.1016/j.bcp.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 133.Akopian AN. Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol. 2011;12:89–94. doi: 10.2174/138920111793937952. [DOI] [PubMed] [Google Scholar]
- 134.Vermeulen W, De Man JG, De Schepper HU, Bult H, Moreels TG, Pelckmans PA, De Winter BY. Role of TRPV1 and TRPA1 in visceral hypersensitivity to colorectal distension during experimental colitis in rats. Eur J Pharmacol. 2013;698:404–412. doi: 10.1016/j.ejphar.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 135.Malykhina AP, Qin C, Greenwood-van Meerveld B, Foreman RD, Lupu F, Akbarali HI. Hyperexcitability of convergent colon and bladder dorsal root ganglion neurons after colonic inflammation: mechanism for pelvic organ cross-talk. Neurogastroenterol Motil. 2006;18:936–948. doi: 10.1111/j.1365-2982.2006.00807.x. [DOI] [PubMed] [Google Scholar]
- 136.Chitkara DK, van Tilburg MA, Blois-Martin N, Whitehead WE. Early life risk factors that contribute to irritable bowel syndrome in adults: a systematic review. Am J Gastroenterol. 2008;103:765–774; quiz 775. doi: 10.1111/j.1572-0241.2007.01722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brumovsky PR, Gebhart GF. Visceral organ cross-sensitization - an integrated perspective. Auton Neurosci. 2010;153:106–115. doi: 10.1016/j.autneu.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Agostini A, Filippini N, Cevolani D, Agati R, Leoni C, Tambasco R, Calabrese C, Rizzello F, Gionchetti P, Ercolani M, et al. Brain functional changes in patients with ulcerative colitis: a functional magnetic resonance imaging study on emotional processing. Inflamm Bowel Dis. 2011;17:1769–1777. doi: 10.1002/ibd.21549. [DOI] [PubMed] [Google Scholar]
- 139.Coffin B, Bouhassira D, Sabaté JM, Barbe L, Jian R. Alteration of the spinal modulation of nociceptive processing in patients with irritable bowel syndrome. Gut. 2004;53:1465–1470. doi: 10.1136/gut.2003.031310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mayer EA, Aziz Q, Coen S, Kern M, Labus JS, Lane R, Kuo B, Naliboff B, Tracey I. Brain imaging approaches to the study of functional GI disorders: a Rome working team report. Neurogastroenterol Motil. 2009;21:579–596. doi: 10.1111/j.1365-2982.2009.01304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Van Oudenhove L, Coen SJ, Aziz Q. Functional brain imaging of gastrointestinal sensation in health and disease. World J Gastroenterol. 2007;13:3438–3445. doi: 10.3748/wjg.v13.i25.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Smith JK, Humes DJ, Head KE, Bush D, White TP, Stevenson CM, Brookes MJ, Marciani L, Spiller RC, Gowland PA, et al. fMRI and MEG analysis of visceral pain in healthy volunteers. Neurogastroenterol Motil. 2011;23:648–e260. doi: 10.1111/j.1365-2982.2011.01712.x. [DOI] [PubMed] [Google Scholar]
- 143.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth Analg. 2003;97:1108–1116. doi: 10.1213/01.ANE.0000081061.12235.55. [DOI] [PubMed] [Google Scholar]
- 145.Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 146.Lin Y, Roman K, Foust KD, Kaspar BK, Bailey MT, Stephens RL. Glutamate Transporter GLT-1 Upregulation Attenuates Visceral Nociception and Hyperalgesia via Spinal Mechanisms Not Related to Anti-Inflammatory or Probiotic Effects. Pain Res Treat. 2011;2011:507029. doi: 10.1155/2011/507029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 148.Reinold H, Ahmadi S, Depner UB, Layh B, Heindl C, Hamza M, Pahl A, Brune K, Narumiya S, Müller U, et al. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J Clin Invest. 2005;115:673–679. doi: 10.1172/JCI200523618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cao J, Yang X, Liu YN, Suo ZW, Shi L, Zheng CR, Yang HB, Li S, Hu XD. GABAergic disinhibition induced pain hypersensitivity by upregulating NMDA receptor functions in spinal dorsal horn. Neuropharmacology. 2011;60:921–929. doi: 10.1016/j.neuropharm.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 150.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 151.Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27:2019–2031. doi: 10.1185/03007995.2011.614934. [DOI] [PubMed] [Google Scholar]
- 152.Bradesi S. Role of spinal cord glia in the central processing of peripheral pain perception. Neurogastroenterol Motil. 2010;22:499–511. doi: 10.1111/j.1365-2982.2010.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Al-Chaer ED, Kawasaki M, Pasricha PJ. A new model of chronic visceral hypersensitivity in adult rats induced by colon irritation during postnatal development. Gastroenterology. 2000;119:1276–1285. doi: 10.1053/gast.2000.19576. [DOI] [PubMed] [Google Scholar]
- 154.Harrington AM, Brierley SM, Isaacs N, Hughes PA, Castro J, Blackshaw LA. Sprouting of colonic afferent central terminals and increased spinal mitogen-activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J Comp Neurol. 2012;520:2241–2255. doi: 10.1002/cne.23042. [DOI] [PubMed] [Google Scholar]
- 155.Gao J, Wu X, Owyang C, Li Y. Enhanced responses of the anterior cingulate cortex neurones to colonic distension in viscerally hypersensitive rats. J Physiol. 2006;570:169–183. doi: 10.1113/jphysiol.2005.096073. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 156.Azpiroz F. Gastrointestinal perception: pathophysiological implications. Neurogastroenterol Motil. 2002;14:229–239. doi: 10.1046/j.1365-2982.2002.00324.x. [DOI] [PubMed] [Google Scholar]
- 157.Bulmer DC, Grundy D. Achieving translation in models of visceral pain. Curr Opin Pharmacol. 2011;11:575–581. doi: 10.1016/j.coph.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 158.Rao SS, Read NW, Davison PA, Bannister JJ, Holdsworth CD. Anorectal sensitivity and responses to rectal distention in patients with ulcerative colitis. Gastroenterology. 1987;93:1270–1275. doi: 10.1016/0016-5085(87)90255-1. [DOI] [PubMed] [Google Scholar]
- 159.Faure C, Giguère L. Functional gastrointestinal disorders and visceral hypersensitivity in children and adolescents suffering from Crohn’s disease. Inflamm Bowel Dis. 2008;14:1569–1574. doi: 10.1002/ibd.20506. [DOI] [PubMed] [Google Scholar]
- 160.Vergnolle N. Postinflammatory visceral sensitivity and pain mechanisms. Neurogastroenterol Motil. 2008;20 Suppl 1:73–80. doi: 10.1111/j.1365-2982.2008.01110.x. [DOI] [PubMed] [Google Scholar]
- 161.Tillisch K, Labus JS. Advances in imaging the brain-gut axis: functional gastrointestinal disorders. Gastroenterology. 2011;140:407–411.e1. doi: 10.1053/j.gastro.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Larsson MB, Tillisch K, Craig AD, Engström M, Labus J, Naliboff B, Lundberg P, Ström M, Mayer EA, Walter SA. Brain responses to visceral stimuli reflect visceral sensitivity thresholds in patients with irritable bowel syndrome. Gastroenterology. 2012;142:463–472.e3. doi: 10.1053/j.gastro.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, Brant SR, Chakravarti S, Kwon JH. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135:1624–1635.e24. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 164.Zhou Q, Souba WW, Croce CM, Verne GN. MicroRNA-29a regulates intestinal membrane permeability in patients with irritable bowel syndrome. Gut. 2010;59:775–784. doi: 10.1136/gut.2009.181834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brain O, Owens BM, Pichulik T, Allan P, Khatamzas E, Leslie A, Steevels T, Sharma S, Mayer A, Catuneanu AM, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39:521–536. doi: 10.1016/j.immuni.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 166.Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology. 2013;38:898–906. doi: 10.1016/j.psyneuen.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 167.Sharma A, Lelic D, Brock C, Paine P, Aziz Q. New technologies to investigate the brain-gut axis. World J Gastroenterol. 2009;15:182–191. doi: 10.3748/wjg.15.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pellissier S, Dantzer C, Canini F, Mathieu N, Bonaz B. Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology. 2010;35:653–662. doi: 10.1016/j.psyneuen.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 169.Wilder-Smith CH. The balancing act: endogenous modulation of pain in functional gastrointestinal disorders. Gut. 2011;60:1589–1599. doi: 10.1136/gutjnl-2011-300253. [DOI] [PubMed] [Google Scholar]