

Abstract
A method for carrying out the intramolecular Schmidt reaction of alkyl azides and ketones using a substoichiometric amount of catalyst is reported. Following extensive screening, the use of the strong hydrogen bond donating solvent hexafluoro-2-propanol was found to be consistent with low catalyst loadings, which range from 2.5 mol% for favorable substrates to 25 mol% for more difficult cases. Reaction optimization, broad substrate scope, and preliminary mechanistic studies of this improved version of the reaction are described.
INTRODUCTION
The intramolecular Schmidt reaction is a useful method for the preparation of lactams from azidoalkyl ketones1,2 that has been applied to alkaloid synthesis and natural product-inspired libraries.3 One limitation of the reaction has been the requirement of excess Lewis or Brønsted acid1a,4 in order to achieve complete conversion, which often renders it unsuitable for strongly acid-sensitive substrates and limits scalability. In addition, a version of this reaction that would employ vastly smaller amounts of metal may well be cleaner and more efficient, even as it minimized the generation of metal waste.5 Two representative examples are shown in Figures 1a and 1b. Indeed, we are unaware of any examples that proceed to high conversion with less than a full equivalent of promoter. This can be attributed to strong product inhibition, which is intrinsic to any reaction that converts a ketone to an amide. The first step in a hypothetical catalytic cycle for the intramolecular Schmidt reaction is the activation of a substrate S with a Lewis or Brønsted acid LA to form complex S–LA (Figure 1c).6 The tethered azide then attacks the activated carbonyl, forming the azidohydrin intermediate A, which upon antiperiplanar bond migration and nitrogen extrusion results in the formation of a product P. The lactam produced is strongly Lewis-basic and sequesters the catalyst in an unproductive manner. We propose that this unfavorable catalyst-product interaction results in product inhibition deterring the progress of reaction and necessitating the use of super-stoichiometric amount of catalyst. 4a,7
Figure 1.

(a) and (b) Examples of intramolecular Schmidt reactions requiring >1 equiv catalyst and (c) hypothetical catalytic cycle displaying product inhibition.
A fundamental challenge in designing a catalytic variant for this reaction lies in the inherent strength of the complex formed between the catalyst and the product, which is a hard acid–hard base interaction. Related reactions such as the Beckmann rearrangement and Ritter reaction, which generate amide or lactam products, have also suffered in the past from the requirement of a stoichiometric amount of strong acids and harsh reaction conditions.8,10 The role of lactam in product inhibition has been demonstrated for Beckmann rearrangement using a microchemical system.9 However, recent catalytic developments for these reactions have allowed for the use of substoichiometric amounts of Brønsted or Lewis acid, improving efficiency and expanding scope of those processes.8,10 The use of ionic liquids11 and extensive screening of catalysts and solvents led to the realization of these catalytic reactions. We envisioned that catalysis in the intramolecular Schmidt reaction might be more efficient if condition were identified wherein a ligand, solvent, or additive is capable of competing with the catalyst in forming a complex with the Lewis-basic lactam, thus allowing catalyst turnover. Herein, we disclose a first report of the catalytic intramolecular Schmidt reaction that is superior in essentially every way to the version that we and others have been exploring since 1991.1,2d,4
RESULTS AND DISCUSSIONS
Screening
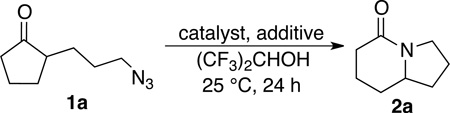
We sought to replace the stoichiometric Schmidt reaction by identifying conditions that would (1) require low, sub-stoichiometric amounts of catalyst, (2) be mild, efficient, and proceed at room temperature, and (3) would have broad substrate scope. We initially focused on catalyst and additive screening. Early on, we found that 10–25 mol% of scandium(III) triflate could efficiently promote the reaction of 1c to 2c, but only at unacceptably high temperatures (Scheme 1; see Supporting Information for details of these and all other early attempts). Moreover, these reaction conditions were plagued with extremely limited substrate scope and low yields. For example, higher catalyst loadings were generally necessary for cyclopentanone 1a (we had in the interim found that MeCN was a better solvent than H2O, either alone or with phase-transfer catalysts) and the reaction of 1d under the same conditions failed (<5% product yields). Reactions of substrates like 1a or 1d require longer reaction times than 1c in the stoichiometric reaction and are often poorer yielding as well.1,12
Scheme 1.

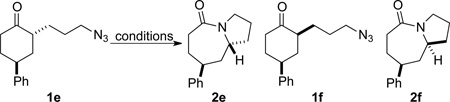
Based on these preliminary results, we decided to expand our search by focusing on three screening parameters: solvent, catalyst, and temperature. trans-4-Phenyl-2-(3-azidopropyl)cyclohexanone 1e was chosen as a test example to probe several issues known to arise in intramolecular Schmidt reactions (Scheme 2). The trans isomer was primarily chosen to probe for epimerization (known to be a problem in some applications),13 which could lead to the thermodynamically more stable cis ketone 1f; the read-out for this process would be the detection of lactam 2f following ring expansion. In addition, trans-1e is capable of generating either a fused lactam 2e or a bridged isomer (3e) by migration of different α-carbons.1b Finally, the phenyl chromophore in 1e allowed faster analyses and quantification of reaction mixtures by UPLC (Supporting Information).
Scheme 2.

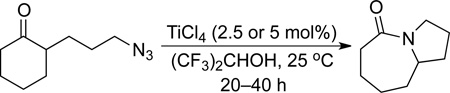
Figure 2 depicts the results of preliminary screening of reaction conditions (see Supporting Information for details). Examination of 23 different solvents was first carried out using 20 mol% of Sc(OTf)3 at 150 °C. Only five solvents (nitromethane, benzonitrile, acetic acid, trifluoroethanol (TFE), and the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate) gave product in high yields. When the Sc(OTf)3 loading was reduced to 10 mol%, only TFE resulted in complete conversion. We then focused our attention on catalyst screening using 10 mol% of catalyst with TFE as a solvent at 80 °C. In total, 51 catalysts were screened, which included 44 Lewis acids that represented 31 different elements and 7 Brønsted acids. Of these, a number of transition metals such as TiCl4, ZrCl4, and Fe(OTf)3, some post-transition metals like In(OTf)3 and Bi(OTf)3, and metalloids such as SiCl4 and SbCl5 have results that were good enough for further screening. Further evaluation at 10 mol% loading of these selected catalysts in TFE at lower temperatures (50 and 25 °C) revealed TiCl4 and SiCl4 to be most effective. The identification of TiCl4 was notable, as it has been a catalyst of choice for many stoichiometric intramolecular Schmidt reactions.1b,4,14
Figure 2.

Screening flowchart. See Supporting Information for details. Transition metals are depicted in deep red, post-transition metals in green, and metalloids in blue.
Identification of TFE as nearly unique in permitting catalyst turnover prompted us to more completely examine the effect of solvents using 1e as the substrate and 10 mol% of TiCl4 as catalyst (Table 1). Again, TFE was observed to give the best results with respect to both conversion and stereochemical retention (cf. entries 1–3 with entry 4). The results with TFE prompted us to consider other fluorinated alcohols, specifically hexafluoro-2-propanol (HFIP). Compared to their non-fluorinated alcohol analogues, TFE and HFIP have low nucleophilicity, low pKa, high ionizing power, high polarity, ability to solvate anions, and are strong hydrogen bond donors.15 Accordingly, they are often used as solvent, co-solvent, or a Lewis acid substitute15e,16 in oxidations,17 or in ring opening reactions of oxiranes, cycloaddition, and deprotection reactions.15b,l5d,l5f Their utility has been attributed to the strong hydrogen bond donor ability of these solvents.15b,c,17b,18 Moreover, the use of these solvents to denature proteins and induce α-helical secondary structures provided some ancillary expectation that they might prove useful in modifying the ability of our product lactams to coordinate with acid promoters.19 Owing to the strong H-bond donor ability and high ionizing power of HFIP compared to TFE; HFIP often provides superior results both in reaction rate enhancement15b,15e,16,20 and as a helix-inducing co-solvent.19a
Table 1.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | catalyst | catalyst loading (mol%) |
solvents | additives | temp (°C) |
time (h) |
% yield (2e:2f)c | % recovery (1e:1f)d |
| 1 | TiCl4 | 10 | CH2Cl2 | - | 25 | 18 | 6% (40:60) | 84% (10:90) |
| 2 | TiCl4 | 10 | i-PrOH | - | 37 | 18 | trace | 86% (3:97) |
| 3 | TiCl4 | 10 | CH3CN | - | 37 | 18 | 41% (15:85) | 47% (1:99) |
| 4 | TiCl4 | 10 | CF3CH2OH | - | 25 | 18 | 79% (82:18)d | trace |
| 5 | none | - | (CF3)2CHOH | - | 37 | 18 | ND | 93% (98:2) |
| 6 | TiCl4 | 10 | CH3CN | (CF3)2CHOHe | 25 | 18 | 34% (10:90) | 61% (5:95) |
| 7 | TiCl4 | 10 | (CF3)2CHOH | - | 25 | 12 | 91% (98:2)f | ND |
| 8 | TiCl4 | 5 | (CF3)2CHOH | - | 25 | 38 | 89% (99:1)f | trace |
| 9 | TiCl4g | 5 | (CF3)2CHOH | - | 25 | 38 | 86% (98:2) | trace |
| 10 | TiCl4h | 5 | (CF3)2CHOH | - | 25 | 38 | 89% (98:2) | ND |
| 11 | TiCl4 | 5 | (CF3)2CHOH | DTBMPi | 25 | 38 | 52% (98:2) | 19% (98:2) |
| 12 | TiCl4 | 5 | (CF3)2CHOH | DTBMPj | 25 | 38 | 21% (99:1) | 50% (96:4)b |
| 13 | SiCl4 | 5 | (CF3)2CHOH | - | 25 | 38 | 86% (98:2)f | ND |
| 14 | Sc(OTf)3 | 5 | (CF3)2CHOH | - | 25 | 38 | 28% (97:3) | 61% (98:2) |
| 15 | HCl | 10 | (CF3)2CHOH | - | 25 | 38 | 40% (96:4)f | 46% (98:2) |
| 16 | HCl | 20 | (CF3)2CHOH | - | 25 | 38 | 78% (97:3)f | trace |
| 17 | CF3COOH | 10 | (CF3)2CHOH | - | 25 | 38 | 62% (97:3) | 34% (98:2) |
| 18 | (S)-BNDHP | 5 | (CF3)2CHOH | - | 25 | 38 | 32% (96:4)k | 62% (98:2) |
| 19 | Ti(iOPr)4 | 10 | (CF3)2CHOH | - | 25 | 38 | trace | 93% (95:5) |
To a solution of substrate 1e (0.1 mmol) in solvent (0.5 mL) at room temperature was added a catalyst under nitrogen or argon atmosphere unless otherwise mentioned (see Supporting Information for the complete optimization table). Throughout, 1.0 M solutions of TiCl4 or SiCl4 in CH2Cl2 were used. 2.0 M solution of HCl in diethyl ether was used. ND = Not detected.
Concentration ca. 0.2 M unless otherwise mentioned.
Isolated yield after preparative TLC purification; ratio determined by 1H NMR.
Isolated yield after preparative TLC purification; ratio determined by UPLC of the crude reaction mixture.
1 equiv of (CF3)2CHOH was added.
Bridged lactam 3e was also isolated in ca. 1–4% yield.
Concentration ca. 0.4 M.
Concentration ca. 0.1 M.
10 mol% of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) was used as a Brønsted acid scavenger.
20 mol% of DTBMP was used.
(S)-BNDHP = (S)- (+)-1,1’-Binaphthyl-2,2’-diyl hydrogenphosphate. No kinetic resolution was observed.
Using HFIP as a substitute for TiCl4 in a control experiment did not afford any product and substrate 1e was recovered almost quantitatively (entry 5). Using one equivalent of HFIP as an additive with CH3CN as a solvent did not improve the yield (entry 6). However, when HFIP was used as solvent in combination with 10 mol% of TiCl4, complete conversion and negligible epimerization was observed with increased catalyst turnover compared to TFE (cf. entries 7 and 4). Lowering the catalyst loading to 5 mol% of TiCl4 produced similar results as with 10 mol% of TiCl4 but at the expense of longer reaction times (entry 8). Changing the concentration of reaction mixture had minimal effect on yield (entries 9 and 10).
We speculated that reaction of HFIP with TiCl4 might generate HCl in situ along with Ti[OCH (CF3)2]4. If so, then 5 mol% of TiCl4 should be capable of generating 20 mol% of HCl in situ. To test this hypothesis, we ran the reaction in the presence of 10 and 20 mol% of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) as a proton scavenger (entries 11 and 12). Significant catalyst inhibition was observed resulting in lower yields but reaction to some extent was still observed when 20 mol% of DTBMP was used. This could mean that the catalytically active species is in situ generated HCl or that DTBMP, being a base, is having some other deleterious effect on the reaction.21 Reaction in HFIP with SiCl4 provided lactam in good yield but Sc(OTf)3 provided product in only 28% yield (entries 13 and 14). The reaction with Brønsted acids (5–20 mol%) delivered comparative lower yield of the product than 5 mol% of TiCl4 (entries 15–18). Interestingly, reaction with 20 mol% of HCl in ether gave a lower yield compared to 5 mol% of TiCl4 (cf. entries 8 and 16). The use of a chiral phosphoric acid4b neither provided good yield nor led to any degree of kinetic resolution (entry 18). Reaction with Ti(iOPr)4 resulted in only a trace amount of product with quantitative recovery of substrate 1e (entry 19). Although this supported our supposition that in situ generated HCl could be the active catalyst, it was hard to reconcile with the reduced yield obtained with HCl added as a solution in ether, possibly due to concentration errors in the commercial product (see Table 4 and associated discussion for more on this point).
Table 4.
 | |||||
|---|---|---|---|---|---|
| entry | Catalysts | catalyst (mol%) |
additive | additive (mol%) |
NMR ratio of 2a:lac |
| 1 | TiCl4 | 20 | - | - | 95:5 |
| 2 | TiCl4 | 10 | - | - | 59:41 |
| 3 | TiF4 | 10 | - | - | 26:74 |
| 4 | TiBr4 | 10 | - | - | 46:54 |
| 5 | Ti(iOPr)4 | 10 | - | - | 0:100 |
| 6 | SiCl4 | 10 | - | - | 58:42 |
| 7 | SbCl5 | 10 | - | - | 45:55 |
| 8 | NbCl5 | 10 | - | - | 44:56 |
| 9 | WC16 | 10 | - | - | 54:46 |
| 10 | HC1 in etherd | 40 | - | - | 45:55 |
| 11 | Aqueous HCle | 40 | - | - | 59:41 |
| 12 | HC1 in HFIPf | 40 | - | - | 57:43–75:25 (variable) |
| 13 | H2SO4 | 40 | - | - | 76:24 |
| 14 | - | - | CF3SO3H | 20 | 49:51 |
| 15 | TiCl4 | 5 | CF3SO3H | 5 | 42:58 |
| 16 | TiCl4 | 5 | CISO3H | 5 | 49:51 |
| 17 | TiCl4 | 10 | AgOTfg | 20 | 56:44 |
| 18 | TiCl4 | 10 | Al(iOPr)3 | 20 | 50:50 |
| 19 | TiCl4 | 10 | Silica gelh | - | 47:53 |
| 20 | TiCl4 | 10 | CH3COCli | 40 | 95:5 |
| 21 | - | - | CH3COCli | 40 | 58:42 |
| 22 | - | - | CH3COCli | 40 | 70:30 |
| 23 | - | - | CH3COCli | 70 | 90:10 |
| 24 | - | - | CH3COCli | 80 | 94:6 |
| 25 | - | - | CH3COClj | 80 | 97:3k |
| 26 | - | - | CH3COBrl | 40 | 72:28 |
| 27 | - | - | CH3COBrl | 80 | 98:2m |
| 28 | TiCl4 | 10 | (CH3)3SiCl | 40 | 89:11 |
| 29 | - | - | (CH3)3SiCl | 80 | 92:8 |
| 30 | - | - | (CH3)3SiI | 80 | 13:87n |
To a solution of substrate 1a (0.1 mmol) in (CF3)2CHOH (0.5 mL) at room temperature was added a catalyst and/or an additive under nitrogen atmosphere unless otherwise mentioned. Throughout, 1.0 M solutions of TiCl4, SiCl4, or SbCl5 in CH2Cl2 were used.
Concentration ca. 0.2 M.
1H NMR ratio determined after a brief work-up (see Supporting Information for details).
A 1.0 M solution of HCl in ether (commercial) was used.
Aqueous HCl (37%) was used.
A ca. 0.105–0.116 M solution of HCl in hexafluoro-2-propanol was prepared and used immediately.
TiCl2(OTf)2 was generated in situ from TiCl4 and AgOTf.24
Silica gel (50 mg) was added.
An old (> 5 years since being opened) container of acetyl chloride was used.
A new container of acetyl chloride was used.
The ratio of 2a:1a did not change between 18–24 h.
A new container of acetyl bromide was used.
The ratio of 2a:1a did not change between 18–24 h.
Several other unidentified byproducts/impurities were also observed.
Scope
Having identified conditions that satisfied our goals, we sought to determine the scope of this substoichiometric, catalytic Schmidt reaction. We began with cyclohexanone-derived azidoketones, as previous experience has taught us that these are in general the most facile substrates (Table 2).1 Indeed, the results obtained were in general as good as or better than those obtained using the stoichiometric reactions. Thus, transformations of 1c and cis-1f required only 2.5 mol% of TiCl4 (entries 1 and 2), whereas trans-1e required 5 mol% of TiCl4 and longer reaction time to obtain slightly lower yields of product (entry 3). The reaction of the 1,3-diketone 1b proceeded in higher yield than reported in the literature (entry 4, cf. Figure 1b)4 while the α-ester-substituted 1d, which failed in the preliminary screening (Scheme 1), afforded an excellent yield of 2d using the optimized protocol (entry 5). Other functionalized cyclohexanones such as β-tetralone 1g and allylic azide 1h also provided good yields of the corresponding lactams 2g and 2h (entries 6 and 7).
Table 2.
Initial Substrate Scope for the catalytic intramolecular Schmidt reaction on cyclohexanone-derived azidoketones.a,b
 | |||||
|---|---|---|---|---|---|
| entry | substrate | catalyst loading (mol%) |
time (h) |
product | yieldc (%) |
| 1 |  |
2.5 | 20 | 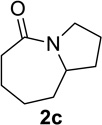 |
94 |
| 2 | 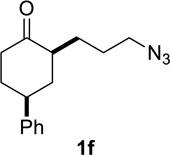 |
2.5 | 20 | 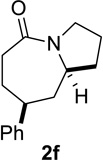 |
98 |
| 3 | 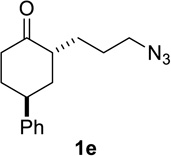 |
5 | 38 | 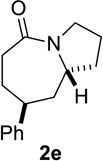 |
87d |
| 4 | 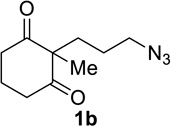 |
5 | 24 | 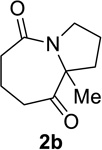 |
94 |
| 5 | 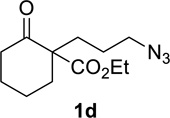 |
5 | 40 | 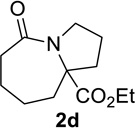 |
94 |
| 10 | 18 | 95 | |||
| 6 | 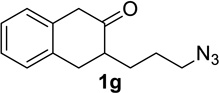 |
5 | 24 | 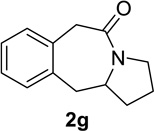 |
89 |
| 7 | 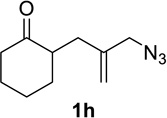 |
5 | 24 | 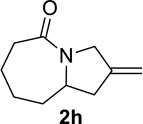 |
84 |
To a solution of a substrate (0.4 mmol) in hexafluoro-2-propanol (2.0 mL) at room temperature was added TiCl4 under nitrogen atmosphere and reaction was allowed to stirred at 25 °C for a designated period unless otherwise mentioned.
Concentration ca. 0.2 M.
Isolated yield.
Bridged lactam 3e was also isolated in ca. 2% yield.
We next examined a broader range of ketone types, including some that we have found challenging under previously established reaction conditions. Although the substrate scope was broad, some recalcitrant substrates generally required higher catalyst loadings compared to cyclohexanonederived azides. For example, cyclopentanone 1a afforded a superior yield of indolizidinone 2a, a structural motif found in many pharmacologically relevant alkaloids, with 20 mol% of TiCl4 (entry 1). The reaction of seven and eight-membered azidoketones afforded lactams of medium-ring sizes in high yields (entries 2 and 3) and the norcamphor-derived 1k provided a good yield of tricyclic lactam 2k with 25 mol% of TiCl4 (entry 4). N-Substituted pyrrolidinones were obtained in good yields from acyclic azidoketones (entries 5 and 6), whereas benzylic azide 1n provided a mixture of two regioisomers 2n and 3n in 4:1 ratio in modest yield with 15 mol% of TiCl4 (entry 7).
Substrate 1o containing a tertiary amine – a possible additional source of catalyst inactivation – required 35 mol% of TiCl4 to provide pyrrolodiazepinone 2o (entry 8).22 Typically, for the intramolecular Schmidt reaction, nitrogen gas evolution is observed immediately upon addition of the catalyst. However, when TiCl4 was added slowly to a solution of substrate 1o in HFIP, a yellow precipitate was initially observed, with effervescence only commencing upon the addition of 25 mol% of TiCl4.23 This observation suggests that the initial 25 mol% of TiCl4, capable of generating 100 mol% of HCl, formed a salt with the basic amine and the remaining 10 mol% of TiCl4 was responsible for the desired transformation into lactam 2o. Azidoaldehyde 1p only required 5 mol% of TiCl4 to provide 3-benzylpyrrolidinone 2p in good yield (entry 9). Unfortunately, extending the tether length between carbonyl and the azide moiety from the usual four to five carbons resulted in a sluggish reaction with only 11% of lactam 2q being obtained, even when 20 mol% of TiCl4 was employed (entry 10). This is consistent with the stringent dependence of the intramolecular Schmidt reaction on tether length observed since the initial discovery of the reaction.1,2d
Given the requirement of relatively high catalyst loading for these less reactive substrates, we sought to further optimize our reaction conditions using substrate 1a (Table 4). After evaluation of a series of Lewis and Brønsted acids, TiCl4 was still found to be the most effective catalyst for this substrate (entries 1–14). However, the combination of TiCl4 with other Lewis or Brønsted acids, while not initially promising (entries 15–20 and 28), ultimately revealed acetyl chloride (CH3COCl) as an effective promoter of this reaction even in the absence of TiCl4 (entries 21–25). Thus, reaction with 80 mol% of acetyl chloride gave comparable results as did 20 mol% of TiCl4 (cf. entries 1 and 25). We realized that this would support the case that HCl is the active catalytic species, provided we could show that HFIP was capable of generating HCl from acetyl chloride (an ironic notion given the low nucleophilicity of HFIP15b). To address this, we combined one 1 equiv of acetyl chloride (AcCl) and 2 equiv of HFIP and monitored the reaction by 1H NMR in CDCl3 (Figure 3; see Supporting Information for details). Within 6 min, ca. 50% conversion to HFIP acetate was observed. The rate slowed down after 20 min and the reaction took 4 h to reach >95% conversion. Conversely, we were not able to obtain any evidence for the in situ generation of HCl from TiCl4.
Figure 3.

Reaction monitoring of acetyl chloride with HFIP for the in situ generation of HCl by 1H NMR.
Additional experiments were carried out to gather further detail about the effect of various sources of H+ on these Schmidt reactions. In our initial survey, we had first tried adding HCl in ether to the HFIP solvent (Table 1, entries 15 and 16, and Table 4, entry 10). Neither that method nor adding aqueous HCl15f (Table 4, entry 11) gave good results in our hands. On the other hand, when HCl gas was separately generated and infused into the HFIP (Table 4, entry 12), a range of results were obtained. The non-reproducibility of these experiments can be blamed on the ease with which the HCl gas escapes the solution, making it difficult to accurately gauge exactly how much acid is present in a particular experiment. For example, markedly reduced yields (on the low end noted in entry 12) were obtained when HCl/HFIP solutions were aged for even a few minutes. We also examined whether HBr, generated by the addition of AcBr to HFIP, was a suitable substitute for HCl and initial evidence suggests that it is (cf. entries 26 and 27 with 22 and 25). We still prefer using AcCl-generated HCl because AcCl is generally easier to handle and more resistant to hydrolysis in air. Moreover, we have observed very little differences in the source of AcCl in the reaction (i.e., freshly opened vs. older bottles of reagent, cf. entries 25 and 24). Taking into account both efficiency and practicality, we prefer using TiCl4 or AcCl as HCl sources among all of the methods tested so far.
We decided to further explore the substrate scope with this new reaction condition in hand that utilizes acetyl chloride as a pro-catalyst. The substrate scope was comparable to that described for TiCl4 and lactams were obtained in good to excellent yields (entries 1–10, Table 5). Although higher amounts of acetyl chloride than TiCl4 were required to achieve complete conversion, the use of acetyl chloride was convenient. In addition, both HFIP and its acetate ester byproduct were volatile, which eased work-up. Finally, no metal waste was produced.
Table 5.
 | |||||
|---|---|---|---|---|---|
| entry | Substrate | catalyst loading (mol%) |
time (h) |
product | yield (%)c |
| 1 | 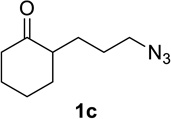 |
10 | 20 | 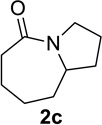 |
95 |
| 2 | 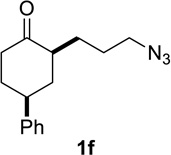 |
10 | 20 | 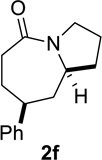 |
98 |
| 3 | 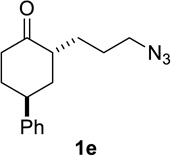 |
20 | 38 | 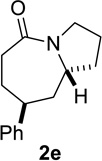 |
90d,e |
| 4 | 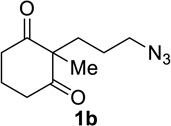 |
20 | 24 | 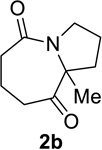 |
95 |
| 5 | 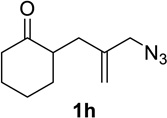 |
20 | 24 | 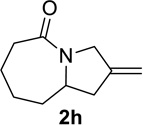 |
77 |
| 6 | 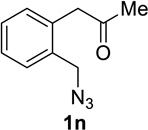 |
60 | 24 | 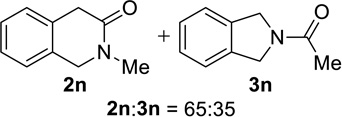 |
68 |
| 7 | 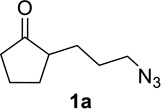 |
80 | 24 | 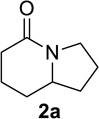 |
90 |
| 8 | 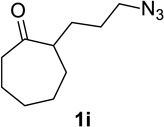 |
80 | 48 | 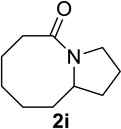 |
87f |
| 9 | 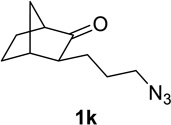 |
100 | 62 | 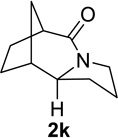 |
92 |
| 10 | 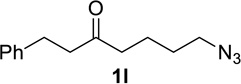 |
100 | 32 | 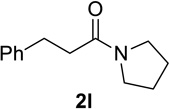 |
96g |
To a solution of a substrate (0.4 mmol) in HFIP (2.0 mL) at room temperature was added CH3COCl under nitrogen atmosphere and reaction stirred at 25 °C for a designated period unless otherwise noted.
Concentration ca. 0.2 M.
Isolated yield.
Bridged lactam 3e was also isolated in ca. 3% yield.
Reaction was ran on 0.1 mmol scale.
Bridged lactam 3i was also isolated in 3% yield (see Supporting Information).
Contains 7% of 1-phenethylpiperidin-2-one 3l (see Supporting Information).
Mechanism
Based on precedent,15b–e,18 we propose the involvement of HFIP as a strong hydrogen bond donor with the lactam carbonyl (Figure 4). As proposed above, we believe that association of a Lewis or Brønsted acid with the Lewis-basic lactam product inhibits the catalytic reaction carried out in CH2Cl2. Hexafluoro-2-propanol solvent can potentially form complexes with the substrate, intermediates, and product. Critically, the hydrogen bonding of HFIP with the lactam carbonyl through the displacement of Lewis or Brønsted acids allows for the regeneration of catalyst – most likely, a proton. In addition, one cannot rule out coordination between HFIP and catalyst to produce a catalytically more reactive species, like [HFIP•H]+.25 We note that cursory pH measurement of the reaction mixture by pH indicator strips (non-bleeding) gave a reading of pH = 4 for the present version, as opposed to pH = 1 for an intramolecular Schmidt reaction carried out with TiCl4 in CH2C12 (the pH of pure HFIP was 5 by this method), suggesting an overall buffering effect of the solvent.
Figure 4.

Proposed catalytic cycle for the intramolecular Schmidt reaction employing HFIP as a solvent.
HFIP has been shown to form aggregates, such as trimers, having potential hydrogen bonds of strengths comparable to those of covalent linkages.15c Such a strong hydrogen bonding could well explain the role of HFIP in the catalysis of the intramolecular Schmidt reaction. Job’s method of continuous variation was used to determine the stoichiometry of binding for HFIP-substrate and HFIP-product complexes (Figure 5).15c,26 Job plots based on 1H NMR data provide good evidence that HFIP forms a 1:1 complex with both substrate la and product 2a. Although the stoichiometry of binding was similar, complexation shift (?δ) of the HFIP hydroxyl resonance for lactam 2a was significantly higher compared to azidoketone la, consistent with the expected stronger complexation of HFIP with lactam over the ketone.
Figure 5.

Job plots for complexation of lactam 2a and azidoketone 1a with HFIP.
In order to gain more insight into the different behaviors of different classes of azidoalkyl ketones, a competition experiment between cyclohexanone- and cyclopentanone-derived 1f and la was performed (Figure 6 and Scheme 3a). Treating an equimolar mixture of 1f and 1a in HFIP with 20 mol% of acetyl chloride resulted in complete conversion of substrate 1f to lactam 2f within 3 h (also see entry 2, Table 5). In sharp contrast, the conversion of la to lactam 2a was only 13% complete after 12 h (also see entry 7, Table 5). These results could be explained by an innate kinetic difference between the substrates, a difference in the degree of product inhibition, or a combination of the two.
Figure 6.

Relative reaction rates for 1f and 1a (see Scheme 3a, below), 1f with 1 equiv of 2f added at the outset of the reaction (Scheme 3b), and 1f with 1 equiv of 2a added at the outset of the reaction (Scheme 3c).
Scheme 3.

With respect to the latter point, we made note of the requirement of different catalyst loadings for different substrate classes. This could be attributed to the difference in basicity of different lactam products, with a more basic lactam requiring higher catalyst loadings.27 In order to demonstrate different degrees of product inhibition with different lactams, 1H NMR experiments were carried out to determine the effect of adding two different product lactams at the outset on a single, relatively fast, reaction. For this, we chose the product of the quicker reaction leading to 2f (and a case that succeeds with 10 mol% of AcCl pro-catalyst) and 2a, the product of a much slower reaction (and one that requires 80 mol% of AcCl to reach completion). In the first case, the facile substrate azidoketone 1f was combined with an equimolar amount of its lactam product 2f and then treated with 20 mol% of acetyl chloride in HFIP (Figure 6 and Scheme 3b). The time it took for quantitative conversion of 1f to 2f was ca. 6 h. In contrast, the reaction of a 1:1 mixture of 1f and 2a with 20 mol% of acetyl chloride in HFIP required >24 h to attain completion (Figure 6 and Scheme 3c). These results suggested significantly more product inhibition by lactam 2a than 2f, which is consistent with the need for higher catalyst loadings with relatively recalcitrant substrates.7c More detailed series of kinetic studies is necessary to fully address the relative roles of kinetics vs. product inhibition and will be reported in due course.
CONCLUSIONS
In summary, we have demonstrated a catalytic intramolecular Schmidt reaction with broad substrate scope and utility. Two versions of the reaction, one using TiCl4 and the other with AcCl, have been identified as having strong synthetic utility that is as good or better than all previous versions of this process. In either case, the strong hydrogen-bonding ability of hexafluoro-2-propanol was critical to the development of these substoichiometric reactions. The discovery of conditions employing acetyl chloride as a pro-catalyst in the presence of hexafluoro-2-propanol provided evidence for HCl being an active catalytic species as well as providing a metal-free catalytic reaction. Prior to this discovery, the primary metal-free variations of the intramolecular Schmidt reaction used either trifluoracetic acid as solvent, or TfOH or ClSO3H as a stoichiometric reagent.
The most favorable examples utilize attractively low loadings of catalyst – as low as 2.5% for the TiCl4-promoted version or 10 mol% of AcCl. Although some of the least cooperative substrates needed as much as 100 mol% of “H+” catalyst added (either via the addition of 25 mol% of TiCl4 or the straightahead addition of 100 mol% of AcCl), we note that these conditions still measure up very favorably to those previously reported for analogous substrates. For example, the reaction of 1a in CH2Cl2 needed 4.5 equiv of TiCl4 to afford a 67% yield,1b while the same reaction carried out with 20 mol% of TiCl4 or 80 mol% of AcCl gave 87% and 90% yields of product, respectively. Although we did not quantitatively compare the purity of products obtained in these various reactions, we note informally that the presently reported procedures tend to provide products requiring little additional purification.
In addition, 1H NMR experiments were performed to exhibit different degree of product inhibition with different lactams. That such structurally similar lactams have substantially different effects on the rate of a given reaction is, minimally, provocative, and might point to a role in understanding the role of product inhibition in this and other reactions that afford lactam or amide products. Future efforts will be directed to extend the scope of this reaction and elucidate further mechanistic details. In the meantime, we consider the method reported herein as the best means of preparatively carrying out this variation of the intramolecular Schmidt reaction.
Supplementary Material
Table 3.
 | |||||
|---|---|---|---|---|---|
| entry | substrate | catalyst loading (mol%) |
time (h) |
product | yield (%)c |
| 1 |  |
10 | 44 | 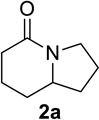 |
60 (78)d |
| 15 | 44 | 79 | |||
| 20 | 24 | 87 | |||
| 2 | 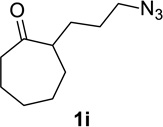 |
5 | 62 | 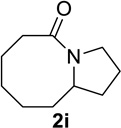 |
34 (85)d |
| 20 | 48 | 86e | |||
| 3 | 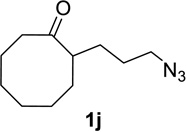 |
25 | 62 | 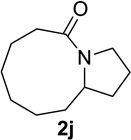 |
90 |
| 4 | 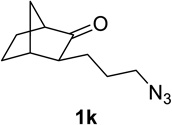 |
10 | 62 | 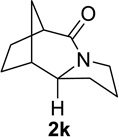 |
43 (78)d |
| 25 | 62 | 87 (90)d | |||
| 5 | 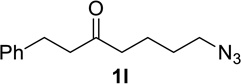 |
20 | 24 | 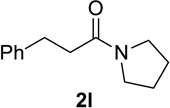 |
79 (96)d,f |
| 25 | 32 | 94f | |||
| 6 | 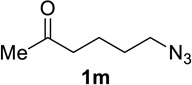 |
10 | 36 | 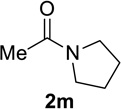 |
73g |
| 15 | 24 | 77(81)d,g | |||
| 20 | 24 | 86g | |||
| 7 | 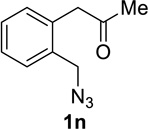 |
15 | 24 | 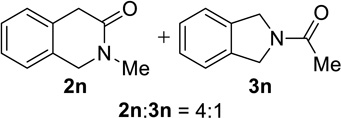 |
64 |
| 8 | 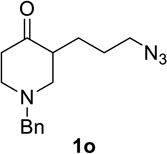 |
35 | 20 | 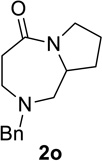 |
90 |
| 9 | 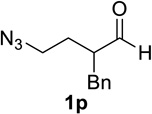 |
5 | 24 | 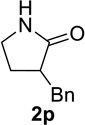 |
86 |
| 10 | 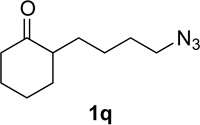 |
20 | 60 | 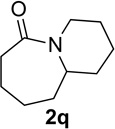 |
11 (20)d |
To a solution of a substrate (0.4 mmol) in hexafluoro-2-propanol (2.0 mL) at room temperature was added TiCl4 under nitrogen atmosphere and reaction was allowed to stir at 25 °C for the designated period unless otherwise noted.
Concentration ca. 0.2 M.
Isolated yield.
Yields in parentheses are based on recovered starting material.
Bridged lactam 3i was also isolated in 2% yield (see Supporting Information).
Contains 7% of 1-phenethylpiperidin-2-one 3l (see Supporting Information).
Contains 3% of N-methyl-2-piperidone 3m (see Supporting Information).
ACKNOWLEDGMENT
We are grateful to the National Institutes of General Medical Sciences (GM-049093) and University of Kansas for financial support. We thank Sarah Neuenswander and Justin Douglas for assistance with NMR, and Ryan Altman and an anonymous reviewer of this manuscript for helpful suggestions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures for new compounds and mechanistic experiments; list of known compounds; additional screening data for reaction optimization experiments; and copies of 1H and 13C NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Aubé J, Milligan GL. J. Am. Chem. Soc. 1991;113:8965–8966. [Google Scholar]; (b) Milligan GL, Mossman CJ, Aube J. J. Am. Chem. Soc. 1995;117:10449–10459. [Google Scholar]
- 2.For reviews, see: Bräse S, Gil C, Knepper K, Zimmermann V. Angew. Chem. Int. Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657. Lang S, Murphy JA. Chem. Soc. Rev. 2006;35:146–156. doi: 10.1039/b505080d. Grecian S, Aube J. In: Organic Azides: Syntheses and Applications. Bräse S, Banert K, editors. Chichester: John Wiley & Sons Ltd; 2010. pp. 191–237. Wrobleski A, Coombs TC, Huh CW, Li S-W, Aube J. Org. React. 2012;78:1–320.
- 3.For selected examples, see: Wendt JA, Aubé J. Tetrahedron Lett. 1996;37:1531–1534. Iyengar R, Schildknegt K, Aubé J. Org. Lett. 2000;2:1625–1627. doi: 10.1021/ol005913c. Wrobleski A, Sahasrabudhe K, Aubé J. J. Am. Chem. Soc. 2004;126:5475–5481. doi: 10.1021/ja0320018. Frankowski KJ, Golden JE, Zeng Y, Lei Y, Aubé J. J. Am. Chem. Soc. 2008;130:6018–6024. doi: 10.1021/ja800574m. Frankowski KJ, Neuenswander B, Aubé J. J. Comb. Chem. 2008;10:721–725. doi: 10.1021/cc800078h. Zhao Y-M, Gu P, Zhang H-J, Zhang Q-W, Fan C-A, Tu Y-Q, Zhang F-M. J. Org. Chem. 2009;74:3211–3213. doi: 10.1021/jo900113s. Ghosh P, Judd WR, Ribelin T, Aubé J. Org. Lett. 2009;11:4140–4142. doi: 10.1021/ol901645j. Kapat A, Nyfeler E, Giuffredi GT, Renaud P. J. Am. Chem. Soc. 2009;131:17746–17747. doi: 10.1021/ja908933s. Chen Z-H, Chen Z-M, Zhang Y-Q, Tu Y-Q, Zhang F-M. J. Org. Chem. 2011;76:10173–10186. doi: 10.1021/jo202042x. Ma A-J, Tu Y-Q, Peng J-B, Dou Q-Y, Hou S-H, Zhang F-M, Wang S-H. Org. Lett. 2012;14:3604–3607. doi: 10.1021/ol301331t.
- 4.(a) Lertpibulpanya D, Marsden SP. Org. Biomol. Chem. 2006;4:3498–3504. doi: 10.1039/b608801e. [DOI] [PubMed] [Google Scholar]; (b) Yang M, Zhao Y-M, Zhang S-Y, Tu Y-Q, Zhang F-M. Chem. Asian J. 2011;6:1344–1347. doi: 10.1002/asia.201100171. [DOI] [PubMed] [Google Scholar]
- 5.(a) Sheldon RA. Pure Appl. Chem. 2000;72:1233–1246. [Google Scholar]; (b) Bolm C, Legros J, Le Paih J, Zani L. Chem. Rev. 2004;104:6217–6254. doi: 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]
- 6.Gutierrez O, Aubé J, Tantillo DJ. J. Org. Chem. 2011;77:640–647. doi: 10.1021/jo202338m. [DOI] [PubMed] [Google Scholar]
- 7.For reviews on reaction kinetics involving product inhibition, see: Blackmond DG. Angew. Chem. Int. Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544. Mathew JS, Klussmann M, Iwamura H, Valera F, Futran A, Emanuelsson EAC, Blackmond DG. J. Org. Chem. 2006;71:4711–4722. doi: 10.1021/jo052409i. For a selected example of product inhibition in a catalytic Diels–Alder reaction, see: Evans DA, Miller SJ, Lectka T, von Matt P. J. Am. Chem. Soc. 1999;121:7559–7573.
- 8.Catalytic Ritter reactions: Sanz R, Martínez A, Guilarte V, Álvarez-Gutiérrez JM, Rodríguez F. Eur. J. Org. Chem. 2007;2007:4642–4645. Guérinot A, Reymond S, Cossy J. Eur. J. Org. Chem. 2012;2012:19–28.
- 9.Zhang JS, Wang K, Lu YC, Luo GS. AlChE J. 2012;58:3156–3160. [Google Scholar]
- 10.For examples of catalytic Beckmann rearrangements, see: Mukaiyama T, Harada T. Chem. Lett. 1991;20:1653–1656. Lee JK, Kim D-C, Eui Song C, Lee S-g. Synth. Commun. 2003;33:2301–2307. Sato S, Hoshino H, Sugimoto T, Kashiwagi K. Chem. Lett. 2010;39:1319–1320. Liu L-F, Liu H, Pi H-J, Yang S, Yao M, Du W, Deng W-P. Synth. Commun. 2011;41:553–560.
- 11.Zicmanis A, Katkevica S, Mekss P. Catal. Commun. 2009;10:614–619. [Google Scholar]
- 12.Nucleophilic addition reactions to cyclopentanones are generally slower than those to cyclohexanones: Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. New York: John Wiley & Sons; 1994. pp. 762pp. 769–771.
- 13.Gracias V, Zeng Y, Desai P, Aubé J. Org. Lett. 2003;5:4999–5001. doi: 10.1021/ol035965c. [DOI] [PubMed] [Google Scholar]
- 14.Gu P, Zhao Y-M, Tu YQ, Ma Y, Zhang F. Org. Lett. 2006;8:5271–5273. doi: 10.1021/ol062116r. [DOI] [PubMed] [Google Scholar]
- 15.(a) Catalán J, Palomar J, Díaz C, de Paz JLG. J. Phys. Chem. A. 1997;101:5183–5189. [Google Scholar]; (b) Bégué J-P, Bonnet-Delpon D, Crousse B. Synlett. 2004;2004:18–29. [Google Scholar]; (c) Berkessel A, Adrio JA, Hüttenhain D, Neudörfl JM. J. Am. Chem. Soc. 2006;128:8421–8426. doi: 10.1021/ja0545463. [DOI] [PubMed] [Google Scholar]; (d) Shuklov IA, Dubrovina NV, Börner A. Synthesis. 2007;2007:2925–2943. [Google Scholar]; (e) Ratnikov MO, Tumanov VV, Smit WA. Angew. Chem. Int. Ed. 2008;47:9739–9742. doi: 10.1002/anie.200803927. [DOI] [PubMed] [Google Scholar]; (f) Palladino P, Stetsenko DA. Org. Lett. 2012;14:6346–6349. doi: 10.1021/ol303124r. [DOI] [PubMed] [Google Scholar]
- 16.Khaksar S, Heydari A, Tajbakhsh M, Vahdat SM. J. Fluorine Chem. 2010;131:1377–1381. [Google Scholar]
- 17.(a) Neimann K, Neumann R. Org. Lett. 2000;2:2861–2863. doi: 10.1021/ol006287m. [DOI] [PubMed] [Google Scholar]; (b) Iskra J, Bonnet-Delpon D, Bégué J-P. Tetrahedron Lett. 2002;43:1001–1003. [Google Scholar]
- 18.Shuklov IA, Dubrovina NV, Barsch E, Ludwig R, Michalik D, Borner A. Chem. Commun. 2009;0:1535–1537. doi: 10.1039/b820730e. [DOI] [PubMed] [Google Scholar]
- 19.(a) Hong D-P, Hoshino M, Kuboi R, Goto Y. J. Am. Chem. Soc. 1999;121:8427–8433. [Google Scholar]; (b) Konno T, Iwashita J, Nagayama K. Protein Sci. 2000;9:564–569. doi: 10.1110/ps.9.3.564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Roccatano D, Fioroni M, Zacharias M, Colombo G. Protein Sci. 2005;14:2582–2589. doi: 10.1110/ps.051426605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cativiela C, García JI, Mayoral JA, Salvatella L. Can. J. Chem. 1994;72:308–311. [Google Scholar]
- 21.During screening of reaction conditions, use of amine base as an additive had a deleterious effect on reaction outcome (see Supporting Information for details).
- 22.Iden HS, Lubell WD. Org. Lett. 2006;8:3425–3428. doi: 10.1021/ol061036k. [DOI] [PubMed] [Google Scholar]
- 23.Reaction mixture was monitored by TLC and no product spot was observed until after the addition of 25 mol% of TiCl4
- 24.Izumi J, Shiina I, Mukaiyama T. Chem. Lett. 1995;24:141–142. [Google Scholar]
- 25.Evans DA, Rovis T, Kozlowski MC, Downey CW, Tedrow JS. J. Am. Chem. Soc. 2000;122:9134–9142. [Google Scholar]
- 26.Jiang J, MacLachlan MJ. Chem. Commun. 2009;0:5695–5697. doi: 10.1039/b914564h. [DOI] [PubMed] [Google Scholar]
- 27.(a) Gorshkova GN, Kolodkin FL, Polishchuk VV, Ponomarenko VA, Sidel'kovskaya FP. Russ. Chem. Bull. 1970;19:506–509. [Google Scholar]; (b) Filgueiras CAL, Huheey JE. J. Org. Chem. 1976;41:49–53. [Google Scholar]; (c) Wan P, Modro TA, Yates K. Can. J. Chem. 1980;58:2423–2432. [Google Scholar]; (d) Cox RA, Druet LM, Klausner AE, Modro TA, Wan P, Yates K. Can. J. Chem. 1981;59:1568–1573. [Google Scholar]; (e) Puffr R, Kubanek V. Lactam Based Polyamides, Vol I: Polymerization, structure and properties. CRC Press, Inc; 1991. [Google Scholar]; (f) Le Questel J-Y, Laurence C, Lachkar A, Helbert M, Berthelot M. J. Chem. Soc., Perkin Trans. 2. 1992;0:2091–2094. [Google Scholar]; (g) El Firdoussi A, Esseffar M, Bouab W, Abboud JLM, Mó O, Yáñez M. J. Phys. Chem. A. 2004;108:10568–10577. [Google Scholar]; (h) Glover SA, Rosser AA. J. Org. Chem. 2012;77:5492–5502. doi: 10.1021/jo300347k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.