

Abstract
Mitochondrial morphology and metabolism play an important role in cellular homeostasis. Recent studies have shown that the fidelity of mitochondrial morphology is important in maintaining mitochondrial shape, number, size, membrane potential, ATP synthesis, mtDNA, motility, signaling, quality control, response to cellular stress, mitophagy and apoptosis. This article provides an overview of the current state of knowledge of the fission and fusion machinery with a focus on the mechanisms underlying the regulation of the mitochondrial morphology and cellular energy state. Several lines of evidence indicate that dysregulation of mitochondrial fission or fusion is associated with mitochondrial dysfunction, which in turn impacts mitophagy and apoptosis. Metabolic disorders are also associated with dysregulation of fission or fusion and the available lines of evidence point to a bidirectional interplay between the mitochondrial fission or fusion reactions and bioenergetics. Clearly, more in-depth studies are needed to fully elucidate the mechanisms that control mitochondrial fission and fusion. It is envisioned that the outcome of such studies will improve the understanding of the molecular basis of related metabolic disorders and also facilitate the development of better therapeutics.
Keywords: Cancer, Fission and Fusion, Metabolic Disorders, Metabolic Stress, Mitochondria, Mitochondrial DNA
1. Mitochondrial dynamics
Mitochondria, in Greek means thread grain (1), are structurally divided into three aqueous compartments including a matrix, an inter-membrane space (IMS) and an intra-cristae space. Mitochondria contain an outer-membrane (MOM), an inner-membrane (IM) and a cristae-membrane (2). Among these sub-compartments, mitochondrial IM is the major site of ATP generation and respiration. The electron carrier, NADH and FADH2, produced during tricarboxylic acid cycle (TCA cycle) or Krebs cycle, are transferred to the electron transport chain (ETC) in IM. The flow of electrons from the matrix to IMS via complexes I, III and IV releases the free energy that fuels the translocation of protons from matrix to IMS via ATP synthase (complex V) with oxidative phosphorylation (oxphos) of ADP to ATP. It makes mitochondria the major site of ATP synthesis.
Mitochondria are considered as the power house of cells and are also involved in various activities including calcium signaling, carbohydrates metabolism, fats and amino acids synthesis, reactive oxygen species (ROS) generation, regulation of cell cycle and cell death. Mitochondrial size, shape and location within a cell play important roles in regulating mitochondrial activity. Mitochondria, as the name suggests are dynamic organelles undergoing balanced fission and fusion (3). Mitochondrial dynamics regulates mitochondrial morphology, mitophagy, apoptosis, oxphos, Ca2+ signaling, mitochondrial DNA (mtDNA) stability, mitochondrial quality, ROS generation and cellular stress response. Being involved in a variety of cellular processes, it is not surprising that mitochondrial dysfunction is linked to various metabolic stresses as well as metabolic and neurodegenerative disorders. This article provides an overview of the current state of knowledge of mitochondrial fission and fusion, and their relationship to metabolic stresses and disorders. For detailed general reviews on these topics, the authors suggest additional excellent articles (4–7) that along with other articles also served as sources for our current review.
2. Proteins involved in the maintenance of mitochondrial morphology
Mitochondrial morphology is maintained, in part, by a balance between the components of mitochondrial fission and fusion (Figures 1 and 2). For example, the expression, availability and activity of the proteins that constitute fission and fusion machineries are tightly controlled. Although the fission and fusion machineries have been studied in various organisms, the focus of this article is the mammalian system.
Figure 1. Mitochondrial fission.
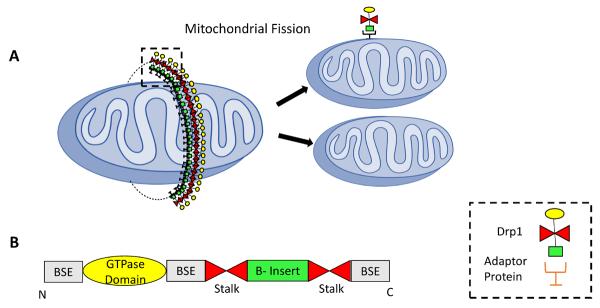
(A) Mitochondrial Fission involves formation of a Drp1 ring across the mitochondria with the help of adaptor proteins. This ring further constricts to divide the parent mitochondria into daughter mitochondria. (B) Illustration of motifs and domains of Drp1. BSE: Bundle Stalking Element (13)
Figure 2. Mitochondrial Fusion.
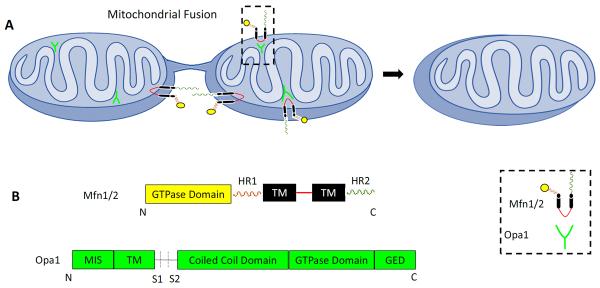
(A) During fusion mitochondria form a hypothetical bridge with the help of interaction between HR domain of mitofusins on the mitochondria involved (B) Domains and motifs of Mfn1/2 and Opa1 (293).
(2A) Mitochondria fission
Mitochondrial fission plays an important role in maintaining mitochondrial quality control. Fission helps in separating the healthy mitochondria from the defective ones. The defective mitochondria are further removed by mitophagy and recycled or degraded. In mammalian system, mitochondrial fission is considered as one-step process because there appears to be a lack of machinery needed for IM fission (8). In the case of mammals, the most well studied components of fission machinery are by Dynamin related protein-1 (Drp1) and fission protein 1 (Fis1) (Figure 1).
Drp1
Drp1 is a mechanochemical enzyme that plays a significant role in apoptosis, calcium signaling (9), and peroxisomal and mitochondrial fission (10). Drp-1 shares similar domains with Dynamin including the GTPase domain, dynamin-2 domain and GTPase effector domain (GED) (11). However, unlike Dynamin, Drp1 contains a variable domain, or B-insert involved in regulation of Drp1 assembly (12). Structurally, Drp1 oligomerizes into an X-shaped dimer on mitochondrial outer membrane (MOM) with the help of two stalk interfaces. The stalk tips constitute two conserved regulatory domains, the GPRP domain and the B-insert domain (13). Drp1 lacks a transmembrane domain, indicating the role of other MOM proteins in Drp1's MOM localization. Drp1 is located in cytosol, peroxisomes and MOM. In the case of the peroxisomes and MOM, Drp1 is concentrated in discrete regions (14, 10). Drp1 is mainly present in the cytosol whereas only 3% is located on mitochondria (14). Therefore, Drp1's activity is mainly dependent on its mitochondrial translocation. Various studies have suggested that Fis1, Mff1, MiD49 and MiD51 (described below) are involved in Drp1's mitochondrial enrollment. Additionally, endoplasmic reticulum (ER)-mitochondria contact points also mark Drp1 binding sites on MOM (15). Once localized to the MOM, Drp1 rims the mitochondria (16) but the exact mechanism of Drp1-mediated fission is not completely understood. Like Dnm1, Drp1 is believed to form multimeric spirals at the constriction site, with GTPase domain pointing away from the membrane (17). Formation of a complete spiral activates the GTPase domain causing GTP hydrolysis. Consequently, the dynamins change its confirmation leading to spiral constriction (18).This suggests that GTP hydrolysis by the GTPase domain drives constriction and scission of outer membrane. Mutation in GTPase domain of Drp1 (K38A) leads to mitochondrial elongation (16) that further supports the role of GTPase domain in Drp1 activity.
Drp1-mediated fission plays an important role is segregation of dysfunctional mitochondria. During apoptosis, Drp1 is also involved in Bax oligomerization on the MOM and cytochrome c release (19, 20). As a positive feedback, Bax and Bak SUMOylate Drp1 and stabilize the MOM-bound Drp1, causing apoptotic mitochondrial fission (21). Drp1 involvement in the variable processes needs precise regulation. Various post-translational modification including phosphorylation, S-nitrosylation, ubiquitination, SUMOylation and O-linked-N-acetyl-glucosamine glycosylation are involved in regulating Drp1 activity (Table 1). Depending upon the type and site of modification, Drp1 post-translational modifications may have an activating or inhibiting effect.
Table 1.
Post-translational modifications regulating Drp1 activity.
| Post-translational Modifications | Proteins involved | References |
|---|---|---|
|
| ||
| Phosphorylation/Dephosphorylation | • Cdk1/cyclinB and PKCδ phosphorylates human Drp1 at Ser616, increasing Drp1 activity | 99, 150, 282, 283, 284, 285, 286; 287 |
| • CAMK-Iα- calcium dependent phosphorylation of Drp1 at Ser637 (Ser600 of isoform3), increasing Drp1 activity | ||
| • ROCK1 and PKA -phosphorylates at Ser600, increasing Drp1 activity | ||
| • PKA-Phosphorylates at Ser637, inhibiting Drp1 GTPase activity | ||
| • GSK3β phosphorylates Drp1 at Ser693 and Lys679. Ser693 decreases Drp1 activity. | ||
| • Calcineurin-calcium dependent de phosphorylation at Ser637, increasing Drp1 activity. | ||
|
| ||
| Ubiquitination | • APC/C(Cdh1) ubiquitinates Drp1 during interphase | 288, 289, 97, 228 |
| • Parkin-E3 ubiquitin ligase, ubiquitinates Drp1 for degradation | ||
| • MITOL/MARCH5, MOM protein, ubiquitinates Drp1. Different studies have given different results regarding its effect on Drp1 activity | ||
|
| ||
| SUMOylation | • Ubiquitin carrier protein 9 (Ubc9)-SUMOylates Drp1 in variable domain and helps in Drp1 localization on MOM, increasing fission | 21, 290, 291 |
| • Mitochondrial-anchored protein ligase (MAPL)-SUMOylates Drp1 and increases fission | ||
|
| ||
| • SENP5, SUMO-1 protease, deSUMOylates Drp1 on G2/M transition and increases MOM bound Drp1 stability and therefore increases fission | 102, 291, 152, 292 | |
| • SENP3, SUMO −2/3 protease deSUMOylates Drp1 during oxygen glucose starvation and inhibits Drp1 mitochondrial localization and therefore decreases fission | ||
|
| ||
| S-nitrosylation | • At Cysteine residues in GED domain, increases GTPase activity and therefore fission | 213 |
|
| ||
| O-linked-N-acetyl-glucosamine glycosylation | • O-GlcNAcylation of Drp at Thr 585–586, increases Drp1 mitochondrial translocation and mitochondrial fission | 152 |
hFis1
Human fission protein 1 homologue, Fis1 is another MOM protein. Fis1 binds to MOM with the help of C-terminal transmembrane domain and the C-terminal tail hanging in the IMS (22, 23). Unlike Drp1, Fis1 is distributed evenly on outer mitochondrial membrane surface, which forms punctate appearing complexes (22, 23). Functionally, Fis1, aided by Mdv1 and Caf4, facilitate the binding of Dnm1 (a yeast homologue of Drp1), but not of mammalian Drp1, on the MOM (24). Thus, unlike Dnm1, hFis1 knockdown does not affect mitochondrial localization of Drp1 (25, 26). However, hFis1 interaction with TBC1D15, a protein involved in mitochondrial fission, aids TBC1D15 mitochondrial localization, suggesting hFis1's role as a receptor for mitochondrial fission machinery (27). Alterations in hFis1 expression affect mitochondrial morphology. For example, hFis1 or Drp1 knockdown causes mitochondrial elongation (26), decrease in mtDNA and cellular respiration (28). Whereas, hFis1 overexpression amplifies mitochondrial fragmentation (29) (unlike Mff1 and Drp1) suggesting hFis1 to be a limiting factor in the mitochondrial fission. hFis1 expression is also regulated via ubiquitination by MITOL, a mitochondrial ubiquitin ligase (30).
Other proteins involved in mitochondrial fission
Mitochondrial fission factor-1 (Mff1) is a MOM protein, linked to the mitochondrial fission (31). Mff1 knockdown causes decrease in the mitochondrial Drp1 levels (32), suggesting Mff1 to be a Drp1 MOM receptor. Also, Drp1 phosphorylation affects Drp1 interaction with Mff1, thereby affecting Drp1's mitochondrial translocation. Another set of proteins involved in fission is the mitochondrial dynamics proteins (MiDs), Mid49 and Mid51. MiDs are transmembrane proteins that bind to the MOM with the help of N-terminus and also involved in Drp1 recruitment to MOM independent of Fis1/Mff1 (33). Interestingly, overexpression of MIEF1/MiD51 causes increase in Drp1-MiD51 interaction and Drp1 mitochondrial localization, but also negatively affects GTP binding to Drp1, decreasing Drp1's fission activity (34). This results in increased mitochondrial fusion on MiD49/51 overexpression (33, 34, 6). These studies project Drp1 as a key player in fission and fusion, whose activity is regulated via binding to different components of fission-fusion machinery.
Various other proteins including mitochondrial protein 18kDa (MTP18), ganglioside-induced differentiation associated protein 1 (GDAP1), endophillin B1, Dna-J3 and mitochondrial ubiquitin ligase activator of NFκB (MULAN), DDP/TIMM8a, and OPA 3 are involved in mitochondrial fragmentation (35, 36, 37, 38, 31, 39, 40). Like Mff1, DDP/TIMM8a interaction with Drp1 helps in Drp1 mitochondrial localization and mitochondrial fragmentation, during apoptosis (41, 5). Also, INF-2, an ER protein, involved in actin assembly is also associated with mitochondrial fission (42) and is also found mutated in some cases of Charcot-Marie-Tooth (CMT) disease (43). Interestingly, MTP18 is the only known mitochondrial inner membrane protein linked to fission.
(2B) Mitochondria Fusion
Mitochondrial fusion is reported as a two-step process, involving fusion of the MOM followed by that of the IM (44). The outer membrane fusion requires the MOM proteins mitofusins namely Mfn1 and Mfn2, whereas the IM fusion mainly involves IM-localized protein, Opa1. Sukhorukov et al. (45) have reported that the frequency of fusion depends on the number of mitochondrial tips available and is also suggested that fusion machinery is localized at these tips (46). Therefore, to understand the fusion mechanism, one needs to determine the role of Mfn1, Mfn2 and Opa1 that are believed to constitute mitochondrial fusion machinery (Figure 2).
Mitofusins (Mfn1 and Mfn2)
In mammals, the mitofusins, Mfn1 and Mfn2, exist as transmembrane dynamin-related GTPases (47). Presence of Mfn1 and Mfn2 is a requisite for the inter-mitochondrial fusion as Mfn-deficient mitochondria are unable to fuse with mitochondria that are Mfn-proficient (48).
In humans, Mfn1 and Mfn2 are 63% identical and have common functional domains (48). Mfn1 and Mfn2 contain a GTPase domain at the N-termini, whereas the C-termini contain two hydrophobic heptad regions, HR1 and HR2. Studies have suggested that the region between HR1 and HR2 contains two transmembrane domains. These transmembrane domains confer upon mitofusins a U-shaped structure such that the N and C-termini face towards the cytosol (49, 4). The HR region between any two mitochondria dimerizes (homotypic or heterotypic) to form a coiled-coil structure and homotypic interactions probably place MOM of adjacent mitochondria in juxtaposition, at ~160° (50, 48). This tethering of mitochondria could be followed by GTP hydrolysis, bringing conformation changes, finally causing mitochondrial fusion in a SNARE-like mechanism (51). For this reason, mutations in the GTPase domain alter mitofusins' activity, thereby affecting mitochondrial fusion (50). Furthermore, defect in the Mfn1 or Mfn2 function causes placental lethality in mice (50). Interestingly, Mfn2 shows tissue specific expression whereas Mfn1 shows ubiquitous expression. Moreover, mitochondrial fusion is more severe with the Mfn1 overexpression than that due to Mfn2 overexpression which could be due to increased GTPase activity of Mfn1 than Mfn2 (52, 53). These findings were further confirmed by Cipolat et al. (54) suggesting the role of Mfn1 but not Mfn2 in Opa1-mediated mitochondrial fusion.
Mitofusins activity is regulated by ubiquitination leading to Mitofusins degradation (55). This degradation is mainly induced by stress and involves various proteins such as PINK1, Parkin (56), E3 ligase Huwe1 (57), MULAN (39, 58), mitochondrial ubiquitin ligase (MITOL/MARCH V) (30), stomatin like protein-2 and Bcl-2 family members (59, 60, 61). It has been reported that whereas Mfn2 and Bax interaction plays a role in apoptosis (62), MITOL-Mfn2 interaction helps in the mitochondria-ER association (63, 59) and decrease in Mfn2 levels causes ER stress (reviewed in (60)). Furthermore, during apoptosis, JNK phosphorylated Mfn2 leads to Mfn2 ubiquitin-proteasome degradation by Huwe1 (57). Various mitofusin deubiquitination enzymes have also been studied including an ubiquitin-specific protease 30 (Ubp16/USP30) and the decrease in Usp30 causes mitochondrial elongation (64). Mitofusin activity is also regulated by Mfn-binding protein (MIB), a cytosolic protein which negatively regulates mitofusin activity (65).
Mfn2 plays a significant role in response to stress stimuli, as exposure to cold or exercise induces Mfn2 expression. Stress-induced increase in Mfn2 expression is mediated by amplification in Mfn2 transcription, with the help of PGC1-α (66). Furthermore, mutations in Mfn2 cause mtDNA depletion, suggesting a role of Mfn2 in mtDNA stability (67). Mutations in Mfn2 have also been linked to Charcot-Marie-Tooth disease (CMT type 2A).
Opa1
Opa1, a mitochondrial protein, is named following its identification as a mutated gene in optic atrophy. Opa1 is a dynamin-related GTPase that contains an N terminal mitochondrial import signal (MIS), a transmembrane domain, a coiled coil variable domain, a GTPase domain, a GED and proteolytic cleavage sites (68). Like other dynamin-related GTPases, Opa1 has a variable domain, which in the case of Opa1 is involved in interaction with the cardiolipin, a mitochondrial IM protein (69). Opa1 is localized to mitochondria and lipid droplets (70). In the case of mitochondria, most of the Opa1 is localized in cristae, leaving about 8 % on the IM rim (71), suggesting its role in maintaining cristae morphology. Functionally, Opa1 is mainly associated with the mitochondrial fusion. Coordination between the MOM and IM fusion is possibly mediated by interaction between Opa1 C-terminus and mitofusin loop formed by a U-shaped topology (54). Like mitofusins, Opa1 also helps in maintaining mitochondrial function, calcium homeostasis (72, 73), apoptosis by regulating cytochrome c release (74, 75), mtDNA level (76), mitophagy (77), respiratory capacity (78) and maintenance of mitochondrial membrane potential (MMP). Opa1-mediated mitochondrial fusion usually follows fission. However, in various physiological processes mitofusins and Opa1 lack coordination (reviewed in (5)), suggesting the involvement of other proteins in mitofusins and Opa1 regulation.
The mitochondrial fusion activity of Opa1 involves coordination of long isoform of Opa1 and its cleaved short isoform (79). Opa1 long isoform is grounded to mitochondrial IM and faces IMS, whereas short isoform is soluble in IMS (80). Various studies have suggested a major role for long-isoform in Opa1-mediated mitochondrial fusion than that for short isoform (reviewed in (5)), which makes Opa1 proteolytic cleavage an important step in regulation of Opa1 activity. Opa1 contains three cleavage sites-S1, S2 and S3. Cleavage at S1 is mediated by various proteases including overlapping activity with m-AAA protease (OMA-1), presenilin-associated rhomboid-like (PARL) and mitochondrial matrix-complexes comprising parapleign and/or Afg3L1 and Afg3L2 (m-AAA) (11, 81–84). On the other hand, cleavage at S2/S3 is mediated by mitochondrial IMS protease, YmeL-1, an i-AAA metallo-protease, leading to formation of s-Opa1 (82, 79). Opa1 proteolytic cleavage is regulated by MMP, apoptosis, ATP level and mtDNA stability (83, 85, 78). Moreover, Opa1 proteolytic cleavage is considered as pro-fission marker as PARL-mediated Opa1 cleavage leads to cytochrome c release and apoptosis (81). Interestingly, direct interaction between Opa1 and hypoxia-induced gene domain protein-1a (Higd-1a) prevents cleavage of long-Opa1, mitochondrial fission and apoptosis (86). Furthermore, S1 cleavage forming Opa1 long and short isoform is influenced by MMP and ATP levels (79, 87, 88), which suggests a role for MMP in regulating Opa1 cleavage and therefore, mitochondrial IM fusion. However, Opa1 interaction with Mfn1 and Mfn2 is not affected by MMP (87). Furthermore, Opa1 knockout causes a decrease in mitochondrial IM fusion and matrix mixing but has no effect on MOM fusion (89, 90). These lines of evidence suggest that MOM fusion is independent of MMP. Looking into fusion mechanism, unlike MOM, IM does not require tethering. During fusion, inter-mitochondrial short-MgM1 and long-Mgm1 (the yeast homologue of Opa1), are activated in the presence of cardiolipin and form multimeric complex (91). It may be possible that these activated isoforms form a multimeric structure between two mitochondria that are undergoing fusion, followed by a pull utilizing energy from GTP hydrolysis, which pushes the IM apart. Regarding the developmental role of Opa1, it is of note, that Opa1 depletion in mice is embryonically lethal (92). Furthermore, an ADOA mutation (R290Q and G300E) in Opa1 GTPase domain has been found to disrupt Opa1 fusion activity, causing mitochondrial fragmentation (93).
3. Coordination of fission and fusion
Mitochondrial fission and fusion are cyclic events (77). Recently, Cagalinec et al (94) suggested that the rate of a mitochondrial fission depends on the length of mitochondria, whereas the rate of mitochondrial fusion depends upon mitochondria motility. It has been proposed that in neurons, the probability of the mitochondrial fusion after fission is 0.864 and that of fission after fusion is 0.835 (94). Various mechanisms are involved in fission-fusion coordination. In a healthy cell, mitofusin-mediated MOM fusion causes activation of phospholipase D. Phospholipase D further hydrolyzes cardiolipin of another mitochondria involved in fusion, forming phosphatidic acid D (51). Phosphatidic acid D then activates Lipin1, which is involved in the formation of DAG, a PKC activator. Activated PKC in turn phosphorylates Drp1 at Ser616, causing Drp1 mitochondrial localization, activation and fission. Fission activation is again followed by mitochondrial fusion (88). Interestingly, Drp1-Mfn2 interaction promotes Mfn2 activity and mitochondrial fusion, by limiting HR1-HR2 heterotypic interaction (95). Furthermore, it has been reported that fragmentation induced by deficiency in Mitofusins or Opa1 require Mff1 activity (7, 31).
4. Need for fission and fusion
Mitochondrial fission and fusion are involved in regulating various cellular processes including cell cycle, response to stresses, apoptosis, mitochondrial quality control, mitophagy, mtDNA stability, oxphos and mitochondrial cellular distribution via regulation of mitochondrial motility. Additionally, mitochondrial dynamics are also involved in senescence and intracellular signaling (reviewed in (5)).
(4A) Cell Cycle regulation
Mitochondrial morphology changes with change in the phases of cell cycle. G1/S phase transition needs increase in ATP levels to generate biomolecules. To increase the efficiency of ATP synthesis, mitochondria fuse and elongate (96). To achieve this, anaphase-promoting complex/cyclosome and its coactivator Cdh1 (APC/C (Cdh1)), an E3 ubiquitin ligase, ubiquitinates and degrades Drp1 during interphase, thereby decreasing mitochondrial fission (97). Cellular ATP levels also stabilize cyclin E, suggesting a role of mitochondrial fusion during G1/S and entry into S phase (98). Furthermore, during mitosis mitochondrial fragmentation is also needed to ensure distribution of mitochondria to daughter cells. Mitotic fragmentation of mitochondria involves (i) Drp1 phosphorylation by Cdk1/cyclin B aided by RALA and RALBP1 (99; 100, 101) and (ii) Drp1 deSUMOlyation by SENP5 SUMO protease to increase mitochondrial Drp1 levels (102). Mitochondrial fission-fusion-mediated cell cycle regulation also influences cellular ability to differentiate. Enhanced mitochondrial fusion is expected to cause entry into S phase and therefore, increased cellular division, whereas increased fission will support cellular differentiation (reviewed in (103)).
(4B) Cellular response to stress
Under conditions of stress such as starvation, exposure to UV radiation, actinomycin D treatment and oxidative stress, mitochondria undergo stress-induced hyperfusion (SIMH) and decreased fission (104, 105). Hyperfusion increases mitochondrial ATP synthesis (104) and therefore, could be considered as a protective step against stress.
Oxidative stress has closely been related to increase in mitochondria fusion. It is further supported by mitochondrial fusion induction by oxidized glutathione (GSSG) (106). The amount of damage caused by oxidative stress depends on mitochondrial dynamics to certain extent (107, 108). Interestingly, inhibiting mitochondrial fragmentation reduces mtROS (109) and treatment with N-acetyl-cysteine reduces mitochondrial fragmentation followed by decrease in ROS level under hyperglycemic conditions (110). Cumulative lines of evidence suggest that ROS and mitochondrial fragmentation play a bidirectional role. Defective mitochondrial morphology increases oxidative stress and show association with various metabolic and neurodegenerative disorders.
(4C) Apoptosis induction
Mitochondrial fission and fusion also play a role in apoptosis. Increased mitochondrial fission is believed to favor apoptosis, whereas elevation in fusion suppresses apoptosis (20). Apoptosis could be mediated by the extrinsic or intrinsic pathway. The intrinsic apoptotic pathway involves mitochondrial permeabilization with the help of Bax and Bak oligomerization on the MOM, further causing cytochrome c release. Fission-fusion machinery has been reported to interact with Bcl2 family proteins (3). For example, Drp1 mitochondrial localization is believed to increase during apoptosis, which appears to facilitate increased Bax oligomerization on the MOM, leading to mitochondrial fragmentation (20) and mitochondrial pore opening for cytochrome c release (19). Conversely, a decrease in the Drp1 level is believed to cause delayed cytochrome c release (111). Moreover, Bax-Bak-mediated Drp1 SUMOylation, also causes mitochondrial fission. Clearly, although various reports have suggested the relevance of Drp1 in the pathways to apoptosis, whether Drp1 is absolutely required for cell death remains to be fully elucidated. Further evidence that fission-fusion are linked to regulating cell death can be gleaned by the findings that Opa1-mediated cristae remodeling also regulates cytochrome c release out of mitochondria (81). It is now well-known that cytochrome c release out of mitochondria is a critical step in the pathway to cell death.
(4D) Mitochondrial quality control
Fusion and fission are also implicated in regulating mitochondrial quality control. Mitochondrial fission segregates damaged mitochondria. In most cases, mitochondrial fission results in a polarized and depolarized daughter mitochondria (77). Depolarized mitochondria are unable to fuse due to deficient Opa1 processing, and therefore, become target of mitophagy. Furthermore, fusion helps in mtDNA mutation compensation and continuous fusion-fission cycles also offer homogenous mitochondrial population per cell (112).
(4E) Mitophagy
Mitophagy is autophagic degradation of the defective mitochondria. Mitophagy is believed to predominantly involve PTEN-induced putative kinase protein 1 (PINK1) and Parkin. PINK1 is a serine threonine kinase, whereas Parkin is an E3 ubiquitin ligase. Dysfunctional and depolarized mitochondria are marked by PINK1, which further recruits Parkin, (113, 114) to undergo proteasome-ubiquitin degradation. Parkin localization in depolarized mitochondria also involves interaction with voltage-dependent anion channels (VDACs) (115). To keep the depolarized mitochondria segregated, PINK-1 phosphorylates Mfn2 and Miro, helping Parkin to ubiquitinate Mfn2 and Miro, thereby inhibiting fusion and mitochondrial motility, respectively (116, 113, 117, 118). Mfn2 deficiency in cardiomyocytes prevents Parkin translocation to depolarized mitochondria and mitophagy (61). Furthermore, defects in mitophagy cause an accumulation of dysfunctional mitochondria leading to oxidative stress and various disease states including Parkinson's disease (119, 120).
(4F) Mitochondrial DNA stability
Mitochondrial DNA (mtDNA) undergoes frequent mutations due to close proximity with the ROS generation site i.e., ETC and limited repair mechanism. These mutations make mtDNA unstable. Mitochondrial dynamics plays an important role in compensating for these mutations. As a result, mammalian cells with defects in mitochondrial fusion or fission machinery have less mtDNA content and increase in the rate of mtDNA mutation (67, 28). Mitochondrial fission machinery is also involved in mtDNA distribution as mitochondrial fission proteins Drp1 and Mff1 localize adjacent to mtDNA (121). Mitochondrial fission and fusion events start close to mitochondrial nucleoids (122). This close proximity helps in equal mtDNA distribution to the daughter mitochondria.
(4G) Mitochondrial motility
Fission and fusion help in intracellular mitochondrial movement to serve various purposes including ATP supply and calcium buffering. Neurons are examples to highlight the significance of mitochondrial motility as ATP demand and calcium concentration are not homogenous in neurons. Mitochondrial outer membrane calcium regulated GTPases, Miro1 and Miro2 interact with Kinesin-1 motor on microtubule for mitochondrial movement (123, 124). Furthermore, increase in mitochondrial motility has been linked to increase in mitochondrial fusion which may lead to increase in ATP synthesis (94).
(4H) Oxidative phosphorylation
Mitochondrial morphology and bioenergetics exhibit mutual regulation as will be discussed in the subsequent sections.
5. Mitochondria and cellular energy state
The cellular energy state plays an important role in cell death and survival. A minor decrease in cellular ATP levels induces apoptosis but a sharp decrease induces necrosis (125). In resting states, cells mainly utilize energy for protein synthesis (~25–30%) (126), ion gradient maintenance (~15–58%) (127), gluconeogenesis (~7–8%), urea genesis (~3%), DNA transcription and replication (~10%) (126). Therefore, at the time of stress due to alterations in cellular energy state, cells undergo decreased DNA transcription, decreased protein synthesis, increased protein and mRNA half-life and channel arrest (128). Cellular energy (ATP) is mainly produced by aerobic metabolism involving mitochondrial oxphos, except in cultured cells (129, 130). Mitochondrial oxphos is regulated by various factors including cellular ATP/ADP content, mitochondrial dynamics, mitochondrial membrane potential (MMP), calcium homeostasis, intracellular oxygen pressure, and substrate availability.
Cellular homeostasis involves a balance between energy input and utilization. Inability of cells to utilize the nutrient supply or excessive nutrient supply is expected to cause conditions such as adiposity and obesity, associated with various diseases including type 2 diabetes, atherosclerosis and cancer (131). Mitochondria are the major site of energy production and mitochondrial activity is regulated by mitochondrial morphology. With this idea, various studies have reported interdependence between mitochondrial dynamics and bioenergetics. In 2007, Benard et al. (132) suggested an interdependent relationship between mitochondrial shape/distribution and bioenergetics. Increase or decrease in mitochondrial bioenergetics affects MMP which further regulates mitochondrial morphology. Various studies have reported mitochondrial elongation following cellular starvation, and mitochondrial fragmentation following excessive nutrient availability (133). Moreover, cristae morphology is also regulated by mitochondrial respiratory state and rate of ATP production (134). Interestingly, both hyperpolarization (20) and depolarization (135) result in increased fission. Mechanistically, the MMP and energy state regulate the ratio of short to long Opa1 isoforms and therefore, also regulate mitochondrial fusion. As has been reported, patients with decrease in respiratory chain complex 1 show fragmented mitochondria (136, 132). Likewise, inhibition of ETC complexes in mammalian cells causes mitochondrial fragmentation (137, 138). On the other hand, decrease or increase in mitochondrial fusion by regulation of Mfn2 expression in muscle cells, affect glucose oxidation by causing decrease or increase in oxphos complex subunits, respectively (139). Decrease in Mfn1/2 or Opa1 levels in mammalian cells also cause MMP and mitochondrial respiration reduction (112, 6). Similar results of reduced oxphos and ATP synthesis have been observed following repression of Drp1 expression in HeLa cells (28). Moreover, disruption of MMP has been reported to also lead to mitochondrial fission (140, 141).
(5A) Effect of cellular energy state on mitochondrial fission and fusion
Excessive nutrient supply leads to increase in substrate availability to mitochondrial ETC. A recent study has shown an initial increase in ATP synthesis followed by mitochondrial fragmentation on glucose stimulation (142). The increased ATP levels are responsible for insulin secretion in healthy pancreatic β-cells. However, increase in substrate supply also causes increase in basal proton conductance/proton leak and ROS production. Under high glucose conditions, oxidative stress ensues due to decrease in antioxidants, NADH and glutathione levels on increased conversion of glucose to polyalcohol sorbitol (143). Furthermore, increased proton leak could be responsible for decrease in the amount of ATP produced on oxidation of a unit nutrient (ATP synthesis efficiency), at later stages of glucose stimulation. Another reason for decrease in ATP levels could be reduced stability of ATP synthase dimer observed during hyperglycemia (144). The increased ROS and decreased ATP levels during hyperglycemia is considered as the main cause of pathophysiology of diabetes and obesity, as ROS perturbs insulin secretion ability of pancreatic β cells and increases respiratory uncoupling.
Hyperglycemia or hyperlipidemia-induced ROS production usually follows mitochondrial fragmentation and consequently, inhibition of fission reduces oxidative stress (109, 145). Hyperglycemia causes increase in the level of mitochondrial fission proteins (Drp1 and Fis1) and decrease in fusion proteins (Opa1 and Mfn2) (146). These studies suggest that mitochondrial fragmentation induced by nutrient excess is due to imbalanced mitochondrial fission-fusion machineries. Another mechanism involves reduction in Opa1 activity on hyperpolarization/depolarization, caused by excessive nutrient (147, 6) inhibition of phospholipase activity mediated by excess fatty acids, thereby affecting mitochondrial fusion (51, 95, 148). Furthermore, post-translational modifications of mitochondrial fission-fusion machinery also play a major role in regulating mitochondrial fission-fusion and therefore, hyperglycemic conditions. In support of this, high glucose causes increase in Erk1/2 and CaMMK1α-mediated Drp1 phosphorylation, increasing Drp1 activity (149, 150). Moreover, increase in the glucose availability leads to increase in O-glcNAcylation of proteins (151) such as Drp1. Increased O-glcNAcylation of Drp1 causes decrease in Drp1 Ser637 phosphorylation and increase in Drp1 mitochondrial localization, causing fragmentation under high glucose conditions (152). Inhibition of mitochondrial fragmentation reduces hyperglycemia-induced ROS production and apoptosis of β cells, however it affects insulin secreting ability of pancreatic β cells (153, 142).
Cells undergo energy stress or metabolic stress (defined as energy deficiency) during physical exercise, nutrient depletion, or hypometabolism induced during diapause (154). Low amounts of energy stress at early stages cause increase in fusion and decrease in fission suggesting a protective move where further increase in stress accelerates fission and decreases fusion (88). During limited nutrient supply, cells intend to increase the ATP synthesis capacity and autophagy to use cellular organelles and metabolites as source of energy. To support this idea, HepG2 cells show increase in cytochrome c oxidase activity and mtDNA on glucose starvation (155). Furthermore, mitochondria, the power house, undergo elongation to increase ATP synthesis and reduce mitophagy (96, 148). A similar phenomenon is also observed during stress-induced hyperfusion (104). Studies have suggested a role of SLP-2, a mitochondrial inner membrane protein, which has been reported to interact with prohibitin 1 and 2 (156) in stress-induced mitochondrial hyperfusion (SIMH) (104). Mitochondrial elongation offers increased cristae density helping ATP synthase oligomerization and increased activity (96). Like hyperglycemia, nutrient starvation also affects the fission-fusion machinery. Starvation induces mitochondrial elongation with the help of cAMP amplification causing PKA activation. Activated PKA phosphorylates Drp1, inhibiting its mitochondrial translocation, thereby leaving elongated mitochondria (96, 157). Furthermore, oxygen glucose starvation causes increase in the Mfn1 levels and decrease in the Drp1 levels (158). Oxygen glucose deprivation also leads to increase in Drp1 SUMOylation (by SENP-3 degradation), thereby decreasing Drp1 mitochondrial localization and mitochondrial fission. SUMOylated Drp1 further prevents cytochrome c release inhibiting apoptosis, suggesting a protective mechanism (159). The mitochondrial elongation further protects mitochondria from mitophagy, thereby giving them a chance to recover (157). It has been proposed that long exposure to glucose starvation leads to an uncontrolled decrease in the cellular ATP levels and mitochondrial fusion (132). To support this line of evidence, it has been reported that a decrease in ATP synthesis enhances Opa1 cleavage, thereby negatively regulating mitochondrial fusion (85). In addition, defects in oxphos cause Opa1 degradation, leading to increased mitochondrial fragmentation (83). Another reason for reduced fusion following ATP depletion is believed to be the need for GTP for mitochondrial outer membrane and inner membrane fusion and MMP for inner membrane fusion (160). These studies suggest the role of cellular energy state in regulating mitochondrial dynamics (85).
(5B) Effect of mitochondrial fission and fusion on metabolism
Mitochondrial dynamics and bioenergetics exhibit mutual regulation. Balanced fission-fusion is required for efficient bioenergetic capacity (148). Defective mitochondrial fusion reduces mitochondrial metabolism. In rat skeletal muscle, it has been reported that a reduction in the Mfn2 expression causes decreases in glucose oxidation, complex IV activity, MMP and mtDNA, which further lead to a decrease in respiration (161, 162). These effects due to Mfn2 reduction have been found to be reversed following restoration of Mfn2 expression (112). Furthermore, Liesa et al have suggested that decrease in oxphos following Mfn2 deficiency could result due to disruption of ER-mitochondria calcium flux (5). Additionally, Opa1 deficiency also causes a decrease in MMP and respiration (163), and this decrease in respiration is normalized following Opa1 overexpression (112). Dominant optic atrophy-related mutations in Opa1 affect mitochondrial encoded complex 1 subunits and, thereby ATP synthesis (164). On the other hand, affected mitochondrial fission also alters mitochondrial energy production. Drp1 depletion in HeLa cervical cancer cells leads to increase in the mitochondrial fluidity and associated decreases in the complex IV activity, mitochondrial ATP synthesis and mtDNA (132, 28). Collectively, these studies suggest that mitochondrial morphology is regulated by cellular energy state.
6. Defects in mitochondrial dynamics and metabolic disorders
Defects in mitochondrial morphology have recently been found to be associated with disease states (78, 165, 166, 88), including genetic or acquired metabolic disorders. Mutations in the components of mitochondrial fission and fusion machinery precede various pathophysiological disorders including autosomal dominant optic atrophy (ADOA), Charcot Marie Tooth type 2A, Charcot-Marie-Tooth type 4A and neonatal lethality (167, 168, 169, 170).
At resting state, the brain consumes about ~20% of the metabolic energy (171). Neurons with long processes also depend heavily on the mitochondria, for energy supply and calcium buffering, for synaptic neurotransmitter release (172). Furthermore, mitochondria helps in satisfying the neuronal heterogenic energy demand (173). Consequently, Drp1 deficiency in neurons disrupts mitochondrial synaptic dispersion, affecting synaptic activity (174). This makes neurons a preferential target of defective mitochondrial function and morphology. Collectively, these studies suggest that defective mitochondrial morphology may lead to neurodegenerative disorders. Moreover, neurodegenerative diseases are not only associated with defects in mitochondrial morphology but have also been linked to endoplasmic reticulum (ER) and golgi fragmentation (175). Various drugs have been developed to target mitochondrial defects associated neurodegenerative diseases.
(6A) Autosomal dominant optic atrophy
Autosomal dominant optic atrophy (ADOA) is an autosomal inherited disease, affecting retinal ganglion cells, thereby causing optic nerve atrophy leading to visual impairment. Mutation in the Opa1 gene is the most common cause of ADOA. ADOA could also result due to haploinsufficiency and dominant-negative mechanism (167, 176). Some ADOA related Opa1 mutations are associated with mtDNA deletion causing external opthalmoplegia, ataxia and deafness (177). In order to understand the pathology of ADOA, various researchers have made knock out/knock in mice of fission-fusion proteins. Related studies have suggested a possible pathway underlying 0the ADOA pathophysiology i.e., the inability of the mutated Opa1 to be stimulated by cardiolopin to hydrolyze GTP and therefore, impairing the mitochondrial fusion (178). Opa1 knockdown in retinal ganglion cells is also associated with a decrease in the mtDNA, oxphos, ATP synthesis and disruption of calcium homeostasis (72).
(6B) Charcot Marie tooth type 2A
Charcot Marie tooth type 2A (CMT2A) is the most common hereditary peripheral neuropathy (179). Most of the CMT2A cases involve Mfn2 mutations, mainly in the GTPase domain. These mutations could be gain of function or loss of function. Interestingly, gain or loss of Mfn2 function gives similar phenotype. For example, loss-of-function mutations induce mitochondrial aggregation (168) and gain of function affects mitochondrial motility on axons and therefore, mitochondria axonal distribution (180). Defects in mitochondrial motility have also been linked to CMT2A development (181). However, human skin fibroblast samples obtained from CMT2A patients have been reported to show wild type mitochondrial morphology which could be due to complementary effect of Mfn1 (182, 5). However, CMT2A patients have reduced oxphos (183). Defective oxphos in CMT2A patients has been linked with increase in mtDNA deletion (184, 185).
Interestingly, Mfn2 mutations causing CMT2A has been linked to amyotrophic lateral sclerosis (ALS) suggesting overlapping mechanism in pathogenesis of two diseases (186). Various studies have also reported overlapping symptoms in CMTA-2A and ADOA as in the case of hereditary motor and sensory neuropathy (187–189). These reports would suggests that the signaling pathway/protein affected by mutations in Mfn2 and Opa1 are not mutually exclusive.
(6C) Costeff optic atrophy syndrome
Costeff optic atrophy syndrome is also known as the Type III 3-methylglutaconic aciduria or optic atrophy plus syndrome (190). This disease is mainly associated with mutation in a mitochondrial inner membrane protein, OPA3. Mutation in OPA3 is believed to cause bilateral optic atrophy (191), and familial OPA3 mutant (G93S) is linked also to increased mitochondrial fragmentation (40). Further studies are needed to better understand the mechanism involved in optic atrophy plus syndrome pathogenesis.
(6D) Charcot Marie tooth type 4A
Charcot Marie tooth type 4A (CMT4A) is an inherited peripheral neuropathy that has an association with mutations in GDAP1 (169). To date, more than 40 mutations in GDAP1 have been linked to CMT4A (192). CMT4A patients express reduced levels of wild type GDAP1, glutathione and MMP. GDAP1 deficiency is believed to cause mitochondria and peroxisome elongation (193). Noack et al. reported that the knockdown of wild type GDAP1 causes increased sensitivity to oxidative stress, decrease in glutathione (GSH) levels and decrease in MMP, whereas overexpression of GDAP1 normalizes the above mentioned phenotypes (194). These studies suggest a role for oxidative stress in CMT4A pathogenesis.
(6E) Wolf-Hirschhorn Syndrome
Wolf-Hirschhorn Syndrome (WHS) is characterized by a deletion of WHSC1 gene and/or the entire WHSCR region within which WHSC1 gene resides (195). Leucine zipper-EF-hand containing transmembrane protein 1 (Letm1), localized in WHSCR has been linked with WHS progression. Letm1 is a mitochondrial IM Ca2+/H+ antiporter. Letm1 mRNA and protein levels are reduced in WHS patient fibroblast (196). Letm1 knockdown is believed to cause mitochondrial fragmentation, reduction in ATP synthesis and altered glucose metabolism (196, 197, 198).
(6F) Non-alcoholic fatty liver disease
NAFLD is associated with fat collection in the liver and according to American Liver Foundation, NAFLD affects ~25% of the US population. NAFLD progression has been linked to increased mitochondrial ROS (199), reduced ATP production (200), and abnormal mitochondrial morphology. Excess fat in NAFLD causes increase in lipid/substrate availability for β-oxidation, forming more NADH. Increase in NADH production causes an enhanced rate of ETC, thereby increasing electron leakage from complex I and III leading to elevated levels of superoxide production (201, 6). This can explain the reduced activity of all ETC complexes in patients with nonalcoholic steatohepatitis (NASH), a severe form of NAFLD, have (202).
(6G) Familial amyotrophic lateral sclerosis
Familial amyotrophic lateral sclerosis (FALS) constitutes ~10% of ALS cases. This motor neuron disease is characterized by hypometabolism (203). It is associated with mutations in SOD1 gene (204) and possibly involves SOD1 misfolding (205). SOD1 mutation in motor neurons show decrease in mitochondrial size, number and MMP (204). This defect has been related to deceases in mitochondrial fusion and motility (204). Likewise, ALS transgenic mice, with mutant-SOD1 overexpression, display altered mitochondrial structure, which precedes defects in mitochondrial respiration (206, 207). Further, mutations in TAR DNA-binding protein 43(TDP-43), fused/trans located in liposarcoma (FUS/TLS) protein and transitional ER ATPase protein have also been linked to ALS (208, 209).
(6H) Alzheimer disease
Alzheimer disease (AD) is a neurodegenerative disorder marked by extracellular β-amyloid plaques and intracellular tangles of hyper phosphorylated tau protein (210). According to Swerdlow et al. (211) mitochondrial dysfunction follows AD pathophysiology. In 2008, Wang et al. (212) linked β-amyloid overproduction with increase in number of fragmented mitochondria (clustered in perinuclear region), increase in oxidative stress and losses of MMP and ATP production. This increased mitochondrial fragmentation is associated with increase in expression of Drp1 during AD (212). Accumulated β-amyloid also causes increase in Drp1 S-nitrosylation at Cys644, thereby enhancing Drp1 activity in neurons (213). Likewise, AD patients are characterized by having Aβ-Drp1 mediated mitochondrial fragmentation, mtDNA mutations (214) and decrease in oxphos (215). Further, AD is associated with various metabolic abnormalities including decreases in pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (KGDH), and ETC complex IV (216). In the AD patients, defective oxphos is believed to induce a switch to neuronal glycolysis (217).
(6I) Parkinson's disease
Parkinson's disease (PD) is marked by deprivation of the dopaminergic neurons in substantia niagra and accumulation of lewy bodies (210). Familial PD has been linked to mutations in Pink1, α-synuclein, Parkin, DJ-1, LRRK2, NURR-1, tau and mtDNA (reviewed in 207, 218, 219). Tfam-deficient mice develop PD in adulthood (220) indicating that the mtDNA appears to also have a role in PD pathogenesis. Further, mitochondrial accumulation of α-synuclein disrupts complex 1 activity. Likewise, PD patients have reduced complex I activity in substantia nigra and Parkin knockout mice have decrease in complex I and IV subunits levels, thereby reduced respiratory capacity (221, 222) and metabolic defects. Mitochondrial dysfunction observed during PD could be the result of defective mitophagy. Recently, it has been reported that PINK1 expression inhibits Drp1 mitochondrial translocation, suggesting a possible mechanism responsible for the increase in mitochondrial fragmentation in PD patients that harbor mutated PINK1 (223, 224). It is of note that mitochondrial abnormality associated with mutated PINK1/Parkin could be reversed by the overexpression of Drp1 in Drosophila (225, 226), suggesting mitochondrial fission to be downstream of PINK1/Parkin. Further, mitochondrial spheroids observed during PD could also result due to the inability of mutated Parkin to degrade Mfn2 (227). Mutation in Parkin also decreases Parkin mediated Drp1 and Mfn1/2 ubiquitination and degradation, and therefore, lessens mitophagy, ultimately leading to mitochondrial dysfunction (228, 229, 230). PD patients have also shown increased expression of cell cycle proteins such as Rb, which are involved in up-regulation of Drp1 (231). Oxidative stress has also been shown to induce mitochondrial fragmentation in the cellular model of PD and the latter phenotype could be rectified with antioxidant treatment (232). Collectively, these studies suggest roles for mitochondrial dynamics, mitophagy and oxidative stress in the PD pathophysiology.
(6J) Huntington's disease
Huntington's disease (HD) is mainly caused by expansion of a CAG trinucleotide sequence in huntington (Htt) gene, leading to an increase in the polyglutamine tract (233). HD pathogenesis is believed to involve mitochondrial dysfunction in brain as well as peripheral tissues (207), increase in mitochondrial fragmentation (234), mitochondrial respiration and ATP levels (235). Consistent with these findings, studies on HD mouse model also show reduced complex II and IV activity (236, 237). Another possible mechanism of reduction in mitochondrial activity involves interactions between mutant Htt and p53. These interactions cause an increase in p53 activity leading to a drop in MMP (238). Likewise, HD patients also suffer from decreased body weight (239) and have reduced glucose metabolism rate (240). Collectively, these lines of evidence suggest that metabolic defects are possibly linked to HD pathology. Interestingly, HD patients have increased mitochondrial fragmentation or reduced fusion and that could be due to high levels of fission proteins (Drp1 and Fis1) and low levels of fusion proteins (Mfn1 and Mfn2) (236). Similarly, expression of Htt with extended poly glutamine repeats in HeLa cervical cells cause increase in the mitochondrial fragmentation (241). This increase in fragmentation is believed to be mediated by an increase in the GTPase activity of Drp1 on MOM upon binding with mutated Htt (234, 242, 175, 243). Drp1 S-nitrosylation is reported as one of the key events in mutated HTT pathogenesis of Huntington disease and accordingly, blocking Drp1 S-nitrosylation abrogates mtHtt-mediated toxic effects (244). MtHtt also causes increase in oxidative mtDNA damage (245). Further, mitochondria with damaged DNA may not be able to compensate, due to reduced fusion and thereby causing decreased energy production and metabolic defects. HD patients also have impaired mitochondrial motility due to defective mitochondria-microtubule association (246), this decrease in mitochondrial motility may further decrease mitochondrial fusion. Cagalinec et al (94) suggested that restoring mitochondrial motility by overexpression of Miro1, helps neurons to fix defects in fusion, thereby offer wild type mitochondrial morphology. Interestingly, like various other neurodegenerative disorders, HD is also linked to oxidative stress, as mutant Htt lessens PGC 1 α activity making neurons more prone to oxidative damage (247, 248).
(6K) Type 2 Diabetes and Obesity (Diabesity)
According to American Diabetes Association, one third of the US population is obese and ~85% of those with type 2 diabetes are believed to be obese. Obesity is clearly considered as a risk factor for type 2 diabetes. It is well-established that type 2 diabetes and obesity are metabolic disorders. Type 2 diabetes and obese patients show decrease in mitochondrial size, number, oxphos activity, oxphos subunit expression, ATP synthesis, mtDNA levels, increase in lipid accumulation and altered mitochondrial morphology (249–251). The mitochondrial dysfunction has also been related to insulin resistance either by blocking insulin signaling or through oxidative stress (252). Obesity and type 2 diabetes are associated with hyperglycemia and cardiac steatosis (253), and involve patients' inability either to secret insulin or sense insulin. Hyperglycemia causes increase in substrate input to ETC, amplifying superoxide production. ROS thus produced, are considered as the major culprit involved in diabetic pathogenesis (254–256). The animal mice models with type 1 and type 2 diabetes also show an increase in the ROS levels. Further, inability of ROS to induce UCP3 is believed to result in an increase in oxidative damage in the diabetic patients (257). Increased ROS in diabetic mice also increases tumor metastasis (258). Interestingly, oxidative stress induced during the hyperglycemia could be alleviated by inhibiting mitochondrial fission (6). There are lines of evidence to support that ROS production and mitochondrial fragmentation exhibit a mutual regulatory relationship. Furthermore, exposure to high glucose is believed to also induce mitochondrial fragmentation. Likewise, pancreatic β-cells and skeletal muscles of type 2 diabetes patients show increase in mitochondrial fragmentation (259, 260) which could be explained with decrease in Mfn2 levels in obese and type 2 diabetes patients (66) and increase in Drp1 mitochondrial localization in high glucose stimulated neurons (261). Reduced Mfn2 levels have also been linked to reduced MMP and oxphos (154). Another pathway involved in the repressed mitochondrial networking in type 2 diabetic and obese patients is the decrease in PGC-1 α and NRF1 levels (154, 250). Decrease in Mfn2 levels could be the result of decreased PGC-1 α activity, affecting Mfn2 transcription (262). The reduced Mfn2 levels are expected to worsen the effect of nutrient excess (263).
Diabetic patients are often diagnosed with diabetic cardiomyopathy (DCM) due to accumulation of triglyceride and cholesterol (264) causing ischemia and hypertension. This cardiac steatosis is believed to mark the beginning of diabetes mellitus (263, 265). Diabetic atrial tissues have higher tendency to undergo mitochondrial pore opening and mitochondria-mediated apoptosis (266). Furthermore, artrial tissue of type 2 diabetic patients have defect in fatty acid oxidation and show increased oxidative stress (256). Galloway and Yoon speculated that mitochondrial dysfunction due to oxidative stress could be among the major causes of DCM pathophysiology (6).
(6L) Cancer
Cancer cells are characterized by decrease in oxphos and increase in glycolysis. Decrease oxphos could be due to variety of alterations for example, defective TCA (mutated succinate dehydrogenase, fumarate hydratase), ETC (SCO2), mutations in mtDNA, decrease in mtDNA level, oxidative stress, (267, 268). Overexpression of an oncogenic protein Yorkie/YAP-2 has been reported to cause an increase in transcription of mitochondrial fusion proteins (Opa1 and Marf), thereby leading to an increase in mitochondrial fusion (4, 269). Yorkie/YAP-2 appears to be also involved in cyclin E accumulation during G1/S transition (98). Furthermore, increase in Mfn2 expression in colorectal cell lines is believed to cause cell cycle arrest at G2/M (270). Mfn2 overexpression has also shown anti-proliferative effects in hepatocellular carcinoma and gastric cancer (271, 272).
(6M) Other Disorders
A variety of other disorders are also associated with defective fission-fusion machinery. For example, A395D mutation in Drp1 has been related to abnormal brain development linked to neonatal lethality (170). This heterozygous mutation is believed to cause defects in mitochondria and peroxisome fission, which could be responsible for observed modifications in mitochondrial and peroxisomal oxidation (170, 5, 273). Available evidence also suggests that A395D mutation affects Drp1 assembly (273). Furthermore, defects in mitochondrial fission and fusion have been linked to mitochondrial dysfunction in critically ill patients (274). Mutations in mitochondrial fusion machinery (Opa1 and Mfn2) have also been associated with mtDNA mutation and deficiency (reviewed by (275)).
Because mitochondrial fission and fusion are believed to play important roles in the pathophysiology of various neurodegenerative diseases, drugs are being developed to target fission-fusion. Mitochondria division inhibitor-1 (mdivi-1) is one example of such drugs. Mdivi-1 has been reported to inhibit Drp1 activity by preventing Bax/Bak mediated mitochondrial pore formation (276). Mdivi helps in mitochondrial networking, similar to Mfn2 overexpression, causing cell cycle arrest in cancer cells (277). Mdivi also prevents the oxidative stress followed by mitochondrial fission (278). M1 hydrazone is another drug that helps in increasing mitochondrial fusion by regulation of Mfn activity. Idebenone, an antioxidant that is used in the management of Alzheimer has recently been advanced to clinical trials for possible treatment of ADOA (279). Thiazolidinediones and metformin are drugs that have shown therapeutic potential to ameliorate hyperglycemic complications during type 2 diabetes and such effects are believed to be mediated via reduction in mitochondrial ROS (280, 281) Various other drugs including creatine, CoQ10, NAC and similar other antioxidants are showing promising neuroprotective activity and are currently in clinical trials (207).
7. Conclusion
This article provides the current state of knowledge of the topic by highlighting a relationship between mitochondrial morphology and function, especially energy production. It is becoming increasingly evident that nutrient starvation leads to mitochondrial elongation, whereas excessive nutrient supply causes mitochondrial fission. Accordingly, a balance between fission and fusion helps cells to adapt to conditions of varying nutrient availability. It has also been reported that defects in or inhibition of mitochondrial electron transport chain alter mitochondrial morphology. Oxidative stress has also been linked to bioenergetic defects as well as abnormal mitochondrial structure, and both of the latter events exhibit interdependent relationship. It is also becoming evident that the regulatory pathways affecting mitochondrial morphology, bioenergetics and oxidative stress are interconnected. Therefore, future studies are warranted to fully elucidate the details of these mechanisms particularly in context to fission and fusion. The outcome of such studies is expected to improve the understanding of molecular pathogenesis of the various metabolic disorders and form the basis for future development of better therapeutics.
Acknowledgements
The work in MSS laboratory is supported partly by the NIH grant CA157168
List of Abbreviations
- AD
Alzheimer disease
- ADOA
Autosomal dominant optic atrophy
- BSE
Bundle Stacking Element
- CMT2A
Charcot Marie tooth type 2A
- CMT4A
Charcot Marie tooth type 4A
- Drp1
Dynamin related protein-1
- ETC
Electron transport chain
- FALS
Familial amyotrophic lateral sclerosis
- Fis1
Fission protein 1
- GADP1
Ganglioside-induced differentiation associated protein 1
- GSH
Glutathione
- GED
GTPase effector domain
- HR
Heptad regions
- Htt
Huntington (Htt) gene
- HD
Huntington's disease
- IM
Inner-membrane
- IMS
Inter-membrane space
- MIB
Mfn-binding protein
- mtDNA
Mitochondrial DNA
- MiDs
Mitochondrial dynamics proteins
- MMP
Mitochondrial membrane potential
- MOM
Mitochondrial outer-membrane
- MTP18
Mitochondrial protein 18kDa
- MULAN
Mitochondrial ubiquitin ligase activator of NFκB
- Mfn
Mitofusin
- HAFLD
Non-alcoholic fatty liver disease
- Opa1
Optic atrophy
- Oxphos
Oxidative phosphorylation
- PD
Parkinson's disease
- PINK1
PTEN-induced putative kinase protein 1
- ROS
Reactive oxygen species
- TCA
Tricarboxylic acid cycle
- WHS
Wolf-Hirschhorn Syndrome
Footnotes
Conflict of interests The authors declare no conflicts of interest.
REFERENCES
- 1.Benda C. Arch Anat Physiol. 1898;73:393–398. [Google Scholar]
- 2.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–24. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- 3.Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial Dynamics in Diabetes. Antioxid Redox Signal. 2011;14:439–57. doi: 10.1089/ars.2010.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Liesa M, Palacin M, Zorzano A. Mitochondrial Dynamics in Mammalian Health and Disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 6.Galloway CA, Yoon Y. What comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol. 2012;139:455–63. doi: 10.1085/jgp.201210771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–68. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Westermann B. Molecular Machinery of Mitochondrial Fusion and Fission. J Biol Chem. 2008;283:13501–5. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- 9.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 10.Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M. Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem. 2003;278:8597–605. doi: 10.1074/jbc.M211761200. [DOI] [PubMed] [Google Scholar]
- 11.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–16. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Strack S, Cribbs JT. Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem. 2012;287(14):10990–1001. doi: 10.1074/jbc.M112.342105. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fröhlich C, Grabiger S, Schwefel D, et al. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013;32:1280–92. doi: 10.1038/emboj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smirnova E, Griparic L, Shurland D-L, Bliek AM van der. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol Biol Cell. 2001;12:2245. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER Tubules Mark Sites of Mitochondrial Division. Science. 2011;334:358–62. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon Y, Pitts KR, McNiven MA. Mammalian Dynamin-like Protein DLP1 Tubulates Membranes. Mol Biol Cell. 2001;12:2894. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingerman E, Perkins EM, Marino M, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–7. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Bliek AM, Payne GS. Dynamin Subunit Interactions Revealed. Dev Cell. 2010;18:687–8. doi: 10.1016/j.devcel.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montessuit S, Somasekharan SP, Terrones O, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frank S, Gaume B, Bergmann-Leitner ES, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 21.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–50. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.James DI, Parone PA, Mattenberger Y, Martinou J-C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–9. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 23.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koirala S, Bui HT, Schubert HL, et al. Molecular architecture of a dynamin adaptor: implications for assembly of mitochondrial fission complexes. J Cell Biol. 2010;191:1127–39. doi: 10.1083/jcb.201005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–36. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 26.Lee Y, Jeong S-Y, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onoue K, Jofuku A, Ban-Ishihara R, et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci. 2013;126:176–85. doi: 10.1242/jcs.111211. [DOI] [PubMed] [Google Scholar]
- 28.Parone PA, Da Cruz S, Tondera D, et al. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS ONE. 2008;3:e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117:1201–10. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 30.Yonashiro R, Ishido S, Kyo S, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–26. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–12. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otera H, Mihara K. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases. 2011;2:167–72. doi: 10.4161/sgtp.2.3.16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–73. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J, Liu T, Jin S, et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–78. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–59. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 36.Niemann A, Ruegg M, La Padula V, Schenone A, Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 2005;170:1067–78. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karbowski M, Jeong S-Y, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–39. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elwi AN, Lee B, Meijndert HC, Braun JEA, Kim S-W. Mitochondrial chaperone DnaJA3 induces Drp1-dependent mitochondrial fragmentation. Int J Biochem Cell Biol. 2012;44:1366–76. doi: 10.1016/j.biocel.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Bengtson MH, Ulbrich A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu S-W, Jeong HJ, Choi M, Karbowski M, Choi C. Optic atrophy 3 as a protein of the mitochondrial outer membrane induces mitochondrial fragmentation. Cell Mol Life Sci. 2010;67:2839–50. doi: 10.1007/s00018-010-0365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnoult D, Rismanchi N, Grodet A, et al. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–8. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 42.Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–7. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyer O, Nevo F, Plaisier E, et al. INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med. 2011;365:2377–88. doi: 10.1056/NEJMoa1109122. [DOI] [PubMed] [Google Scholar]
- 44.Malka F, Guillery O, Cifuentes-Diaz C, et al. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005;6:853–9. doi: 10.1038/sj.embor.7400488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sukhorukov VM, Dikov D, Reichert AS, Meyer-Hermann M. Emergence of the Mitochondrial Reticulum from Fission and Fusion Dynamics. PLoS Comput Biol. 2012;8:e1002745. doi: 10.1371/journal.pcbi.1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell. 1997;8:1233–42. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–74. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 48.Koshiba T. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science. 2004;305:858–62. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 49.Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–74. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 50.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi S-Y, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–62. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 52.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mole Cell Bio. 2007;8:870–9. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 53.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–46. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 54.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–32. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cohen MMJ, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM. Ubiquitin-Proteasome-dependent Degradation of a Mitofusin, a Critical Regulator of Mitochondrial Fusion. Mol Biol Cell. 2008;19:2457–64. doi: 10.1091/mbc.E08-02-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gegg ME, Cooper JM, Chau K-Y, Rojo M, Schapira AHV, Taanman J-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–70. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leboucher GP, Tsai YC, Yang M, et al. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–57. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Livnat-Levanon N, Glickman MH. Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim Biophys Acta. 2011;1809:80–7. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 59.Sugiura A, Nagashima S, Tokuyama T, et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol Cell. 2013;51:20–34. doi: 10.1016/j.molcel.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 60.Muñoz JP, Ivanova S, Sánchez-Wandelmer J, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–61. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–5. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karbowski M, Lee Y-J, Gaume B, Jeong S-Y, Frank S, Nechushtan A, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–8. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura N, Hirose S. Regulation of Mitochondrial Morphology by USP30, a Deubiquitinating Enzyme Present in the Mitochondrial Outer Membrane. Mol Biol Cell. 2008;19:1903–11. doi: 10.1091/mbc.E07-11-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eura Y, Ishihara N, Oka T, Mihara K. Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J Cell Sci. 2006;119:4913–25. doi: 10.1242/jcs.03253. [DOI] [PubMed] [Google Scholar]
- 66.Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. The International Journal of Biochemistry & Cell Biology. 2009;41:1846–54. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Chen H, Vermulst M, Wang YE, et al. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell. 2010;141:280–9. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Delettre C, Lenaers G, Griffoin J-M, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–10. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 69.Meglei G, McQuibban GA. The dynamin-related protein Mgm1p assembles into oligomers and hydrolyzes GTP to function in mitochondrial membrane fusion. Biochemistry. 2009;48:1774–84. doi: 10.1021/bi801723d. [DOI] [PubMed] [Google Scholar]
- 70.Pidoux G, Witczak O, Jarnæss E, et al. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 2011;30:4371–86. doi: 10.1038/emboj.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–8. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- 72.Kushnareva YE, Gerencser AA, Bossy B, et al. Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 2013;20:353–65. doi: 10.1038/cdd.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fülöp L, Szanda G, Enyedi B, Várnai P, Spät A. The effect of OPA1 on mitochondrial Ca2+ signaling. PLoS ONE. 2011;6:e25199. doi: 10.1371/journal.pone.0025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamaguchi R, Lartigue L, Perkins G, et al. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–69. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frezza C, Cipolat S, Martins de Brito O, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–89. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 76.Elachouri G, Vidoni S, Zanna C, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011;21:12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Twig G, Elorza A, Molina AJA, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan DC. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu Rev Genet. 2012;46:265–87. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 79.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–55. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olichon A, Emorine LJ, Descoins E, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–6. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- 81.Cipolat S, Rudka T, Hartmann D, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–75. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 82.Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–64. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. The EMBO Journal. 2006;25:2966–77. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–66. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baricault L, Ségui B, Guégand L, et al. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp Cell Res. 2007;313:3800–8. doi: 10.1016/j.yexcr.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 86.An H-J, Cho G, Lee J-O, Paik S-G, Kim YS, Lee H. Higd-1a interacts with Opa1 and is required for the morphological and functional integrity of mitochondria. Proc Natl Acad Sci USA. 2013;110:13014–9. doi: 10.1073/pnas.1307170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guillery O, Malka F, Landes T, et al. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell. 2008;100:315–25. doi: 10.1042/BC20070110. [DOI] [PubMed] [Google Scholar]
- 88.Bliek AM van der, Shen Q, Kawajiri S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–32. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belenguer P, Pellegrini L. The dynamin GTPase OPA1: more than mitochondria? Biochim Biophys Acta. 2013;1833:176–83. doi: 10.1016/j.bbamcr.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 91.DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rahn JJ, Stackley KD, Chan SSL. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE. 2013;8:e59218. doi: 10.1371/journal.pone.0059218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Olichon A, Landes T, Arnauné-Pelloquin L, et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. 2007;211:423–30. doi: 10.1002/jcp.20950. [DOI] [PubMed] [Google Scholar]
- 94.Cagalinec M, Safiulina D, Liiv M, et al. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013;126:2187–97. doi: 10.1242/jcs.118844. [DOI] [PubMed] [Google Scholar]
- 95.Huang P, Galloway CA, Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS ONE. 2011;6:e20655. doi: 10.1371/journal.pone.0020655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gomes LC, Benedetto GD, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–98. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Horn SR, Thomenius MJ, Johnson ES, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 2011;22:1207–16. doi: 10.1091/mbc.E10-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–5. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 100.Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxid Redox Signal. 2012;16:1150–80. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kashatus DF, Lim K-H, Brady DC, Pershing NLK, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol10. 2011;13:1108–15. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–95. 1. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitra K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. BioEssays. 2013;35:955–64. doi: 10.1002/bies.201300011. [DOI] [PubMed] [Google Scholar]
- 104.Tondera D, Grandemange S, Jourdain A, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Redpath CJ, Bou Khalil M, Drozdzal G, Radisic M, McBride HM. Mitochondrial Hyperfusion during Oxidative Stress Is Coupled to a Dysregulation in Calcium Handling within a C2C12 Cell Model. PLoS ONE. 2013;8:e69165. doi: 10.1371/journal.pone.0069165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–15. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Park J, Lee J, Choi C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE. 2011;6:e23211. doi: 10.1371/journal.pone.0023211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park J, Choi C. Contribution of mitochondrial network dynamics to intracellular ROS signaling. Commun Integr Biol. 2012;5:81–3. doi: 10.4161/cib.18257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–8. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–94. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 112.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 113.Ziviani E, Tao RN, Whitworth AJ. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proceedings of the National Academy of Sciences. 2010;107:5018–23. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287:40652–60. doi: 10.1074/jbc.M112.419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang H, Song P, Du L, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–58. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pallanck L. Mitophagy: Mitofusin Recruits a Mitochondrial Killer. Curr Biol. 2013;23:R570–R572. doi: 10.1016/j.cub.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 119.Fedorowicz MA, de Vries-Schneider RLA, Rüb C, et al. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2013 doi: 10.1002/embr.201337294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3:461–91. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ban-Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. PNAS. 2013;110:11863–8. doi: 10.1073/pnas.1301951110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tauber J, Dlasková A, Šantorová J, et al. Distribution of mitochondrial nucleoids upon mitochondrial network fragmentation and network reintegration in HEPG2 cells. Int J Biochem Cell Biol. 2013;45:593–603. doi: 10.1016/j.biocel.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 123.Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–10. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- 124.Saotome M, Safiulina D, Szabadkai G, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA. 2008;105:20728–33. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann N Y Acad Sci. 2010;1201:50–7. doi: 10.1111/j.1749-6632.2010.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–58. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 127.Covi JA, Hand SC. Energizing an invertebrate embryo: bafilomycin-dependent respiration and the metabolic cost of proton pumping by the V-ATPase. Physiol Biochem Zool. 2007;80:422–32. doi: 10.1086/518344. [DOI] [PubMed] [Google Scholar]
- 128.Hand SC, Menze MA. Mitochondria in energy-limited states: mechanisms that blunt the signaling of cell death. J Exp Biol. 2008;211:1829–40. doi: 10.1242/jeb.000299. [DOI] [PubMed] [Google Scholar]
- 129.Gnaiger E, Kemp RB. Anaerobic metabolism in aerobic mammalian cells: information from the ratio of calorimetric heat flux and respirometric oxygen flux. BBA-Bioenergetics. 1990;1016:328–32. doi: 10.1016/0005-2728(90)90164-y. [DOI] [PubMed] [Google Scholar]
- 130.Kuznetsov AV, Janakiraman M, Margreiter R, Troppmair J. Regulating cell survival by controlling cellular energy production: novel functions for ancient signaling pathways? FEBS Letters. 2004;577:1–4. doi: 10.1016/j.febslet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 131.Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid Redox Signal. 2013;19:240–2. doi: 10.1089/ars.2013.5255. [DOI] [PubMed] [Google Scholar]
- 132.Benard G, Bellance N, James D, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–48. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 133.Molina AJA, Wikstrom JD, Stiles L, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–15. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966;30:269–97. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meeusen SL, Nunnari J. How mitochondria fuse. Curr Opin Cell Biol. 2005;17:389–94. doi: 10.1016/j.ceb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 136.Koopman WJH, Visch H-J, Verkaart S, van den Heuvel LWPJ, Smeitink JAM, Willems PHGM. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol, Cell Physiol. 2005;289:C881–890. doi: 10.1152/ajpcell.00104.2005. [DOI] [PubMed] [Google Scholar]
- 137.Plecitá-Hlavatá L, Lessard M, Šantorová J, Bewersdorf J, Ježek P. Mitochondrial oxidative phosphorylation and energetic status are reflected by morphology of mitochondrial network in INS-1E and HEPG2 cells viewed by 4Pi microscopy. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2008;1777:834–46. doi: 10.1016/j.bbabio.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 138.Guillery O, Malka F, Frachon P, Milea D, Rojo M, Lombès A. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord. 2008;18:319–30. doi: 10.1016/j.nmd.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 139.Pich S, Bach D, Briones P, et al. The Charcot–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–15. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 140.Legros F, Lombès A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–54. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ishihara N, Jofuku A, Eura Y, Mihara K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem Biophys Res Commun. 2003;301:891–8. doi: 10.1016/s0006-291x(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 142.Jhun BS, Lee H, Jin Z-G, Yoon Y. Shirihai OS, editor. Glucose Stimulation Induces Dynamic Change of Mitochondrial Morphology to Promote Insulin Secretion in the Insulinoma Cell Line INS-1E. PLoS ONE. 2013;8:e60810. doi: 10.1371/journal.pone.0060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brownlee M. The Pathobiology of Diabetic Complications A Unifying Mechanism. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 144.Domenis R, Bisetto E, Rossi D, Comelli M, Mavelli I. Glucose-modulated mitochondria adaptation in tumor cells: a focus on ATP synthase and inhibitor factor 1. Int J Mol Sci. 2012;13:1933–50. doi: 10.3390/ijms13021933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nakamura T, Ako J, Kadowaki T, et al. Impact of acute hyperglycemia during primary stent implantation in patients with ST-elevation myocardial infarction. J Cardiol. 2009;53:272–7. doi: 10.1016/j.jjcc.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 146.Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia Alters Mitochondrial Fission and Fusion Proteins in Mice Subjected to Cerebral Ischemia and Reperfusion. Transl Stroke Res. 2012;3:296–304. doi: 10.1007/s12975-012-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Duvezin-Caubet S, Jagasia R, Wagener J, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–9. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 148.Liesa M, Shirihai OS. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yu T, Jhun BS, Yoon Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production Through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxid Redox Signal. 2011;14:425–37. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Han X-J, Lu Y-F, Li S-A, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–85. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010;36:423–35. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 152.Gawlowski T, Suarez J, Scott B, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. 2012;287:30024–34. doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Men X, Wang H, Li M, et al. Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. Int J Biochem Cell B. 2009;41:879–90. doi: 10.1016/j.biocel.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 154.Reynolds JA, Hand SC. Differences in isolated mitochondria are insufficient to account for respiratory depression during diapause in artemia franciscana embryos. Physiol Biochem Zool. 2004;77:366–77. doi: 10.1086/420950. [DOI] [PubMed] [Google Scholar]
- 155.Weber K, Ridderskamp D, Alfert M, Hoyer S, Wiesner RJ. Cultivation in glucose-deprived medium stimulates mitochondrial biogenesis and oxidative metabolism in HepG2 hepatoma cells. Biol Chem. 2002;383:283–90. doi: 10.1515/BC.2002.030. [DOI] [PubMed] [Google Scholar]
- 156.Da Cruz S, Parone PA, Gonzalo P, et al. SLP-2 interacts with prohibitins in the mitochondrial inner membrane and contributes to their stability. Biochim Biophys Acta. 2008;1783:904–11. doi: 10.1016/j.bbamcr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 157.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–5. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wappler EA, Institoris A, Dutta S, Katakam PVG, Busija DW. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS ONE. 2013;8:e63206. doi: 10.1371/journal.pone.0063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013;32:1514–28. doi: 10.1038/emboj.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–52. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 161.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 162.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–5. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 163.Olichon A, Baricault L, Gas N, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–6. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 164.Zanna C, Ghelli A, Porcelli AM, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131:352–67. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- 165.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Youle RJ, Bliek AM van der. Mitochondrial Fission, Fusion, and Stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–5. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 168.Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations. J Neurosci. 2007;27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–2. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- 170.Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wanders RJA, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–41. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 171.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 172.Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–9. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 173.Chang DTW, Honick AS, Reynolds IJ. Mitochondrial Trafficking to Synapses in Cultured Primary Cortical Neurons. J Neurosci. 2006;26:7035–45. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Verstreken P, Ly CV, Venken KJT, Koh T-W, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–78. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 175.Knott AB, Bossy-Wetzel E. Impairing the Mitochondrial Fission and Fusion Balance: A New Mechanism of Neurodegeneration. Ann NY Acad Sci. 2008;1147:283–92. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pesch UE, Leo-Kottler B, Mayer S, et al. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum Mol Genet. 2001;10:1359–68. doi: 10.1093/hmg/10.13.1359. [DOI] [PubMed] [Google Scholar]
- 177.Hudson G, Amati-Bonneau P, Blakely EL, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–37. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- 178.Ban T, Heymann JAW, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;9:2113–22. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Karbowski M, Neutzner A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012;123:157–71. doi: 10.1007/s00401-011-0921-0. [DOI] [PubMed] [Google Scholar]
- 180.Chapman AL, Bennett EJ, Ramesh TM, De Vos KJ, Grierson AJ. Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS ONE. 2013;8:e67276. doi: 10.1371/journal.pone.0067276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cartoni R, Martinou J-C. Role of mitofusin 2 mutations in the physiopathology of Charcot–Marie–Tooth disease type 2A. Exp Neurol. 2009;218:268–73. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 182.Amiott EA, Lott P, Soto J, et al. Mitochondrial fusion and function in Charcot-Marie-Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol. 2008;21:115–27. doi: 10.1016/j.expneurol.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Loiseau D, Chevrollier A, Verny C, et al. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann Neurol. 2007;61:315–23. doi: 10.1002/ana.21086. [DOI] [PubMed] [Google Scholar]
- 184.Guillet V, Gueguen N, Verny C, et al. Adenine nucleotide translocase is involved in a mitochondrial coupling defect in MFN2-related Charcot–Marie–Tooth type 2A disease. Neurogenetics. 2010;11:127–33. doi: 10.1007/s10048-009-0207-z. [DOI] [PubMed] [Google Scholar]
- 185.Vielhaber S, Debska-Vielhaber G, Peeva V, et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013;125:245–56. doi: 10.1007/s00401-012-1036-y. [DOI] [PubMed] [Google Scholar]
- 186.Marchesi C, Ciano C, Salsano E, et al. Co-occurrence of amyotrophic lateral sclerosis and Charcot-Marie-Tooth disease type 2A in a patient with a novel mutation in the mitofusin-2 gene. Neuromuscular Disord. 2011;21:129–31. doi: 10.1016/j.nmd.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 187.Züchner S, De Jonghe P, Jordanova A, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–81. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- 188.Banchs I, Casasnovas C, Montero J, Martínez-Matos JA, Volpini V. Two Spanish families with Charcot-Marie-Tooth type 2A: clinical, electrophysiological and molecular findings. Neuromuscul Disord. 2008;18:974–8. doi: 10.1016/j.nmd.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 189.Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Votruba M, Marshall V, Thiselton D. Mutations in the OPA3 gene are not found in patients with inherited optic atrophy from the UK. Invest Ophthalmol Vis Sci. 2004;45:2462. [Google Scholar]
- 191.Anikster Y, Kleta R, Shaag A, Gahl WA, Elpeleg O. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am J Hum Genet. 2001;69:1218–24. doi: 10.1086/324651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cassereau J, Chevrollier A, Bonneau D, et al. A locus-specific database for mutations in GDAP1 allows analysis of genotype-phenotype correlations in Charcot-Marie-Tooth diseases type 4A and 2K. Orphanet J Rare Dis. 2011;6:87. doi: 10.1186/1750-1172-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Huber N, Guimaraes S, Schrader M, Suter U, Niemann A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 2013;14:545–52. doi: 10.1038/embor.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Noack R, Frede S, Albrecht P, et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum Mol Genet. 2012;21:150–62. doi: 10.1093/hmg/ddr450. [DOI] [PubMed] [Google Scholar]
- 195.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf–Hirschhorn syndrome. Trends Genet. 2005;2:188–95. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 196.Dimmer KS, Navoni F, Casarin A, et al. LETM1, deleted in Wolf–Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201–14. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- 197.Tamai S, Iida H, Yokota S, et al. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J Cell Sci. 2008;121:2588–600. doi: 10.1242/jcs.026625. [DOI] [PubMed] [Google Scholar]
- 198.Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf–Hirschhorn syndrome. PNAS. 2013;110:E2249–E2254. doi: 10.1073/pnas.1308558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Begriche K, Massart J, Robin M-A, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497–507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- 200.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–64. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 201.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pérez-Carreras M, Del Hoyo P, Martín MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 203.Hatazawa J, Brooks RA, Dalakas MC, Mansi L, Di Chiro G. Cortical motor-sensory hypometabolism in amyotrophic lateral sclerosis: a PET study. J Comput Assist Tomogr. 1988;12:630–6. doi: 10.1097/00004728-198807000-00019. [DOI] [PubMed] [Google Scholar]
- 204.Magrané J, Sahawneh MA, Przedborski S, Estévez ÁG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci. 2012;32:229–42. doi: 10.1523/JNEUROSCI.1233-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pickles S, Destroismaisons L, Peyrard SL, et al. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet. 2013;22:3947–59. doi: 10.1093/hmg/ddt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sasaki S, Warita H, Abe K, Iwata M. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005;110:48–56. doi: 10.1007/s00401-005-1021-9. [DOI] [PubMed] [Google Scholar]
- 207.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21:904–19. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.De Vos KJ, Mórotz GM, Stoica R, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2012;21:1299–311. doi: 10.1093/hmg/ddr559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342:619–30. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20(Suppl 2):S265–79. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2008;18:737–52. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cho D-H, Nakamura T, Fang J, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–5. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hamblet NS, Ragland B, Ali M, Conyers B, Castora FJ. Mutations in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes detected in Alzheimer's disease using single-strand conformation polymorphism. Electrophoresis. 2006;27:398–408. doi: 10.1002/elps.200500420. [DOI] [PubMed] [Google Scholar]
- 215.Chandrasekaran K, Hatanpää K, Brady DR, Rapoport SI. Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer's disease. Exp Neurol. 1996;142:80–8. doi: 10.1006/exnr.1996.0180. [DOI] [PubMed] [Google Scholar]
- 216.Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci. 2000;903:204–21. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- 217.Hedskog L, Zhang S, Ankarcrona M. Strategic role for mitochondria in Alzheimer's disease and cancer. Antioxid Redox Signal. 2012;16:1476–91. doi: 10.1089/ars.2011.4259. [DOI] [PubMed] [Google Scholar]
- 218.Büeler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp Neurol. 2009;218:235–46. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 219.Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson's disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012 Jun;121:830–9. doi: 10.1111/j.1471-4159.2012.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord. 2009;15(Suppl 3):S185–188. doi: 10.1016/S1353-8020(09)70811-9. [DOI] [PubMed] [Google Scholar]
- 221.Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, et al. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol. 1994 Dec;36(6):876–81. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- 222.Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 223.Yang Y, Ouyang Y, Yang L, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–5. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhao Y, Chen F, Chen S, Liu X, Cui M, Dong Q. The Parkinson's disease-associated gene PINK1 protects neurons from ischemic damage by decreasing mitochondrial translocation of the fission promoter Drp1. J Neurochem. 2013 doi: 10.1111/jnc.12340. [DOI] [PubMed] [Google Scholar]
- 225.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci. 2008;105:1638–43. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liu W, Acín-Peréz R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci USA. 2011;108:12920–4. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ding W-X, Guo F, Ni H-M, et al. Parkin and Mitofusins Reciprocally Regulate Mitophagy and Mitochondrial Spheroid Formation. J Biol Chem. 2012;287:42379–88. doi: 10.1074/jbc.M112.413682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem. 2011;118:636–45. doi: 10.1111/j.1471-4159.2011.07318.x. [DOI] [PubMed] [Google Scholar]
- 230.Rakovic A, Grünewald A, Kottwitz J, et al. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE. 2011;6:e16746. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, Achim CL. Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol. 2003;62:68–74. doi: 10.1093/jnen/62.1.68. [DOI] [PubMed] [Google Scholar]
- 232.Solesio ME, Prime TA, Logan A, et al. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson's disease. Biochim Biophys Acta. 2013;1832:174–82. doi: 10.1016/j.bbadis.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 233.Imarisio S, Carmichael J, Korolchuk V, et al. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 234.Song W, Chen J, Petrilli A, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–82. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Milakovic T, Johnson GVW. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–82. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 236.Shirendeb U, Reddy AP, Manczak M, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–55. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Aidt FH, Nielsen SMB, Kanters J, et al. Dysfunctional mitochondrial respiration in the striatum of the Huntington's disease transgenic R6/2 mouse model. PLoS Currents. 2013;5 doi: 10.1371/currents.hd.d8917b4862929772c5a2f2a34ef1c201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bae B-I, Xu H, Igarashi S, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 239.Djoussé L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington's disease. Neurology. 2002;59:1325–30. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- 240.Feigin A, Leenders KL, Moeller JR, et al. Metabolic network abnormalities in early Huntington's disease: an [(18)F]FDG PET study. J Nucl Med. 2001;42:1591–5. [PubMed] [Google Scholar]
- 241.Wang X, Su B, Siedlak SL, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008;105:19318–23. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Panov AV, Gutekunst C-A, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 243.Shirendeb UP, Calkins MJ, Manczak M, et al. Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum Mol Genet. 2012;21:406–20. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Haun F, Nakamura T, Shiu AD, Cho D-H, Tsunemi T, Holland EA, et al. S-Nitrosylation of Dynamin-Related Protein 1 Mediates Mutant Huntingtin-Induced Mitochondrial Fragmentation and Neuronal Injury in Huntington's Disease. Antioxid Redox Signal. 2013;19:1173–84. doi: 10.1089/ars.2012.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Siddiqui A, Rivera-Sánchez S, Castro M del R, et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radic Biol Med. 2012;53:1478–88. doi: 10.1016/j.freeradbiomed.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Orr AL, Li S, Wang C-E, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–92. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 248.St-Pierre J, Drori S, Uldry M, et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 249.Krssak M, Falk Petersen K, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–6. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 250.Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–71. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12:537–77. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.McGavock JM, Lingvay I, Zib I, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–5. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 254.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 255.Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53(Suppl 1):S110–118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 256.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J Am Coll Cardiol. 2009;54:1891–8. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Schrauwen P, Hesselink MK, Blaak EE, et al. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2001;50:2870–3. doi: 10.2337/diabetes.50.12.2870. [DOI] [PubMed] [Google Scholar]
- 258.Ikemura M, Nishikawa M, Kusamori K, Fukuoka M, Yamashita F, Hashida M. Pivotal role of oxidative stress in tumor metastasis under diabetic conditions in mice. J Control Release. 2013;170:191–7. doi: 10.1016/j.jconrel.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 259.Anello M, Lupi R, Spampinato D, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48:282–9. doi: 10.1007/s00125-004-1627-9. [DOI] [PubMed] [Google Scholar]
- 260.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–50. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 261.Leinninger GM, Backus C, Sastry AM, Yi Y-B, Wang C-W, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 262.Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–91. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- 263.Sebastián D, Hernández-Alvarez MI, Segalés J, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–8. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Regan TJ, Lyons MM, Ahmed SS, et al. Evidence for Cardiomyopathy in Familial Diabetes Mellitus. JClin Invest. 1977;60:885–99. doi: 10.1172/JCI108843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Rijzewijk LJ, van der Meer RW, Smit JWA, et al. Myocardial Steatosis Is an Independent Predictor of Diastolic Dysfunction in Type 2 Diabetes Mellitus. J Am Coll Cardiol. 2008;52:1793–9. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 266.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. AJP: Heart Circ Physiol. 2010;300:H118–H124. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 268.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006 Aug 7;25(34):4663–74. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 269.Nagaraj R, Gururaja-Rao S, Jones KT, et al. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Gene Dev. 2012;26:2027–37. doi: 10.1101/gad.183061.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Cheng X, Zhou D, Wei J, Lin J. Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines. Neoplasma. 2013;60:620–6. doi: 10.4149/neo_2013_080. [DOI] [PubMed] [Google Scholar]
- 271.Wang W, Lu J, Zhu F, Wei J, Jia C, Zhang Y, et al. Pro-apoptotic and anti-proliferative effects of mitofusin-2 via Bax signaling in hepatocellular carcinoma cells. Med Oncol. 2012;29:70–6. doi: 10.1007/s12032-010-9779-6. [DOI] [PubMed] [Google Scholar]
- 272.Zhang G-E, Jin H-L, Lin X-K, et al. Anti-tumor effects of mfn2 in gastric cancer. Int J Mol Sci. 2013;14:13005–21. doi: 10.3390/ijms140713005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chang C-R, Manlandro CM, Arnoult D, et al. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem. 2010;285:32494–503. doi: 10.1074/jbc.M110.142430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Vanhorebeek I, Gunst J, Derde S, et al. Mitochondrial Fusion, Fission, and Biogenesis in Prolonged Critically Ill Patients. JCEM. 2012;97:E59–E64. doi: 10.1210/jc.2011-1760. [DOI] [PubMed] [Google Scholar]
- 275.Ranieri M, Brajkovic S, Riboldi G, et al. Mitochondrial Fusion Proteins and Human Diseases. Neurology Research International. 2013;2013:293893. doi: 10.1155/2013/293893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cassidy-Stone A, Chipuk JE, Ingerman E, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Rehman J, Zhang HJ, Toth PT, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–86. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Qiu X, Cao L, Yang X, et al. Role of mitochondrial fission in neuronal injury in pilocarpine-induced epileptic rats. Neuroscience. 2013;245:157–65. doi: 10.1016/j.neuroscience.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 279.Barboni P, Valentino ML, Morgia CL, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain. 2013;136:e231–e231. doi: 10.1093/brain/aws280. [DOI] [PubMed] [Google Scholar]
- 280.Fujisawa K, Nishikawa T, Kukidome D, et al. TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem Biophys Res Commun. 2009;379:43–8. doi: 10.1016/j.bbrc.2008.11.141. [DOI] [PubMed] [Google Scholar]
- 281.Kukidome D, Nishikawa T, Sonoda K, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–7. [PubMed] [Google Scholar]
- 282.Qi X, Disatnik M-H, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol Biol Cell. 2011;22:256–65. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–44. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Chang C-R, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–9. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chen CH, Hwang SL, Howng SL, Chou CK, Hong YR. Three rat brain alternative splicing dynamin-like protein variants: interaction with the glycogen synthase kinase 3beta and action as a substrate. Biochem Biophys Res Commun. 2000;268:893–8. doi: 10.1006/bbrc.2000.2197. [DOI] [PubMed] [Google Scholar]
- 287.Chou C-H, Lin C-C, Yang M-C, et al. GSK3beta-Mediated Drp1 Phosphorylation Induced Elongated Mitochondrial Morphology against Oxidative Stress. PLoS ONE. 2012;7:e49112. doi: 10.1371/journal.pone.0049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–22. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, et al. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009;23:3917–27. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–54. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Anderson CA, Blackstone C. SUMO wrestling with Drp1 at mitochondria. EMBO J. 2013;32:1496–8. doi: 10.1038/emboj.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568–81. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]