

Abstract
Whether proper heat shock preconditioning can reduce liver injury and accelerate liver repair after acute liver injury is worth study. So mice received heat shock preconditioning at 40°C for 10 minutes (min), 20 min or 30 min and recovered at room temperature for 8 hours (h) under normal feeding conditions. Then acute liver injury was induced in the heat shock-pretreated mice and unheated control mice by intraperitoneal (i.p.) injection of carbon tetrachloride (CCl4). Hematoxylin and eosin (H&E) staining, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels and the expression levels of heat shock protein 70 (HSP70), cytochrome P450 1A2 (CYP1A2) and proliferating cell nuclear antigen (PCNA) were detected in the unheated control mice and heat shock-pretreated mice after CCl4 administration. Our results showed that heat shock preconditioning at 40°C for 20 min remarkably improved the mice’s survival rate (P<0.05), lowered the levels of serum AST and ALT (P<0.05), induced HSP70 (P<0.01), CYP1A2 (P<0.01) and PCNA (P<0.05) expression, effectively reduced liver injury (P<0.05) and accelerated the liver repair (P<0.05) compared with heat shock preconditioning at 40°C for 10 min or 30 min in the mice after acute liver injury induced by CCl4 when compared with the control mice. Our results may be helpful in further investigation of heat shock pretreatment as a potential clinical approach to target liver injury
Keywords: heat shock pretreatment, acute liver injury, CCl4, HSP70, CYP1A2, PCNA
Introduction
Acute liver failure with massive hepatocellular loss occurs due to various causes. Because liver failure still exhibits high mortality despite intensive care, effective therapeutic approaches are needed1. The carbon tetrachloride (CCl4)-induced acute liver injury model is similar to the human disease from the standpoint of morphology and biochemical aspects of collagen metabolism. A single dose or very few doses result in acute liver damage that has been widely employed to study morphological and biochemical features of cellular lesions, especially steatosis and necrosis2. At an early stage after CCl4 administration, transient increases in the expression of the stress-inducible HSP70 gene occur3. HSP70 knockout mice have a higher degree of necrosis and neutrophilic infiltration than normal mice. So HSP70 could play an important role in the cytoprotection of the liver against hepatotoxic agents2. Heat shock preconditioning induces HSP72 in the rat liver with fibrosis and provides significantly increased tolerance to warm ischemia-reperfusion injury4. Hagiwara et al. (2007)5 reported that thermal pretreatment is associated with the induction of HSP70 protein synthesis, which subsequently attenuates tissue damage in experimental lung fibrosis.
Heat shock can cause cell death if cellular defense mechanisms are insufficient to cope with the stress. This is particularly obvious when the temperature increases well above that of the normal environment and/or exposure time is prolonged. An important feature of HSPs is their role in the cytoprotection and repair of cells and tissues with regard to the harmful effects of stress6,7. The major families of mammalian stress proteins, HSP90 and HSP70, as well as the smaller HSP28 family, have all been well characterized5. HSP70 overexpression confers myocardial protection, as observed by resistance to myocardial ischemic stress and reperfusion damage8,9,10. In a rodent model for adult respiratory distress syndrome, heat shock-induced HSP70 accumulation within the lung has been associated with decreased pulmonary inflammation and prevention of lethality11. The cytoprotective role of HSP70 has also been documented in the areas of metabolic disorders12, and infection13. These observations suggest new therapeutic strategies relying upon the development of methods that are able to increase the expression of HSPs. Furthermore, it has been shown that the production of HSPs could protect the organism against a second exposure to otherwise lethal hyperthermia, which has been described as the thermotolerance phenomenon14.
Whether proper heat shock preconditioning can reduce liver injury and accelerate liver repair after acute liver failure induced by CCl4 is worth study. Our previous study showed that heat shock at a lower temperature (heat shock at 40°C for 20 min) significantly promotes hepatocyte proliferation and improves metabolic efficiency in the mouse liver, while heat shock at a higher temperature (heat shock at 46°C for 20 min) remarkably inhibits hepatocyte proliferation, promotes hepatocyte apoptosis and induces liver injury15. So we selected a proper temperature and time to study whether proper heat shock preconditioning could reduce liver injury and accelerate liver repair after acute liver failure induced by CCl4.
Materials and Methods
Animals
Male BALB/c mice are sensitive to temperature, and it is easy to induce acute liver injury in them using CCl4; so male BALB/c mice (approximately 6–8 weeks old, 22 ± 2 g) were purchased from the Experimental Animals Center of Henan Province and maintained in an air-conditioned animal room at 25°C with free access to water and food under 12 h light/dark cycles for the experiments. All animals were allowed to adapt to the environment for 1 week before the experiment and were fed laboratory chow. All protocols conformed to the guidelines of the National Animal Care and Use Committee of China. All animals received care in compliance with the Principles of Laboratory Animal Care.
Heat shock preconditioning and acute liver injury induced by CCl4
Our previous work suggested that heat shock at 40°C for 20 min is a proper condition for heat shock preconditioning because it could effectively promote hepatocyte proliferation and improves the metabolic efficiency in the mouse liver15. So mice were anesthetized with urethane (1.4 g/kg, i.p.) and divided into two groups. In the heat shock group (HS20 group, n=90), mice received heat shock preconditioning at 40°C for 20 min and subsequent CCl4 (analytical reagent, from Tianjin Kaitong Chemical Reagent Co., Ltd; Tianjin, China) administration. The mice in the control group (n=90) were only injected with CCl4 to induce acute liver injury. Briefly, mice in the HS20 group were placed in a temperature-controlled ventilated and humidified chamber to raise the rectal temperature to 40°C for 20 min, which was monitored by a digital thermometer in the rectum. The animals were then allowed to recover at room temperature in normal feeding conditions. At 8 h after heat shock pretreatment, 0.1% CCl4 (1 μl CCl4 in 1 ml mineral oil) was administered to the mice in group HS20 and the control group by i.p. injection of 10 mL/Kg. Blood was drawn via the orbital vein at 0, 3, 6, 12, 24, 30, 36, 42, 48 and 54 h in the mice of the two groups after CCl4 injection. The coagulated blood was left to clot at room temperature for approximately 15 to 30 min. After it was completely clotted, it was rimmed using an applicator stick and then centrifuged for approximately 5–10 min at 2500 rpm. Then the supernatant fluid was separated. Serum AST and ALT activities were determined with a commercial assay kit (Nanjing Jiancheng Biological Technology, Inc., Nanjing, China) at 0, 3, 6, 12, 24, 30, 36, 42, 48 and 54 h in the mice of the two groups after CCl4 injection. Enzyme activities were expressed as an international unit per liter (IU/L). The mice at each time point in each group were divided into three subgroups 1, 2 and 3. Each subgroup contained three mice. We also performed an experiment in which mice received heat shock preconditioning at 40°C for 10 min (HS10 group, n=90) or 30 min (HS30 group, n=90) and subsequent CCl4 administration. In addition, we performed a parallel experiment and only evaluated the survival rate in heat shock-pretreated mice and control mice after CCl4 injection.
Histologic examination
Liver specimens were obtained from 9 mice at each time point. Each liver specimen from each mouse was divided into two sections. One was used for histologic examination, and the other was used for Western blot detection. Samples of liver were fixed in 10% formaldehyde for 24 h and then dehydrated and embedded in paraffin. Six-micrometer sections were cut from each paraffin-embedded tissue and stained with hematoxylin and eosin (H&E). To evaluate the degree of necrosis after acute liver injury, we created an injury grading score (Grade 0–4) based on severity of necrotic lesions in the liver parenchyma16. The grades were as follows: Grade 0, no pathological change; Grade 1, presence of degenerated hepatocytes with only rare foci of necrosis; Grade 2, small area of mild centrilobular necrosis around the central vein; Grade 3, area of mild centrilobular necrosis severer than Grade 2; and Grade 4, centrilobular necrosis severer than Grade 3. Every group contains three subgroups 1, 2 and 3. Each subgroup contains three mice.
Immunohistochemistry for HSP70, CYP1A2 and PCNA
Six-micrometer sections were cut from each paraffin-embedded tissue as prepared above. The sections of liver from the mice in different groups were immunostained with a monoclonal antibody to mouse HSP70, CYP1A2 and PCNA (dilution 1:300) (Santa Cruz) as described by Xu et al. (2000)17. The signal was detected using the Polink-2 plus polymer HRP detection system (Zhongshan, Beijing, China) using DAB. A negative control was carried out on each slide by omitting the primary antibody. Sections were examined microscopically for specific staining and photographs were taken using a digital image-capture system (Olympus, Tokyo, Japan). Numbers of positive cells from the control and preheated mice were 12 mm2 tissue sections counted for each mouse.
Western blot of HSP70, CYP1A2 and PCNA
HSP70 play important roles in stress response, and CYP1A2 principally participates in metabolizing chemicals and environmental toxins in the liver. PCNA has been shown to be a good marker to distinguish proliferating cells17,18. So the expression levels of HSP70, CYP1A2 and PCNA were detected by Western blot to compare liver stress response, metabolism and regeneration in heat shock-pretreated mice and control mice after CCl4 administration. Protein samples of 70 μg from the mice in the different groups were adjusted to the composition of the electrophoresis sample buffer (50 mM Tris, pH 6.8, 10% glycerol, 5% beta-mercaptoethanol, 2% SDS, 0.1% bromphenol blue) and boiled for 5 min prior to analysis. SDS-PAGE (10% polyacrylamide gels) in 1 mm slab gel was performed as described by Sambrook and Russell (2001)19. The proteins were transferred from the gel to nitrocellulose membranes. Then the membrane was probed with a monoclonal antibody to mouse HSP70, CYP1A2 or PCNA (Santa Cruz), respectively. The signal was detected by a horseradish peroxidase detection system using DAB (Sigma). Protein bands were quantified with the Gel-Pro Analyzer 4.0 software (Media Cybernetics Inc., Bethesda, MD, USA), and the intensities of the bands were normalized against β-actin. Every experiment was repeated three times.
Statistical analysis
The data were presented as means ± SEM of 9 animals per group at each time point. Statistical comparisons were made using one-way and two-way (without interaction) ANOVA with the Tukey post-hoc test for multiple comparisons. All statistical analyses were performed using SPSS 14.0 (SPSS Inc., Chicago, IL, USA). Survival rates of mice were analyzed using the log-rank test and expressed as Kaplan-Meier survival curves.
Results
Alterations in the serum AST and ALT levels and survival rate in the mice
Figure 1 shows that the maximum levels of serum AST and ALT were observed at 24 h post-CCl4 injection in the control group, but at 30 h in the mice with heat shock pretreatment at 40°C for 20 min (HS20 group). In addition, the peak values were approximately 1.9-fold higher in the control group than in the HS20 group. In the control group, the AST and ALT values decreased from 30 h post-CCl4 injection, reaching the basal value at 54 h, while the AST and ALT values decreased from 36 h post-CCl4 injection, reaching the basal value at 48 h observed in the HS20 group. In addition, the AST and ALT levels in the control group were significantly higher than those in the HS20 group at each time point from 3 h to 48 h post CCl4 injection (P<0.01). The AST and ALT levels in the HS10 group and HS30 group were lower than those in the control group at some time points post CCl4 injection (P<0.05), but the time points at which the peak levels of serum AST and ALT were reached in the HS10 group and HS30 group were similar to those in the control group post CCl4 injection (Fig. 1). The preheated animals showed a marked increase in survival rate (94.4%) in the HS20 group compared with the unheated control group (74.4%) at 54 h after CCl4 injection (P<0.05). But the survival rates of the HS10 group (80%) and HS30 group (80%) were not significantly improved compared with the unheated control group at 54 h after CCl4 injection (P>0.05) (Fig. 2).
Fig. 1.

Serum AST (A) and ALT (B) levels in the mice of the control and pretreatment groups at 0, 3, 6, 12, 24, 30, 36, 42, 48 and 54 h after administration of 0.1 ml/10 g 0.1% CCl4 (1 μl CCl4 in 1 ml mineral oil). Data are expressed as means ± SEM (n=9). HS10 group, HS20 group and HS30 group: mice received heat shock preconditioning with 40°C for 10, 20 and 30 min, respectively and then recovered at room temperature for 8 h under normal feeding conditions; 0.1% CCl4 was subsequently administered to the mice. Control group: mice were only administered 0.1% CCl4. * P<0.05; ** P<0.01; significant difference in the HS group as compared with the control group. # P<0.05 when compared with the HS10 and HS30 groups.
Fig. 2.

The effect of heat shock pretreatment on the survival rate of mice in which liver injury was induced by CCl4. A, B, C and D, respectively, represent the survival rates of mice in the control group, HS10 group, HS20 group and HS30 group at 0, 3, 6, 12, 24, 30, 36, 42, 48 and 54 h after exposure to 0.1 ml/10 g 0.1% CCl4 (1 μl CCl4 in 1 ml mineral oil). The results were analyzed using the log-rank test and expressed as Kaplan-Meier survival curves. The data show that the survival rate of the HS20 group was significantly improved compared with the HS10 group and the HS30 group when compared with the control group (P<0.05).
Histological changes in heat shock-pretreated mice and control mice with acute liver injury induced by CCl4
To evaluate liver injury degree, H&E staining was performed in the liver of mice. Table 1 and Fig. 3 show that heat shock pretreatment at 40°C for 20 min significantly reduced liver injury degree compared with the control mice when acute liver injury was induced by CCl4 (P<0.01). Heat shock pretreatment at 40°C for 20 min not only postponed the peak of liver injury but also accelerated repair of liver injury (Table 1) (P<0.01). But heat shock pretreatment at 40°C for 10 min or 30 min did not significantly ameliorate liver injury induced by CCl4 (P>0.05) (Table 1 and Fig. 3).
Table 1. Liver Injury Degree.

Fig. 3.
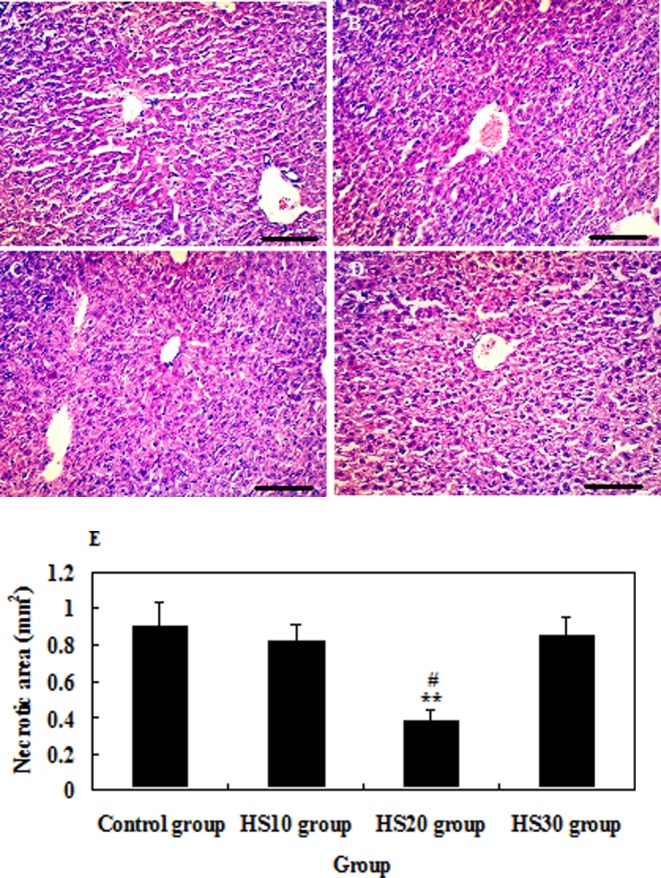
Histologic examination of liver injury in mice by H&E staining at 24 h after CCl4 administration. A, B, C and D, respectively, represent the degree of liver injury of mice in the control group, HS10 group, HS20 group and HS30 group after CCl4 administration. E: Necrotic areas. Representative findings from counting with tissue sections that were at least 10 mm2 for each mouse. Scale bar: 50 µm.
The expression of HSP70, CYP1A2 and PCNA in the liver of mice
The expression of HSP70 in the liver of the HS20 group was significantly higher than those in the control group (P<0.01) and HS10 and HS30 groups (P<0.01) from 0 h to 54 h after CCl4 injection. But the expression of HSP70 in the HS10 and HS30 groups was only higher than that in the control group from 0 h to 3 h after CCl4 injection (P<0.05) (Figs. 4, 7A and 7B). From 0 h to 54 h after CCl4 administration, the expression of CYP1A2 in the HS20 group was always higher than that in the control group (P<0.01) and was higher than that in the HS10 and HS30 groups at subtotal time points (P<0.05), except at 12 h and 30 h after CCl4 administration. The expression of CYP1A2 in the HS10 and HS30 groups was higher than that in the control group at major time points (P<0.05), except at 24 h and 36 h after CCl4 injection (Figs. 5, 7A and 7C). The expression of PCNA in the liver of heat shock-pretreated mice reached the minimum level at 12 h and then increased at 24 h to 48 h when liver regeneration started after liver injury. On the other hand, the expression of PCNA in the liver of the control mice reached the minimum level at 24 h and then increased from 30 h to 54 h after liver injury. In addition, the expression level of PCNA in the heat shock-pretreated mice was significantly higher than that in the control mice after CCl4 administration at each time point from 0 h to 54 h (P<0.05). But heat shock pretreatment at 40°C for 10 min or 30 min did not significantly induce the expression of PCNA (P>0.05) after liver injury induced by CCl4 when compared with the control mice (Figs. 6, 7A and 7D).
Fig. 4.
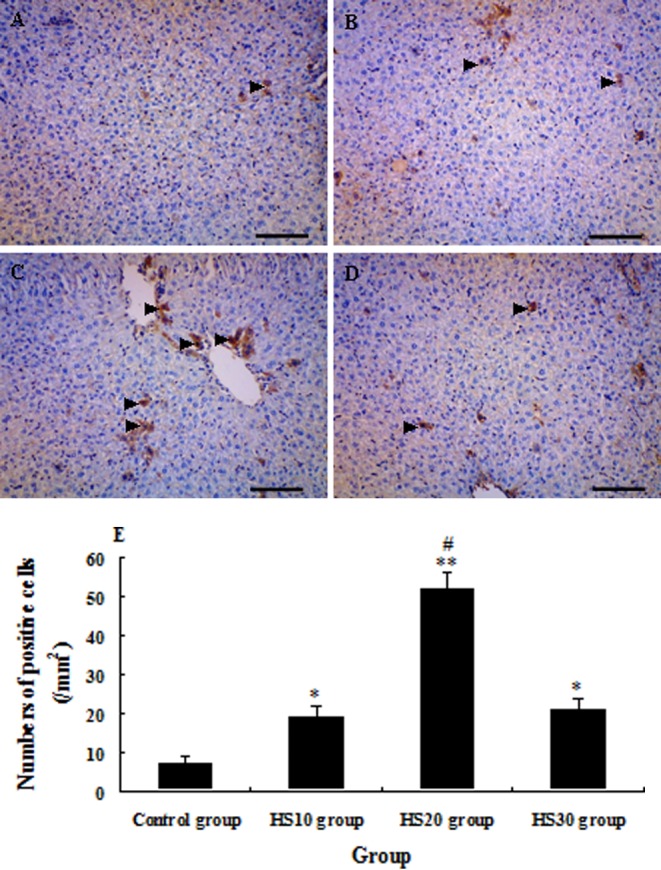
Photomicrographs of immunohistochemical staining of HSP70 in the liver of unheated control mice and heat shock-pretreated mice at 0 h after CCl4 administration. The arrowheads indicate the HSP70-positive cells in the liver of mice in the control group (A), HS10 group (B), HS20 group (C) and HS30 group (D). E: Numbers of HSP70 + cells in the liver of mice in the control group and preheated groups; tissue sections that were at least 12 mm2 were counted for each mouse. (Scale bar: 50 µm).
Fig. 7.

The expression of HSP70, CYP1A2 and PCNA in the liver of unheated control mice and heat shock-pretreated mice after CCl4 administration. The expression of HSP70, CYP1A2 and PCNA was detected by Western blot (A). The protein bands were quantified for HSP70 (B), CYP1A2 (C) and PCNA (D) with the Gel-Pro Analyzer 4.0 software (Media Cybernetics Inc.) (B), and the intensities of the bands were normalized against β-actin. AU represents arbitrary unit. Every experiment was repeated three times. All data were presented as the mean ± standard error of the mean (SEM). ##P<0.01; significant difference when compared with the HS10 and HS30 groups.
Fig. 5.
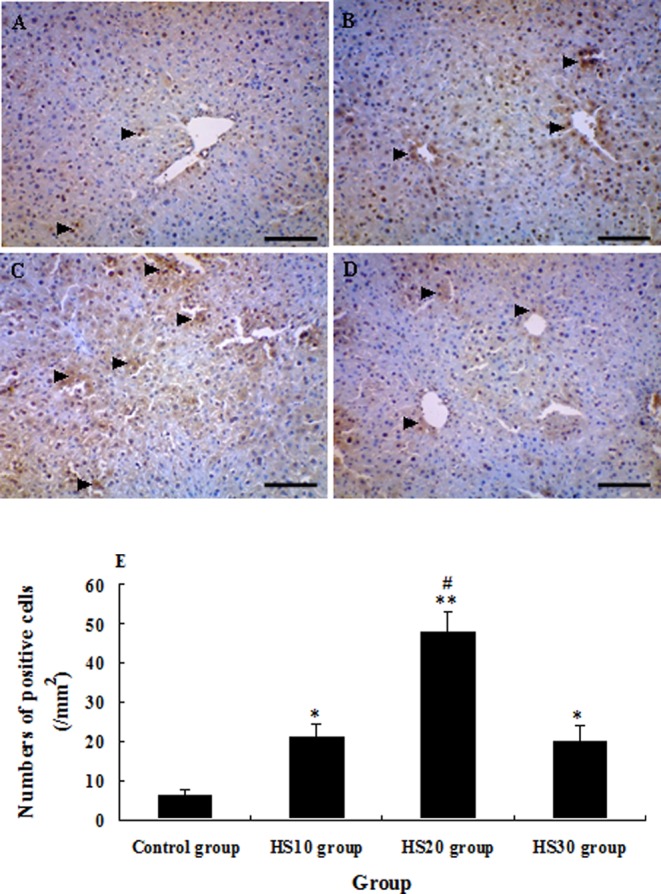
Photomicrographs of immunohistochemical staining of CYP1A2 in the liver of unheated control mice and heat shock-pretreated mice at 0 h after CCl4 administration. The arrowheads indicate the CYP1A2-positive cells in the liver of mice in the control group (A), HS10 group (B), HS20 group (C) and HS30 group (D). E: Numbers of CYP1A2 + cells in the liver of mice in the control group and preheated groups; tissue sections that were at least 12 mm2 were counted for each mouse. (Scale bar: 50 µm).
Fig. 6.

Photomicrographs of immunohistochemical staining of PCNA in the liver of unheated control mice and heat shock-pretreated mice at 0 h after CCl4 administration. The arrowheads indicate the PCNA-positive cells in the liver of mice in the control group (A), HS10 group (B), HS20 group (C) and HS30 group (D). E: Numbers of PCNA+ cells in the liver of mice in the control group and preheated groups; tissue sections that were at least 12 mm2 were counted for each mouse. (Scale bar: 50 µm).
Discussion
Our results suggested that proper heat shock pretreatment, such as 40°C for 20 min, could significantly induce HSP70, CYP1A2 and PCNA expression (Figs. 4–7), reduce liver injury and accelerate liver repair (Table 1 and Fig. 3) after liver injury induced by CCl4 in mice.
It has been reported that thermal pretreatment is associated with the induction of HSP70 protein synthesis, which subsequently attenuates acute lung injury induced by lipopolysaccharide in rats5. But whether heat shock pretreatment can reduce acute liver injury induced by CCl4 in mice has not been studied. Based on the results of analysis in a previous study15 (Li et al., 2012), we think that heat shock at 40°C for 20 min is an optimal thermal pretreatment to induce HSP70 expression and improve liver function compared with heat shock at 42°C, 44°C and 46°C for 20 min. So mice received heat shock preconditioning at 40°C for 20 min (HS20 group) and subsequent CCl4 administration. Figure 1 shows that heat shock preconditioning at 40°C for 20 min effectively lowered serum AST and ALT levels in the mice with acute liver injury induced by CCl4 compared with heat shock preconditioning at 40°C for 10 min or 30 min as compared with the control mice (P<0.01), which indicated that heat shock preconditioning at 40°C for 20 min could effectively reduce liver injury induced by CCl4 (Table 1 and Fig. 3) and improve the survival rate (Table1 and Fig. 2) in the mice. Saad et al. (1995)14 reported that heat exposure associated with HSP induction has a significant protective effect against warm ischemic liver injury. Yang et al. (1998)20 reported that heat shock protein expression protects against death following exposure to heatstroke in rats. Although the HSPs were associated with the phenomenon of thermotolerance, cross protection against other kinds of cellular stress, the underlying mechanism of this protection is not clear. The function of HSP has been explained by an improved transportation of repair proteins across cellular and subcellular membranes, preventing intracellular calcium overload and an increased catalase activity. It has been speculated that HSPs reduce the oxidative injury caused by oxygen free radicals that occurs during the early phase of reperfusion14,21. So we think that heat shock pretreatment significantly induced HSP70 expression (Figs. 4, 7A and 7B), which is the cellular response to oxidative stress, and provided protection against oxygen distress in acute liver injury induced by CCl4 in the mice.
Our results also showed that heat shock pretreatment at 40°C for 20 min could significantly induced CYP1A2 expression compared with heat shock pretreatment at 40°C for 10 min or 30 min when compared with control mice (P<0.01) (Figs. 5, 7A and 7C). CYP1A2 is one of the major CYPs in the human liver (approximately 13%) and metabolizes a variety of clinically important drugs. This enzyme also metabolizes several important endogenous compounds including steroids, retinols, melatonin, uroporphyrinogen and arachidonic acid. Like many of other CYPs, CYP1A2 is subject to induction and inhibition by a number of compounds22. Our results demonstrated that proper heat shock preconditioning (40°C for 20 min) could effectively induce CYP1A2 expression to improve the metabolic efficiency of the liver, which may be helpful in resisting the damage induced by CCl4 in the liver of mice.
Our results indicated that heat shock preconditioning at 40°C for 20 min significantly induced PCNA expression in the liver cells of mice compared with heat shock pretreatment at 40°C for 10 min or 30 min when compared with control mice (P<0.05) (Figs. 6, 7A and 7D). PCNA is a subunit of the mammalian DNA polymerase delta and is synthesized primarily during the S phase of the cell cycle23. It is a relay or anchoring molecule that functions as a molecular integrator for proteins involved in control of the cell cycle, DNA replication, DNA repair and cell death24,25. PCNA has been shown to be a good marker to distinguish proliferating cells17,18. So heat shock preconditioning at 40°C for 20 min significantly promoted hepatocellular proliferation and accelerated the liver repair after acute liver failure induced by CCl4. Analysis of the above results suggested that both high levels of HSPs and proper heat shock-timing between heat shock pretreatment and CCl4-induced liver injury were critical for optimal protection. Heat shock preconditioning at 40°C for 20 min may be helpful in improving the metabolic efficiency of the liver, lowering serum AST and ALT levels and accelerating liver repair after acute liver failure induced by CCl4. But heat shock preconditioning at 40°C for 10 min could not sufficiently induce the expression of HSP70, CYP1A2 and PCNA. The stimulation of heat shock preconditioning at 40°C for 30 min may be too heavy to induce tolerance of heat in mice.
In conclusion, our study showed that proper heat shock pretreatment ameliorated liver injury and accelerated liver repair after acute liver injury induced by CCl4 in mice, which may be helpful in further investigation of heat shock pretreatment as a potential clinical approach targeting liver injury.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (#U1204802 and #81201558) to SQL, Program for Science & Technology Innovation Talents in Universities of Henan Province (#13HASTIT025) to SQL and Foundation for Henan Province’s Key Project to Tackle Key Problems of Science and Technology (#122102310030). The authors thank all the members in the laboratory when this work was carried out. The experiments complied with the current laws of China.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives (by-nc-nd) License <http://creativecommons.org/licenses/by-nc-nd/3.0/>.
References
- 1.Yoneyama H, Kai Y, Koyama J, Suzuki K, Kawachi H, Narumi S, and Ichida T. Neutralization of CXCL10 accelerates liver regeneration in carbon tetrachloride-induced acute liver injury. Med Mol Morphol. 40: 191–197 2007. [DOI] [PubMed] [Google Scholar]
- 2.Song JY, Li L, Ahn JB, Park JG, Jo JS, Park DH, Jang HK, Jang JJ, and Lee MJ. Acute liver toxicity by carbon tetrachloride in HSP70 knock out mice. Exp Toxicol Pathol. 59: 29–34 2007. [DOI] [PubMed] [Google Scholar]
- 3.Schiaffonati L, and Tiberio L. Gene expression in liver after toxic injury: analysis of heat shock response and oxidative stress-inducible genes. Liver. 17: 183–191 1997. [DOI] [PubMed] [Google Scholar]
- 4.Shimabukuro T, Yamamoto Y, Kume M, Kimoto S, Okamoto R, Morimoto T, and Yamaoka Y. Induction of heat shock response: effect on the rat liver with carbon tetrachloride-induced fibrosis from ischemia-reperfusion injury. World J Surg. 22: 464–468 1998. [DOI] [PubMed] [Google Scholar]
- 5.Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T, and Yoshioka H. Association between heat stress protein 70 induction and decreased pulmonary fibrosis in an animal model of acute lung injury. Lung. 185: 287–293 2007. [DOI] [PubMed] [Google Scholar]
- 6.Sachidhanandam SB, Lu J, Low KS, and Moochhala SM. Herbimycin A attenuates apoptosis during heat stress in rats. Eur J Pharmacol. 474: 121–128 2003. [DOI] [PubMed] [Google Scholar]
- 7.Fournier S, Kinkead R, and Joseph V. Influence of housing conditions from weaning to adulthood on the ventilatory, thermoregulatory, and endocrine responses to hypoxia of adult female rats. J Appl Physiol. 112: 1474–1481 2012. [DOI] [PubMed] [Google Scholar]
- 8.Mestril R, Chi SH, Sayen MR, O’Reilly K, and Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest. 93: 759–767 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, and Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 95: 1446–1456 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plumier JC, Ross BM, Currie RW, Angelidis CE, Kazlaris H, Kollias G, and Pagoulatos GN. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 95: 1854–1860 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villar J, Edelson JD, Post M, Mullen JB, and Slutsky AS. Induction of heat stress proteins is associated with decreased mortality in an animal model of acute lung injury. Am Rev Respir Dis. 147: 177–181 1993. [DOI] [PubMed] [Google Scholar]
- 12.Williams RS, Thomas JA, Fina M, German Z, and Benjamin IJ. Human heat shock protein 70 (HSP70) protects murine cells from injury during metabolic stress. J Clin Invest. 92: 503–508 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amici C, Giorgi C, Rossi A, and Santoro MG. Selective inhibition of virus protein synthesis by prostaglandin A1: a translational block associated with HSP70 synthesis. J Virol. 68: 6890–6899 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saad S, Kanai M, Awane M, Yamamoto Y, Morimoto T, Isselhard W, Minor T, Troidl H, Ozawa K, and Yamaoka Y. Protective effect of heat shock pretreatment with heat shock protein induction before hepatic warm ischemic injury caused by Pringle’s maneuver. Surgery. 118: 510–516 1995. [DOI] [PubMed] [Google Scholar]
- 15.Li SQ, Li RF, Xi SM, Hu S, Jia ZQ, Li SP, Wen XL, Song YK, Li S, Li SP, Wei FB, and Chen XL. Systematical analysis of impacts of heat stress on the proliferation, apoptosis and metabolism of mouse hepatocyte. J Physiol Sci. 62: 29–43 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu RZ, Xiang D, Xie C, Li JJ, Hu JJ, He HL, Yuan YS, Gao J, Han W, and Yu Y. Protective effect of recombinant human IL-1Ra on CCl4-induced acute liver injury in mice. World J Gastroenterol. 16: 2771–2779 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu CS, Lu AL, Xiong L, and Li GW. Changes of ACP, AKP, HSC70/HSP68 and PCNA in growth and development of rat Liver. Dev Reprod Biol. 9: 1–14 2000 [Google Scholar]
- 18.Assy N, Gong Y, Zhang M, Pettigrew NM, Pashniak D, and Minuk GY. Use of proliferating cell nuclear antigen as a marker of liver regeneration after partial hepatectomy in rats. J Lab Clin Med. 131: 251–256 1998. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook J, and Russell DW. Molecular Cloning: A Laboratory Manual, 4th ed. Cold Spring Harbor Laboratory Press. 256–1260. 2001 [Google Scholar]
- 20.Yang YL, Lu KT, Tsay HJ, Lin CH, and Lin MT. Heat shock protein expression protects against death following exposure to heatstroke in rats. Neurosci Lett. 252: 9–12 1998. [DOI] [PubMed] [Google Scholar]
- 21.Hillman AR, Vince RV, Taylor L, McNaughton L, Mitchell N, and Siegler J. Exercise-induced dehydration with and without environmental heat stress results in increased oxidative stress. Appl Physiol Nutr Metab. 36: 698–706 2011. [DOI] [PubMed] [Google Scholar]
- 22.Zhou SF, Yang LP, Zhou ZW, Liu YH, and Chan E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 11: 481–494 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Celis JE, Madsen P, Celis A, Nielsen HV, and Gesser B. Cyclin (PCNA, auxiliary protein of DNA polymerase delta) is a central component of the pathway(s) leading to DNA replication and cell division. FEBS Lett. 220: 1–7 1987. [DOI] [PubMed] [Google Scholar]
- 24.Kelman Z. PCNA: structure, functions and interactions. Oncogene. 14: 629–640 1997. [DOI] [PubMed] [Google Scholar]
- 25.Morrow PW, Tung HY, and Hemmings HC. Rapamycin causes activation of protein phosphatase-2A1 and nuclear translocation of PCNA in CD4+ T cells. Biochem Biophys Res Commun. 323: 645–651 2004. [DOI] [PubMed] [Google Scholar]