

Abstract
Microwave promoted novel and efficient one-step parallel synthesis of dibenzopyranones and heterocyclic analogues from bromo arylcarboxylates and o-hydroxyarylboronic acids via Suzuki-Miyaura cross coupling reaction is described. Spontaneous lactonization gave dibenzopyranones and heterocyclic analogues bearing electron donating and withdrawing groups on both aromatic rings in good to excellent yields.
Keywords: Suzuki-Miyaura cross coupling, microwave based synthesis, benzopyranones
Introduction
Dibenzopyranone is a privileged scaffold in many natural products1 such as alternariol, graphislactones, autumnariol, autumnariniol, altenuisol and biologically active compounds.2 Such lactones have been used as intermediates in the synthesis of several pharmaceutically interesting compounds including progesterone, androgen, glucocorticoid modulators,3 and endothelial cell proliferation inhibitors.4 Furthermore, dibenzopyranones occur naturally in many food sources including citrus fruits, herbs, and vegetables.5 There are several methods available for the synthesis of dibenzopyranones. The most popular involves Suzuki-Miyaura cross coupling followed by Lewis acid6 or metal7 mediated lactonization. More recently the Diels-Alder cycloaddition of 4-cyanocoumarins,8 tert-butyllithium-mediated cyclization of bromobenzylfluorophenyl ethers,9 and ruthenium-catalysed cyclotrimerization of aryl diynes10 have been reported. In 2002, Abbott Laboratories reported a practicable and scalable synthesis of glucocorticoid receptor A-224817.0 through a Negishi cross coupling.11 However, none of these methods are suitable for the parallel synthesis approach to generating large numbers of analogues for drug discovery research. The disadvantages include low overall yields, long reaction times, low temperature conditions, multi-step sequences, and the need to purify intermediates. Although the previous method of coupling of o-methoxyphenylboronic acids with bromo arylcarboxylates 1 followed by borontribromide6 or hydroiodic acid mediated lactonization12 has potential for parallel synthesis, but suffers from the following drawbacks: a) it is two step procedure requiring minimum the purification of two intermediates, b) alkoxy and alkoxyester substituents undergo demethylation in the presence of borontribromide,13 and c) BBr3-mediated lactonization6 requires the reaction to be performed at -78 °C, while HI-mediated lactonization requires to be conducted at 110 °C.12
Recently, we reported13 that dibenzopyranone analogues act as CB2 agonists and that they are being investigated for the treatment of neuropathic pain, inflammation, and Alzheimer's diseases. As part of an ongoing lead optimization project, we required a facile route to prepare a library of dibenzopyranones and heterocyclic analogues 3. We have, therefore, investigated a straightforward route for dibenzopyranones 3 via tandem Suzuki coupling of bromo arylcarboxylates 1 with o-hydroxyarylboronic acids 2 followed by lactonization (Scheme 1).
Scheme 1. Suzuki-Miyaura cross coupling of bromo arylcarboxylates 1 and o-hydroxyarylboronic acids 2.

Also, we utilized microwave assisted organic synthesis, which has attracted increasing attention because of its potential to accelerate drug discovery research.15 Reaction times of many organic transformations are often dramatically reduced from hours to minutes or seconds. Moreover, there have been several reports of microwave-assisted syntheses of combinatorial libraries.16 We now report a novel and efficient one step parallel synthesis of a library of dibenzopyaranones and heterocyclic analogues 3 via Suzuki-Miyaura cross coupling of bromo arylcarboxylates 1 with o-hydroxylarylboronic acids 2.
Results and Discussion
To develop an efficient methodology for the synthesis of dibenzopyranones and heterocyclic analogues 3, using bromo arylcarboxylates 1 and o-hydroxylarylboronic acids 2, various catalysts, ligands, bases and solvents were first screened for the Suzuki-Miyaura cross coupling reaction (Table 1). At the outset of the studies, commercially available methyl 2-bromo-5-methoxybenzoate 1{1}, methyl 2-bromo-4-fluorobenzoate 1{2}, and o-hydroxyphenylboronic acid 2{1} were selected as representative reactants (Table 1). We initially examined the reaction conditions reported for chloro(di-2-norbornylphosphino)(2′-dimethylamino-1,1′-biphenyl-2yl)palladium (II), an air-stable catalyst that is highly active for C-C and C-N coupling reactions with aryl chlorides.17 When a solution of 1{1} or 1{2} (1 equiv), 2{1} (1.2 equiv), potassium fluoride (5 equiv), and 5 mol% of the above catalyst in dioxane was heated in a microwave reactor at 130 °C for 15 min, the desired product i.e., 8-methoxy-6H-benzo[c]chromen-6-one 3{1} or 9-fluoro-6H-benzo[c]chromen-6-one 3{2}, was obtained in 54 and 48% isolated yields, respectively (entry 1, Table 1). Coupling under Buchwald's conditions using highly active palladium catalyst, i.e.,18 potassium fluoride, Pd(OAc)2 and (o-biphenyl)P(t-Bu)2 in THF at room temperature, afforded 3{1} and 3{2} in 45% and 47% yields, respectively (entry 2, Table 1). To improve the yields for the desired products 3, we screened several catalysts such as Pd(PCy3)2Cl2, PdCl2(PPh3)2, Pd(PPh3)4, 1,1′-bis(diphenylphosphino) ferrocene dichloropalladium (II); bases such as NaOAc, Cs2CO3, CsF, KF; ligand (o-biphenyl)P(t-Bu)2) with different equivalents and combinations of reagents and microwave irradiation temperatures in THF, NMP, DME or dioxane. The results of formation of products 3{1} and 3{2} are shown in Table 1. From the screening results, the best conditions for coupling of 1{1} or 1{2} with 2{1} were found to be the use of Pd(PPh3)4 as a catalyst in the presence of Cs2CO3 in DME at 125 °C under microwave irradiation for 15 min (Table 1, entry 6).
Table 1.
Optimization of Suzuki-Miyaura cross coupling reaction conditions.
 | |||
|---|---|---|---|
| Entry | Conditions | Yielda (%) | |
| 3{1} | 3{2} | ||
| 1 | Chloro(di-2-norbornylphosphino)(2′-dimethylamino-1,1′-biphenyl-2yl)palladium (II) (5 mol %), KF (5 equiv), dioxane, 130 °C, 15 min | 54 | 48 |
| 2 | Pd(OAc)2 (1 mol%), (o-biphenyl)P(t-Bu)2 (2 mol %), KF (3 equiv), THF, rt, 24 h | 45 | 47 |
| 3 | PdCl2(PPh3)2 (10 mol %), Cs2CO3 (4 equiv), DME, H2O, 130 °C, 15 min | 38 | 41 |
| 4 | 1,1′-bis(diphenylphosphino) ferrocene dichloropalladium (II) (10 mol %), NaOAc (5 equiv), DME, H2O, 150 °C, 15 min | 31 | 29 |
| 5 | Pd(PCy3)2Cl2 (5 mol %), CsF (2 equiv), NMP, 130 °C, 15 min | 31 | 32 |
| 6 | Pd(PPh3)4 (10 mol %), Cs2CO3 (4 equiv), DME, H2O, 125 °C, 15 min | 98 | 96 |
Isolated product after purification by silica gel column chromatography.
Utilising the above optimized conditions, Suzuki-Miyaura cross coupling of various bromo arylcarboxylates 1 (Figure 1) with o-hydroxyarylboronic acids 2 (Figure 2) was conducted and the results are summarized in Table 2. The coupling of o-hydroxyarylboronic acids 2 with bromo arylcarboxylates 1 bearing a range of electron donating and electron withdrawing groups gave corresponding benzopyaranones 3 in good to excellent yields (entries 1-19, Table 2). In addition, bromothiophene carboxylates (entry 20, 21), bromopyridine carboxylates (entries 22 and 23), and fused bromoaryl carboxylates (entries 24 and 25) also reacted well to give the corresponding products (entries 20-25, Table 2). Clearly, the tandem coupling followed by lactonization is a facile protocol to generate a library of compounds 3.
Figure 1.

Diversity of bromo arylcarboxylates 1
Figure 2.

Diversity of o-hydroxyarylboronic acids 2
Table 2.
Synthesis of a library of benzopyranone and heterocyclic analogues 3.
 | ||||
|---|---|---|---|---|
| Entry | 1 | 2 | Product | Yielda (%) |
| 1 | 1{1} | 2{1} |  |
98b,19 |
| 2 | 1{2} | 2{1} |  |
96 |
| 3 | 1{3} | 2{1} |  |
88 |
| 4 | 1{4} | 2{1} |  |
88b,6a |
| 5 | 1{5} | 2{1} |  |
80 |
| 6 | 1{6} | 2{1} |  |
88b,6a |
| 7 | 1{7} | 2{1} |  |
83 |
| 8 | 1{8} | 2{1} |  |
80 |
| 9 | 1{9} | 2{1} |  |
85 |
| 10 | 1{10} | 2{1} |  |
90 |
| 11 | 1{11} | 2{1} |  |
92 |
| 12 | 1{12} | 2{2} |  |
89 |
| 13 | 1{13} | 2{1} |  |
92b,6a |
| 14 | 1{14} | 2{1} |  |
84 |
| 15 | 1{15} | 2{1} | 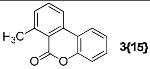 |
92 |
| 16 | 1{16} | 2{1} |  |
86 |
| 17 | 1{17} | 2{1} |  |
68 |
| 18 | 1{18} | 2{1} |  |
86 |
| 19 | 1{2} | 2{3} |  |
94 |
| 20 | 1{19} | 2{1} |  |
85 |
| 21 | 1{20} | 2{1} |  |
84 |
| 22 | 1{21} | 2{1} |  |
95 |
| 23 | 1{22} | 2{1} |  |
83 |
| 24 | 1{23} | 2{1} |  |
87 |
| 25 | 1{24} | 2{1} |  |
94 |
| 26 | 1{8} | 2{4} |  |
88 |
| 27 | 1{7} | 2{5} |  |
69 |
| 28 | 1{2} | 2{6} |  |
71 |
| 29 | 1{7} | 2{7} |  |
85 |
| 30 | 1{4} | 2{9} |  |
92 |
| 31 | 1{11} | 2{8} |  |
70 |
| 32 | 1{7} | 2{10} |  |
72 |
Isolated product after purification by silica gel column chromatography.
Known compounds, see references 6a and 19.
Conclusion
A simple, rapid and versatile one-step synthetic route to dibenzopyranones 3 and their heterocyclic analogues has been developed. The tandem C-C bond formation via Suzuki-Miyaura cross coupling followed by lactonization affords the desired dibenzopyranones 3 in a manner suitable for parallel synthesis. The protocol is complementary to the existing syntheses for dibenzopyranones in terms of number of synthetic steps, ease of accessibility of starting materials, and suitability for combinatorial format.
Experimental Section
General
Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. All palladium catalysts were purchased from Strem chemicals, Inc., and boronic acids were purchased from Combi-Blocks Inc., unless otherwise mentioned. 1H NMR spectra were recorded on a Varian Inova 500 MHz and 400 MHz spectrometers; the chemical shifts are reported in parts per million (δ), and the coupling constants are reported in hertz. IR spectra were obtained on a Bruker ALPHA ATR spectrometer. The high resolution mass spectral (HRMS) data (EI) were obtained on a Micromass 70-VSE instrument. The final compounds were purified with WATERS preparative LC/MS autopurification system, unless otherwise mentioned. The microwave reactions were conducted in a Emrys optimizer from Personal Chemistry, and generally 10 reactions were performed sequentially for parallel library synthesis.
Methyl 2-bromo-5-(trifluoromethyl)benzoate (1{7})
To a solution of 2-bromo-5-(trifluoromethyl) benzoic acid (5.00 g, 18.58 mmol) in methanol (20 mL) was added thionyl chloride (1.0 mL, 13.7 mmol) and the resulting mixture was refluxed for 8h. After cooling to room temperature, the solvent was concentrated to dryness under reduced pressure. The residue was dissolved in dichloromethane (40 mL), washed with saturated aqueous sodium bicarbonate solution (50 mL) followed by water (100 mL). The organic layer was dried over anhydrous Na2SO4, and the filtrate was concentrated to dryness under reduced pressure to give 1{7} (4.98 g, 95% yield). 1H-NMR (CDCl3, 400 MHz) δ 8.07 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.59-7.56 (m, 1H), 3.97 (s, 3H).
General procedure for the preparation of 2-hydroxyarylboronic acids (1{27} -1{32})
To a solution of corresponding methoxy boronicacids (1 mmol) in anhydrous dichloromethane (15 mL), was added boron tribromide (1.2 mmol) at -78°C under nitrogen atmosphere. The resulting solution was stirred for 15 minutes at -78°C. The cooling bath was removed and the reaction mixture was continued to stir for 30 minutes at room temperature. The reaction was quenched with anhydrous methanol (1 mL) at -78°C, concentrated in vacuo to dryness. The desired corresponding o-hydroxy boronic acids were confirmed by LC/MS and used as it is without further purification.
Illustrative example: Preparation of 8-methoxy-6H-benzo[c]chromen-6-one (3{1})
19 A solution of methyl 3-methoxybenzoate (1{1}, 0.5 mmol), 2-hydroxyarylboronic acid (2{1}, 0.65 mmol, 1.3 equiv), cesium carbonate (2 mmol, 4 equiv) in a mixture of dimethoxyethane (5 mL) and water (0.75 mL) was degassed with argon for 5 minutes. Tetrakis(triphenylphosphine)palladium (0) (10 mol%) was added, the reaction tube was sealed. The reaction was heated in a microwave (Emrys optimizer from Personal Chemistry) at 125 °C (power 300 W) for 15 minutes. After cooling at room temperature, the reaction mixture was diluted with water (30 mL), extracted with dichloromethane (2 × 15 mL). The combined organic layers were passed through a phase separator and concentrated in vacuo. The residue was purified with WATERS preparative LC/MS autopurification system to afford the title compound in 98% yield. 1H-NMR (CDCl3, 500 MHz) δ 8.05 (d, J = 8.5 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 3.0 Hz, 1H), 7.45-7.31 (m, 4H), 3.95 (s, 3H); IR (cm-1) 1707.9, 1610.1, 1479.8, 1276.1, 1024.4, 824.1, 742.1; LC/MS 226.98 (M+1); HRMS (EI): m/z calcd for C14H10O3 226.0630, found 226.0635.
9-Fluoro-6H-benzo[c]chromen-6-one (3{2})
Yield 96%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.44-8.41 (m, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.74-7.72 (m, 1H), 7.54-7.50 (m, 1H), 7.38-7.34 (m, 2H), 7.29-7.25 (m, 1H); IR (cm-1) 1731.6, 1611.2, 1420.8, 1300.8, 1187.5, 877.3, 742.1; LC/MS 215.0 (M+1); HRMS (EI): m/z calcd for C13H7FO2 214.0430, found 214.0422.
8-Fluoro-6H-benzo[c]chromen-6-one (3{3})
Yield 88%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.14-8.12 (m, 1H), 8.06-8.03 (m, 1H), 8.02-7.99 (m, 1H), 7.57-7.53 (m, 1H), 7.50-7.47 (m, 1H), 7.38-7.34 (m, 2H); LC/MS 214.9 (M+1); HRMS (EI): m/z calcd for C13H7FO2 214.0430, found 214.0437.
8-Methyl-6H-benzo[c]chromen-6-one (3{4})
6a Yield 88%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.18 (s, 1H), 8.02-7.98 (m, 2H), 7.63-7.61 (m, 1H), 7.46-7.43 (m, 1H), 7.35-7.30 (m, 2H), 2.49 (s, 3H); LC/MS 211.0 (M+1); HRMS (EI): m/z calcd for C14H10O2 210.0681, found 210.0682.
8-(Trifluoromethoxy)-6H-benzo[c]chromen-6-one (3{5})
Yield 80%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.25 (s, 1H), 8.19 (d, J = 8.5 Hz, 1H), 8.04 (d, J = 8.0, 1H), 7.69-7.66 (m, 1H), 7.54-7.51 (m, 1H), 7.41-7.36 (m, 2H); LC/MS 280.9 (M+1); HRMS (EI): m/z calcd for C14H7F3O2 280.0347, found 280.0388.
8-Nitro-6H-benzo[c]chromen-6-one (3{6})
6a Yield 88%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 9.25 (d, J = 2.5 Hz, 1H), 8.09 (d, J = 9.0, 2.5 Hz, 1H), 8.31 (d, J = 8.5 Hz, 1H), 8.14-8.12 (m, 1H), 7.65-7.62 (m, 1H), 7.46-7.42 (m, 2H); LC/MS 241.9 (M+1); HRMS (EI): m/z calcd for C13H7NO4 241.0375, found 241.0358.
8-(Trifluoromethyl)-6H-benzo[c]chromen-6-one (3{7})
Yield 83%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.70 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.11 (dd, J = 8.0, 1.5 Hz, 1H), 8.05 (dd, J = 8.5, 2.0 Hz, 1H), 7.60-7.56 (m, 1H), 7.43-7.39 (m, 2H); LC/MS 264.9 (M+1); HRMS (EI): m/z calcd for C14H7F3O2 264.0398, found 264.0407.
Methyl 6-oxo-6H-benzo[c]chromene-8-carboxylate (3{8})
Yield 80%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 9.08 (s, 1H), 8.47 (d, J = 8.5 Hz, 1H), 8.23 (d, J = 8.5 Hz, 1H), 8.13-8.11 (m, 1H), 7.57-7.55 (m, 1H), 7.42-7.39 (m, 2H), 3.99 (s, 3H); LC/MS 254.9 (M+1); HRMS (EI): m/z calcd for C15H10O4 254.0579, found 254.0570.
6H-Benzo[c]chromen-6-one (3{9})
Yield 85%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.42 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 7.5 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.86-7.83 (m, 1H), 7.61-7.58 (m, 1H), 7.51-7.48 (m, 1H), 7.39-7.33 (m, 2H); IR (cm-1) 1725.5, 1604.9, 1431.6, 1302.1, 1262.2, 1076.2, 743.9; LC/MS 196.9 (M+1); HRMS (EI): m/z calcd for C13H8O2 196.0529, found 196.0524.
Methyl 6-oxo-6H-benzo[c]chromene-9-carboxylate (3{10})
Yield 90%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.79 (s, 1H), 8.47 (d, J = 8.5 Hz, 1H), 8.20-8.15 (m, 2H), 7.53-7.51 (m, 1H), 7.40-7.37 (m, 2H), 4.03 (s, 3H); LC/MS 254.9 (M+1); HRMS (EI): m/z calcd for C15H10O4 254.0579, found 254.0571.
9-Chloro-6H-benzo[c]chromen-6-one (3{11})
Yield 92%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.35 (d, J = 8.5 Hz, 1H), 8.64 (d, J = 2.0 Hz, 1H), 8.02-8.00 (m, 1H), 7.56-7.51 (m, 2H), 7.40-7.35 (m, 2H); LC/MS 230.9, 232.9 (M+1); HRMS (EI): m/z calcd for C13H7ClO2 230.0140, found 230.0134.
2-Chloro-8-methoxy-6H-benzo[c]chromen-6-one (3{12})
Yield 89%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 7.97 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.41 (dd, J = 8.5, 2.5 Hz, 1H), 7.37 (dd, J = 8.5, 2.5 Hz, 1H), 7.30 (d, J = 8.5 Hz, 1H); LC/MS 260.9, 263.0 (M+1); HRMS (EI): m/z calcd for C14H9ClO3 260.0240, found 260.0225.
9-Methyl-6H-benzo[c]chromen-6-one (3{13})
6a Yield 92%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.27 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 1.0 Hz, 1H), 7.89 (s, 1H), 7.48-7.45 (m, 1H), 7.39-7.31 (m, 2H), 2.56 (s, 3H); LC/MS 211.0 (M+1); HRMS (EI): m/z calcd for C14H10O2 210.0680, found 210.0674.
8, 9-Dimethoxy-6H-benzo[c]chromen-6-one (3{14})
Yield 84%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 7.96 (d, J = 8.00 Hz, 1H), 7.75 (s, 1H), 7.61-7.28 (m, 2H), 7.37-7.31 (m, 2H); LC/MS 257.0 (M+1); HRMS (EI): m/z calcd for C15H12O4 256.0735, found 256.0729.
7-Methyl-6H-benzo[c]chromen-6-one (3{15})
Yield 92%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.04-7.98 (m, 2H), 7.67-7.64 (m, 1H), 7.46-7.43 (m, 1H), 7.38-7.37 (m, 1H), 7.33-7.28 (m, 2H), 2.87 (s, 3H); IR (cm-1) 1716.2, 1461.1, 1242.9 1206.4, 1049.0, 785.1, 751.5; LC/MS 211.0 (M+1); HRMS (EI): m/z calcd for C14H10O2 210.0680, found 210.0675.
10-Methyl-6H-benzo[c]chromen-6-one (3{16})
Yield 86%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.39-8.30 (m, 2H), 7.66-7.64 (m, 1H), 7.49-7.46 (m, 2H), 7.42-7.39 (m, 1H), 7.35-7.32 (m, 1H), 2.91 (s, 3H); LC/MS 211.0 (M+1); HRMS (EI): m/z calcd for C14H10O2 210.0680, found 210.0682.
7-Chloro-6H-benzo[c]chromen-6-one (3{17})
The residue was purified with Biotage SP1 flash autopurification system to afford the white solid in 68% yield, 1H-NMR (CDCl3, 500 MHz) δ 8.52 (d, J = 3.0 Hz, 1H), 7.82 (dd, J = 8.0, 1.5 Hz, 1H), 7.79-7.78 (m, 1H), 7.43-7.40 (m, 1H), 7.35-7.33 (m, 1H), 7.30-7.26 (m, 1H); LC/MS 230.9, 232.9 (M+1); HRMS (EI): m/z calcd for C13H7ClO2 230.0134, found 230.0126.
8,9-Difluoro-6H-benzo[c]chromen-6-one (3{18})
Yield 86%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.21-8.18 (m, 1H), 7.92-7.86 (m, 2H), 7.55-7.51 (m, 1H), 7.40-7.36 (m, 2H); LC/MS 232.9 (M+1); HRMS (EI): m/z calcd for C13H6F2O2 232.0335, found 232.0342.
2,9-Difluoro-6H-benzo[c]chromen-6-one (3{19})
Yield 94%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.47-8.44 (m, 1H), 7.67-7.62 (m, 2H), 7.39-7.31 (m, 2H), 7.27-7.23 (m, 1H); LC/MS 232.9 (M+1); HRMS (EI): m/z calcd for C13H6F2O2 232.0335, found 232.0343.
4H-Thieno[3,4-c]chromen-4-one (3{20})
Yield 85%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.52 (d, J = 3.0 Hz, 1H), 7.82 (dd, J = 8.0, 1.5 Hz, 1H), 7.79-7.78 (m, 1H), 7.43-7.40 (m, 1H), 7.35-7.33 (m, 1H), 7.30-7.26 (m, 1H); LC/MS 202.9 (M+1); HRMS (EI): m/z calcd for C11H6O2S 202.0088, found 202.0083.
4H-Thieno[2,3-c]chromen-4-one (3{21})
Yield 84%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 7.94 (d, J = 5.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 5.0 Hz, 1H), 7.53-7.45 (m, 2H), 7.38-7.34 (m, 1H); LC/MS 202.9 (M+1); HRMS (EI): m/z calcd for C11H6O2S 202.0088, found 202.0093.
5H-Chromeno[4,3-b]pyridin-5-one (3{22})
Yield 95%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 9.03-9.02 (m, 1H), 8.63-8.57 (m, 2H), 7.60-7.57 (m, 1H), 7.54-7.51 (m, 1H), 7.42-7.38 (m, 2H); LC/MS 197.9 (M+1); HRMS (EI): m/z calcd for C12H7NO2 197.0476, found 197.0482.
5H-Chromeno[4,3-c]pyridin-5-one (3{23})
Yield 83%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 9.58 (s, 1H), 8.88-8.87 (m, 1H), 8.21-8.16 (m, 2H), 7.58-7.55 (m, 1H), 7.44-7.41 (m, 2H); LC/MS 198.0 (M+1); HRMS (EI): m/z calcd for C12H7NO2 197.0476, found 197.0469.
6H-Naphtho[2,1-c]chromen-6-one (3{24})
Yield 87%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.87 (d, J = 7.5 Hz, 1H), 8.52 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.01-7.94 (m, 2H), 7.75-7.68 (m, 2H), 7.56-7.49 (m, 2H), 7.42-7.39 (m, 1H); LC/MS 246.9 (M+1); HRMS (EI): m/z calcd for C17H10O2 246.0680, found 246.0689.
6H-[1]Benzothieno[2,3-c]chromen-6-one (3{25})
Yield 94%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.65-8.62 (m, 1H), 8.49 (d, J = 8.0 Hz, 1H), 8.03-8.01 (m, 1H), 7.65-7.61 (m, 2H), 7.58-7.52 (m, 2H), 7.47-7.44 (m, 1H); LC/MS 252.9 (M+1); HRMS (EI): m/z calcd for C15H8O2S 252.0245, found 252.0241.
Methyl 3-fluoro-6-oxo-6H-benzo[c]chromene-8-carboxylate (3{26})
Yield 88%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 9.05 (s, 1H), 8.46 (dd, J = 7.0, 2.0 Hz, 1H), 8.13-8.12 (m, 2H), 7.14-7.12 (m, 2H), 3.99 (s, 3H); LC/MS 273.2 (M+1); HRMS (EI): m/z calcd for C15H9FO4 272.0430, found 272.0428.
2-Methyl-8-(trifluoromethyl)-6H-benzo[c]chromen-6-one (3{27})
The residue was purified with Biotage SP1 flash autopurification system to afford the white solid in 69% yield, 1H-NMR (CDCl3, 500 MHz) δ 8.67 (s, 1H), 8.24-8.23 (d, J = 8.0 Hz, 1H), 8.03-8.02 (d, J = 2.0, 1H), 7.87 (s, 1H), 7.35-7.27 (m, 2H), 2.49 (s, 3H); LC/MS 279.9 (M+1); HRMS (EI): m/z calcd for C15H9F3O2 278.0554, found 278.0545.
4-Chloro-9-fluoro-6H-benzo[c]chromen-6-one (3{28})
Yield 71%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.47-8.44 (m, 1H), 7.88 (dd, J = 7.0, 1.0 Hz, 1H), 7.74 (dd, J = 7.5, 2.5 Hz, 1H), 7.59 (dd, J = 6.0, 1.5 Hz, 1H), 7.34-7.28 (m, 2H); LC/MS 249.8 (M+1); HRMS (EI): m/z calcd for C13H6ClFO2 248.0040, found 248.0029.
2-Methoxy-8-(trifluoromethyl)-6H-benzo[c]chromen-6-one (3{29})
Yield 85%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.69 (s, 1H), 8.19 (d, J = 8.5 Hz, 1H), 8.04 (d, J = 2.0 Hz, 1H), 7.51 (s, 1H), 7.34 (m, 1H), 7.14-7.12 (m, 1H); LC/MS 295.1 (M+1); HRMS (EI): m/z calcd for C15H9F3O3 294.0503, found 294.0512.
3-Chloro-8-methyl-6H-benzo[c]chromen-6-one (3{30})
Yield 92%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.19 (s, 1H), 7.97-7.94 (m, 2H), 7.65-7.64 (m, 1H), 7.36-7.28 (m, 2H), 2.50 (s, 3H); LC/MS 245.6, 247.2 (M+1); HRMS (EI): m/z calcd for C14H9ClO2 244.0291, found 244.0306.
9-Chloro-1,2-difluoro-6H-benzo[c]chromen-6-one (3{31})
Yield 70%, white solid, 1H-NMR (CDCl3, 500 MHz) δ 8.51-8.50 (m, 1H), 8.40 (d, J = 8.0 Hz, 1H), 7.64 (dd, 7.0, 2.0 Hz, 1H), 7.37-7.33 (m, 1H), 7.19-7.17 (m, 1H); LC/MS 267.8, 269.9 (M+1); HRMS (EI): m/z calcd for C13H5ClF2O2 266.9946, found 265.9955.
2-Hydroxy-8-(trifluoromethyl)-6H-benzo[c]chromen-6-one (3{32})
The residue was purified with Biotage SP1 flash autopurification system to afford the white solid in 72% yield, 1H-NMR (CD3OD, 500 MHz) δ 9.33-9.30 (m, 1H), 9.24 (s, 1H), 9.05-9.03 (m, 1H), 8.48-8.47 (m, 1H), 8.13-8.11 (m, 1H), 7.90-7.87 (m, 1H); LC/MS 281.1 (M+1); HRMS (EI): m/z calcd for C14H7F3O3 280.0428, found 280.0431.
Supplementary Material
Acknowledgments
This research work was supported by Grants DA007215 and DA003801 from the National Institute of Health and National Institute of Drug Abuse. This paper is dedicated to Prof. John R. Scheffer, University of British Columbia on the occasion of his 70th birthday. One of the authors (KV) would like to thank Prof. J. Narasimha Moorthy, Indian Institute of Technology, Kanpur for helpful suggestions.
Footnotes
Supporting Information Available. General experimental details, yields, and compound characterization data (1H NMR, IR and MS analysis) of all final products. This information is available free of charge via the internet at http://pubs.acs.org.
Reference and Notes
- 1.(a) Koch K, Podlech J, Pfeiffer E, Metzler M. J Org Chem. 2005;70:3275–3276. doi: 10.1021/jo050075r. [DOI] [PubMed] [Google Scholar]; (b) Abe H, Nishioka K, Takeda S, Arai M, Takeuchi Y, Harayama T. Tetrahedron Lett. 2005;46:3197–3200. [Google Scholar]; (c) Sidwell WTL, Fritz H, Tamm C. Helv Chim Acta. 1971;54:207–215. [Google Scholar]; (d) Raistrick H, Stilkings CE, Thomas R. Biochemistry. 1953;55:421–433. doi: 10.1042/bj0550421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pero RW, Harvan D. Tetrahedron Lett. 1973;12:945–948. [Google Scholar]
- 2.(a) Garino C, Bihel F, Pietrancosta N, Laras Y, Quelever G, Woo I, Klein P, Bain J, Boucher JL, Kraus JL. Bioorg Med Chem Lett. 2005;15:135–138. doi: 10.1016/j.bmcl.2004.10.018. [DOI] [PubMed] [Google Scholar]; (b) Sun W, Cama LD, Birzin ET, Warrier S, Locco L, Mosley R, Hammond ML, Rohrer SP. Bioorg Med Chem Lett. 2006;16:1468–1472. doi: 10.1016/j.bmcl.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 3.(a) Edwards JP, West SJ, Marschke KB, Mais DE, Gottardis MM, Jones TK. J Med Chem. 1998;41:303–310. doi: 10.1021/jm9705770. [DOI] [PubMed] [Google Scholar]; (b) Hamann LG, Higuchi RI, Zhi L, Edwards JP, Wang XN, Marschke KB, Kong JW, Farmer LJ, Jones TK. J Med Chem. 1998;41:623–639. doi: 10.1021/jm970699s. [DOI] [PubMed] [Google Scholar]; (c) Coghlan MJ, Kym PR, Elmore SW, Wang AX, Luly JR, Wilcox D, Stashko M, Lin CW, Miner J, Tyree C, Nakane M, Jacobson P, Lane BC. J Med Chem. 2001;44:2879–2885. doi: 10.1021/jm010228c. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt JM, Tremblay GB, Page M, Mercure J, Feher M, Dunn-Dufault R, Peter MG, Redden PR. J Med Chem. 2003;46:1289–1292. doi: 10.1021/jm034007d. [DOI] [PubMed] [Google Scholar]
- 5.Myrray R, Mendez J, Brown S. The Natural Coumarins: Occurrence, Chemistry and Biochemistry. John Wiley & Sons; New York: 1982. pp. 97–111. [Google Scholar]
- 6.(a) Zhou QJ, Worm K, Dolle RE. J Org Chem. 2004;69:5147–5149. doi: 10.1021/jo049343w. [DOI] [PubMed] [Google Scholar]; (b) Kemperman GJ, Ter Horst B, Van de Goor D, Roeters T, Bergwerff J, van der Eem R, Basten J. Eur J Org Chem. 2006;14:3169–3174. [Google Scholar]; (c) Hussain I, Nguyen VTH, Yawer TT, Fiscer C, Reinke H, Langer P. J Org Chem. 2007;72:6255–6258. doi: 10.1021/jo070608r. [DOI] [PubMed] [Google Scholar]; (d) Alo BI, Patil PA, Sharp MJ, Siddiqui MA, Snieckus V. J Org Chem. 1991;56:3763–3768. [Google Scholar]
- 7.Thasana N, Worayuthakarn R, Kradanrat P, Hohn E, Young L, Ruchirawat S. J Org Chem. 2007;72:9379–9382. doi: 10.1021/jo701599g. [DOI] [PubMed] [Google Scholar]
- 8.Jung ME, Allen DA. Org Lett. 2009;11:757–760. doi: 10.1021/ol802792g. [DOI] [PubMed] [Google Scholar]
- 9.Sanz R, Fernandez Y, Castroviejo MP, Perez A, Fananas FJ. Eur J Org Chem. 2007;1:62–69. [Google Scholar]
- 10.Teske JA, Deiters A. Org Lett. 2008;10:2195–2198. doi: 10.1021/ol800589e. [DOI] [PubMed] [Google Scholar]
- 11.Ku YY, Grieme T, Raje P, Morton HE, Rozema M, King SA. J Org Chem. 2003;68:3238–3240. doi: 10.1021/jo0268613. [DOI] [PubMed] [Google Scholar]
- 12.Jilani JA. Chem, Pap. 2007;61:410–412. [Google Scholar]
- 13.Khanolkar AD, Lu D, Ibrahim M, Duclos R, Jr, Thakur GA, Malan TP, Jr, Porreca F, Veerappan V, Tian X, George C, Parrish DA, Papahatjis DP, Makriyannis A. J Med Chem. 2007;50:6493–6500. doi: 10.1021/jm070441u. [DOI] [PubMed] [Google Scholar]
- 14.McOmie JFW, Watts ML, West DE. Tetrahedron. 1968;24:2289–2292. [Google Scholar]
- 15.(a) Dallinger D, Kappe O. Chem Rev. 2007;107:2563–2591. doi: 10.1021/cr0509410. [DOI] [PubMed] [Google Scholar]; (b) Lidstrom P, Tierney J, Wathey B, Westman J. Tetrahedron. 2001;57:9225–9283. [Google Scholar]
- 16.(a) Kennedy JP, Williams L, Bridges DN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]; (b) Lew A, Krutzik PO, Hart ME, Chamberlin R. J Comb Chem. 2002;4:95–105. doi: 10.1021/cc010048o. [DOI] [PubMed] [Google Scholar]
- 17.Liu B, Moffett KK, Joseph RW, Dorsey BD. Tetrahedron Lett. 2005;46:1779–1782. [Google Scholar]
- 18.Wolfe JP, Singer RA, Yang BH, Buchwald SL. J Am Chem Soc. 1999;121:9550–9561. [Google Scholar]
- 19.Zhang W, Pugh G. Tetrahedron Lett. 2001;42:5613–5615. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.