

Abstract
The innate immune response plays a critical role in pathogen clearance. However, dysregulation of innate immunity contributes to acute inflammatory diseases such as sepsis and many chronic inflammatory diseases including asthma, arthritis, and Crohn’s disease. Pathogen recognition receptors including the Toll-like family of receptors play a pivotal role in the initiation of inflammation and in the pathogenesis of many diseases with an inflammatory component. Studies over the last 15 years have identified complex innate immune signal transduction pathways involved in inflammation that have provided many new potential therapeutic targets to treat disease. We are investigating several novel genes that exert spatial and in some cases temporal regulation on innate immunity signaling pathways. These novel genes include Tbc1d23, a RAB-GAP that inhibits innate immunity. In this review, we will discuss inflammation, the role of inflammation in disease, innate immune signal transduction pathways, and the use of spatiotemporal regulators of innate immunity as potential targets for discovery and therapeutics.
Keywords: Innate immunity, Inflammation, RAB, Toll-like receptor, Spatiotemporal
Inflammation and disease
The innate immune response regulates the process of inflammation, which plays a pivotal role in pathogen clearance (Fig. 1). However, inflammation can also lead to tissue damage and if over-activated acutely, inflammation can lead to diseases such as sepsis (Fig. 1). Chronic inflammation contributes to the pathogenesis of many different human diseases such as cancer, diabetes, atherosclerosis, chronic obstructive pulmonary disease (COPD), asthma, allergy, and inflammatory bowel disease (IBD) (Fig. 1) [1–12]. For example, while sepsis initiates with an infection (often due to healthcare-associated procedures or opportunistic infections in immune compromised individuals), unregulated inflammation and immune dysfunction ultimately are the cause of mortality in septic patients. There is an estimated fatality rate of 210,000 patients in the United States each year associated with sepsis [13]. Morbidity and mortality related to sepsis costs an estimated $17 billion in treatment every year, and the incidence of sepsis, like many other immune diseases, has increased greatly over the past few decades [14–16]. Clearly, novel therapeutic approaches are necessary to improve patient outcome and relieve economic costs for sepsis and other inflammatory diseases [17, 18]. It is therefore critical to identify mediators of inflammation in these diseases to identify potential therapeutic targets to improve disease outcome.
Fig. 1.
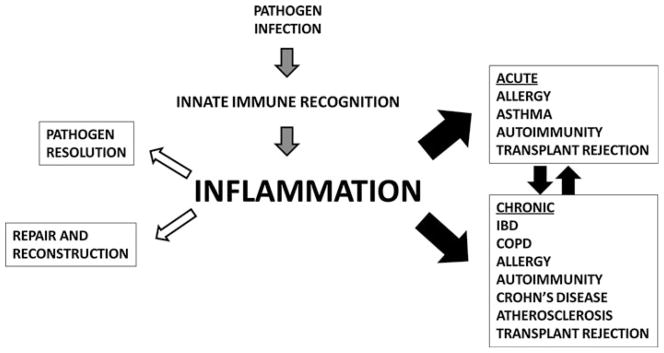
The innate immune response and disease. The innate immune response plays a critical role in fighting infection (left side), but a dysregulated response can contribute, at least in part, to the pathogenesis of many diseases (right side)
The innate immune response
Host defense against infection is controlled by two branches of the immune system, innate immunity and adaptive immunity. Innate immunity is the first line of defense, with all the cells and mechanisms poised to act immediately upon infection. These mechanisms include passive barriers such as skin, pH, and mucus, and active defense mechanisms provided by resident phagocytic cellular populations such as neutrophils and macrophages. The innate immune response can distinguish self from non-self, although with less specificity than the adaptive response. The adaptive immune response to a newly encountered pathogen occurs after 3–8 days; this response is against specific antigens present in pathogens and provides the host with cellular and humoral memory to these specific epitopes. The innate immune response is capable of recognizing specific pathogen-associated molecular patterns (PAMPs) present in a wide array of pathogens [19, 20], but unlike adaptive immunity, is unable to develop memory to those PAMPs. Besides initiating inflammation, the innate immune response is critical for a robust adaptive immune response. Recognition of PAMPs on a pathogen by the innate immune system is mediated through pattern recognition receptors (PRRs) present on both lymphoid and non-lymphoid cells that alert the host to an infection.
Innate immunity pathogen recognition receptors
Pathogen recognition receptors (PRRs) play an important role in innate immunity by recognizing conserved pathogen-specific PAMPs. Several families of PRRs have been identified including Rig-I-like receptors (RLRs), NOD-like receptors (NLRs), and Toll-like receptors (TLRs) [19, 20]. RLRs and NLRs recognize cytosolic PAMPs, while TLRs recognize both extracellular and intracellular PAMPs. Humans have ten functional TLRs, and mice have twelve known TLRs that can be divided by their subcellular localization. TLRs 3, 7, 8, and 9 are located intracellularly [21] and recognize nucleic acid PAMPs such as dsRNA (3), ssRNA (7 and 8), and unmethylated DNA (9) (Fig. 2). TLRs present on the cell surface include heterodimers of TLR2/1 (recognize diacylated lipopeptides), TLR2/6 (tri-acylated lipopeptides), and homodimers of TLR5 (bacterial flagellin) (Fig. 2) [21]. Unique among the PRRs, TLR4, which recognizes lipopolysaccharide (LPS) from Gram-negative bacteria, functions at both the cell surface and intracellularly (Fig. 2) [22].
Fig. 2.

Subcellular localization and trafficking of TLRs. The schematic depicts TLRs that are located on the cell surface (such as TLR5, TLR2/1, and TLR2/6) and other TLRs that are present in the endolysosome (TLRs 3, 7, 8, 9). TLR4 is present on the surface and the endosome. Arrows depict the direction of transport of the TLRs and some of the known transport genes (Rab10, Rab7b, and Unc93b)
TLR signaling
The initial response to PAMPs is mediated through the assembly of several signaling complexes and a series of post-translational modifications of resident signaling proteins that transduce signals from PRRs to transcription factors. The most common post-translational protein modifications that activate or inhibit signaling proteins include phosphorylation and ubiquitination [23]. These signal transduction pathways have been reviewed in great detail elsewhere [19, 20, 24–32], and we review them only briefly here, focusing on one of the best characterized pathways, the TLR4-mediated response to LPS [33]. TLR4 is a type I transmembrane protein that binds LPS using its extracellular domain in conjunction with its co-receptor MD-2. Binding of LPS to the extracellular domain of TLR4 induces dimerization of TLR4, which in turn induces a conformational change in the cytosolic domain of TLR4. This conformation change allows recruitment of Toll/IL-1 receptor (TIR) domain-containing adaptor proteins such as TIRAP and MyD88. These adaptor molecules then recruit several interleukin-1 receptor-associated kinases (IRAKs) and TNF receptor-associated factor 6 (Traf6). The IRAKs are kinases that phosphorylate each other and other proteins. Traf6 is a ubiquitin ligase that transfers K63-linked ubiquitin to activate proteins including TGFβ-activated kinase 1 (TAK1). TAK1 serves several key functions in the TLR4 response pathway including the activation of the MAP kinases p38, JNK, and ERK. Additionally, TAK1 phosphorylates NEMO (also called IKKγ) which in turn phosphorylates IKKα/β. IKKβ phosphorylates IkB, which is then targeted for K48-linked ubiquitination and degradation by the proteasome [30]. In unstimulated cells, IκB is bound to the transcription factor NFκB and sequesters NFκB in the cytoplasm; however, when IκB is degraded, NFκB re-localizes to the nucleus to activate transcription of pro-inflammatory cytokines. TLR4 also signals from the endosome through TRIF and TRAF3, which induces translocation of interferon response factor 3 (IRF3) to the nucleus to produce type I IFNs to enhance inflammation. Mutations have been identified in many of these genes that affect human disease, and several are being used as targets for the potential development of therapeutics [34–36].
Temporal regulation of innate immunity signaling
Regulation of innate immunity through positive and negative feedback loops can occur at multiple levels, and this regulation is critical both to generate a robust response to fight infection and to limit the response to prevent tissue damage. In general, the innate immune response can be segregated into three overlapping temporal phases [37]. In the first phase, constitutively expressed PRRs, signal transduction proteins, and cytosolically sequestered transcription factors such as NFκB are present and poised to respond rapidly to PAMP challenge. Stimulation with PAMP leads to rapid translocation of transcription factors into the nucleus and almost immediate production of important immune response products such as cytokines. This first phase of the innate immune response induces production of de novo proteins including additional transcription factors that regulate the second phase of the response, which occurs 2–8 h post-activation [37]. These transcription factors, which either enhance or diminish the response, include CEBPδ, which binds the IL-6 promoter and enhances IL-6 transcription, and ATF3, which inhibits IL-6 production by blocking CEBPδ binding [38]. Production of negative regulators like ATF3 is critical to limit the inflammatory response and prevent disease. The third phase of inflammatory gene expression is characterized by chromatin remodeling, which provides putative lineage commitments or provides an end to the response [37]. Positive and negative factors antagonize one another and often compete to determine the appropriate degree of response as well as final outcome in a temporal fashion. This ensures proper initiation of the inflammatory response but also ensures that inflammation is self-limiting and will not cause disease.
Spatial regulation of innate immunity signaling
Substantial research over the last 15 years has identified many proteins involved in innate immunity signal transduction pathways, thereby identifying human disease genes and potential targets for treating disease. Recently, the subcellular location and trafficking of these innate immunity proteins has gained substantial attention [22]. The identification of proteins that control trafficking of innate immune signaling proteins provides an additional layer of important regulatory factors that could represent human disease genes and therapeutic targets.
This trafficking has been best studied for the TLR family of PRRs. TLRs 3, 7, 8, and 9 localize to the endolysosome, while the other TLRs are located on the cell surface (Fig. 2) (The one exception to this is TLR4, which is present on the surface and inside the cell) [21, 22]. This compartmentalization and trafficking of TLRs is important. For example, if TLR9 is artificially targeted to the cell surface instead of the endolysosome, it has a diminished ability to recognize viral DNA but an enhanced ability to recognize self-DNA [39]. Thus, TLR9’s intracellular location facilitates recognition of pathogens while restricting recognition of self and possible autoimmune disease [40, 41].
The trafficking of intracellular TLRs to the endolysosome is regulated by Unc93b, which facilitates trafficking of TLR 3, 7, 8 and 9 from the endoplasmic reticulum to the endolysosome, which is critical for ligand recognition [42, 43]. Mutations in TLR3, which recognizes dsRNA from some viruses, have been identified in patients with recurrent herpes simplex virus 1 (HSV)-induced encephalitis [44]. Similarly, mutations in Unc93b also result in susceptibility to HSV-1 encephalitis in humans [45]. Thus, mutations in either the signaling protein (TLR3) or the trafficking protein (Unc93b) both cause the same disease in humans.
Other human immune diseases also may be affected by trafficking of immune proteins. Trafficking of cytokines is implicated in Griscelli syndrome (which is characterized by albinism and immunodeficiency) [46–48] and Crohn’s disease [49]. Unc93b is upregulated in patients with active systemic lupus erythematosus (SLE), suggesting that it could play a role in SLE [50]. Thus, identifying genes that control trafficking of immune signaling proteins could open up new opportunities for diagnosis of and treatment for many diseases.
Comparative genomics for the identification of novel spatiotemporal regulators of innate immunity
Our laboratory has used a comparative genomics RNAi screening approach in simple model systems to identify novel regulators of innate immunity [51]. These model systems include mouse macrophages, an important phagocytic innate immune cell [52], and the soil nematode Caenorhabditis elegans, which has an innate immune system but not an adaptive immune system, making it useful for innate immune studies [53–55]. One advantage of these models is the ability to perform rapid genomic-scale RNAi screens in a reasonable time frame. Once candidate innate immune regulators are identified in these simple model systems, they are validated in mammalian disease models. These mammalian models include testing knockout mice in sepsis and inflammatory lung disease models and investigating the association of polymorphisms in candidate genes with inflammatory disease in human patient populations. Thus far, we have identified dozens of candidates that affect the innate immune response in mouse macrophages [51, 56–58] and we are now investigating three genes in knockout mice that could affect spatiotemporal regulation of innate immunity signaling. One of these genes is Tbc1d23, a regulator of RABs [51, 59].
Role of RABs in innate immunity
The functions of the small GTPase RAB proteins in vesicular trafficking have been extensively studied [60] but have only recently been investigated in innate immunity. RABs are members of the RAS GTPase superfamily; more than 60 RABs are present in mammals. RAB proteins are compartmentalized to different organelles and are further segregated into microdomains. RABs play a critical role in vesicular trafficking; when RAB proteins are activated, they enhance the functions of their effector proteins, which can vary from vesicular membrane fusion to kinase activity. RAB proteins exist in two different states, either bound to guanine triphosphate (GTP), the active state, or bound to guanine diphosphate (GDP), the inactive state. GTPase-activating proteins (GAPs) facilitate hydrolysis of the GTP on their cognate RABs, thereby inactivating the protein, and guanine nucleotide exchange factors (GEFs) reactivate RABs by exchanging GDP for GTP on their cognate RABs [61–63]. RAB effector proteins have been shown to be involved in several different processes of vesicular function such as fusion, coating, and motility. Recently, several studies have highlighted the role of Rab proteins in TLR trafficking. Three RABs are known to affect TLR4 movement: RAB10 controls movement of TLR4 to the cell surface [64], Rab11a traffics TLR4 into the cell [65], and RAB7b traffics TLR4 to the lysosome [66]. Inhibition of Rab10 decreases TLR4 signaling, leading to decreased inflammatory gene expression [64], while inhibition of Rab7b stabilizes TLR4, leading to increased signaling and increased inflammatory gene expression [66]. Rabs also have been shown to activate PI3 kinase, which is involved in several different aspects of both innate and adaptive immunity [67–72]. Finally, mutations in Rab27a have been shown to cause Griscelli syndrome in humans [46, 47].
The Tbc1d23 RAB-GAP inhibits innate immunity signaling in spatiotemporal fashion
We identified Tbc1d23 as a novel innate immunity regulator in our comparative genomics RNAi screens. Mutation of the Tbc1d23 ortholog in C. elegans renders the nematodes more susceptible to pathogenic but not non-pathogenic bacteria [51]. Based on these results, we generated a Tbc1d23 knockout mouse and also engineered macrophages that overexpress Tbc1d23. Studies in these mice and cells have demonstrated that Tbc1d23 inhibits the mammalian innate immune response in spatiotemporal fashion [59]. The knockout mouse and macrophages derived from these mice had an enhanced response to many TLR agonists including LPS while macrophages overexpressing Tbc1d23 had weaker response (Fig. 3). This inhibition was regulated temporally, as cytokine production was only affected several hours after stimulation (Fig. 4). Consistent with the effects early but not late was the observation that initial activation events following LPS challenge were not affected by Tbc1d23. One clue to how this could be occurring was the observation that Tbc1d23 inhibited activation of the Xbp1 transcription factor, which acts in both ER stress and TLR signaling pathways [73–76]. Interestingly, Xbp1, like Tbc1d23, affects late but not early inflammatory gene expression [59, 73]. Thus, understanding the regulation of these genes could provide insight into the later phase of inflammatory gene expression regulation.
Fig. 3.
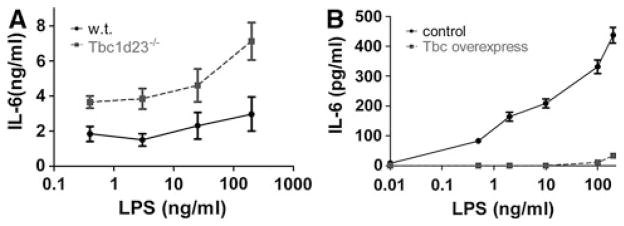
Tbc1d23 inhibits the innate immune response to LPS. a Bone marrow-derived macrophages from Tbc1d23−/− mice (dashed line) and from their wild-type siblings (solid line) were exposed to the indicated LPS doses for 6 h, and cytokine production was monitored by ELISA. b Macrophages overexpressing Tbc1d23 (dashed line) and control macrophages (solid line) were exposed to the indicated LPS doses for 6 h, and cytokine production was monitored by ELISA. Based on data from [59]
Fig. 4.
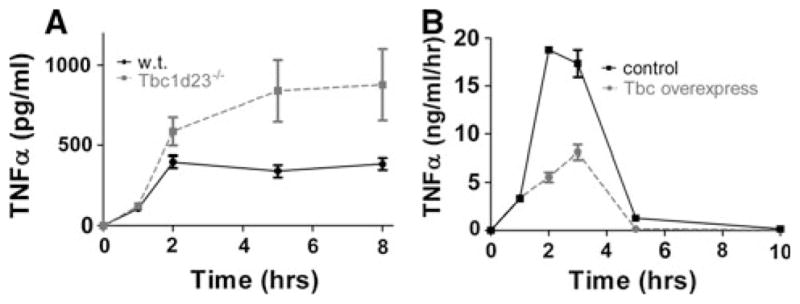
Tbc1d23 inhibits late but not early LPS-induced cytokine production. a Bone marrow-derived macrophages from Tbc1d23−/− mice (dashed line) and from their wild-type siblings (solid line) were exposed to LPS for the indicated times, and cytokine production was measured by ELISA. b Macrophages overexpressing Tbc1d23 (dashed line) and control macrophages (solid line) were exposed to LPS for the indicated times. At every time point, all of the media were removed for ELISA analysis and fresh media with LPS were added back to the cells. Thus, the data in a depict cytokine accumulation over time, while the data in b depict the differential cytokine production over time. In both experiments, whether Tbc1d23 was overexpressed or deleted, TNFα production was similar for the first 1–2 h (this data and data in [59]) following stimulation but not at later times. Based on data from [59]
Tbc1d23 contains the conserved Tbc domain present in RAB-GAPs. By mutating a conserved arginine in this domain, we showed that the Tbc domain is required for Tbc1d23 function, as the mutation in this domain abrogated the ability of Tbc1d23 to inhibit cytokine production without affecting localization or expression. This suggests that Tbc1d23 regulates innate immunity as a RAB-GAP, although to demonstrate this formally, it will be necessary to identify the cognate RAB target and test this biochemically. These data suggest that Tbc1d23 acts by controlling the activity of a yet to be identified RAB (the RABs already known to regulate TLR signaling do not fit the criteria for the Tbc1d23 cognate RAB). We therefore infer that Tbc1d23 is controlling the trafficking of an innate immune signaling protein by controlling RAB activity. This signaling protein could be in TLR signaling pathways upstream of the Xbp1 transcription factor.
Conclusion
Diseases with an inflammatory component such as sepsis and COPD are major causes of mortality and a major economic burden on today’s healthcare system. With the increase in immune disease in the past few decades, new therapeutic approaches and targets are necessary to improve patient outcome. In principle, any novel spatiotemporal regulator of innate immunity could represent a novel target for the development of therapeutics for many diseases. For example, Tbc1d23 overexpression does not affect the initiation of inflammation but does affect maintenance of inflammatory gene expression. Tbc1d23 overexpression should inhibit its cognate RAB, so we would predict that inhibition of this RAB, once it is identified, would largely abolish maintenance of inflammation without affecting the initial inflammatory response. Such a gene would be an intriguing target for the development of therapeutics to treat chronic inflammatory diseases without completely abolishing the necessary antimicrobial response.
Acknowledgments
This work was supported by grants R21ES 019256 from the National Institute of Environmental Health Sciences and RG-169529-N from the American Lung Association.
Contributor Information
Francisco Victorino, Integrated Department of Immunology, National Jewish Health and the University of Colorado School of Medicine, 1400 Jackson St., Denver, CO 80206, USA. Center for Genes, Environment, and Health, National Jewish Health, 1400 Jackson St., Denver, CO 80206, USA.
Scott Alper, Email: alpers@njhealth.org, Integrated Department of Immunology, National Jewish Health and the University of Colorado School of Medicine, 1400 Jackson St., Denver, CO 80206, USA. Center for Genes, Environment, and Health, National Jewish Health, 1400 Jackson St., Denver, CO 80206, USA.
References
- 1.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory microenvironment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Arcaroli J, Fessler MB, Abraham E. Genetic polymorphisms and sepsis. Shock. 2005;24(4):300–12. doi: 10.1097/01.shk.0000180621.52058.e1. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett NW, McLean GR, Chang YS, Johnston SL. Genetics and epidemiology: asthma and infection. Curr Opin Allergy Clin Immunol. 2009;9(5):395–400. doi: 10.1097/ACI.0b013e32833066fa. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri N, Dower SK, Whyte MK, Sabroe I. Toll-like receptors and chronic lung disease. Clin Sci (Lond) 2005;109(2):125–33. doi: 10.1042/CS20050044. [DOI] [PubMed] [Google Scholar]
- 5.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 7.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115(11):3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rutgers SR, Postma DS, ten Hacken NH, Kauffman HF, van Der Mark TW, Koeter GH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax. 2000;55(1):12–8. doi: 10.1136/thorax.55.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith JD, Peng DQ, Dansky HM, Settle M, Baglione J, Le Goff W, et al. Transcriptome profile of macrophages from atherosclerosis-sensitive and atherosclerosis-resistant mice. Mamm Genome. 2006;17(3):220–9. doi: 10.1007/s00335-005-0099-7. [DOI] [PubMed] [Google Scholar]
- 10.Tsan MF. Toll-like receptors, inflammation and cancer. Semin Cancer Biol. 2006;16(1):32–7. doi: 10.1016/j.semcancer.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Ishimori N, Korstanje R, Rollins J, Paigen B. Identifying novel genes for atherosclerosis through mouse-human comparative genetics. Am J Hum Genet. 2005;77(1):1–15. doi: 10.1086/431656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Bai C, Li K, Adler KB, Wang X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir Med. 2008;102(7):949–55. doi: 10.1016/j.rmed.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Wenzel RP. Treating sepsis. N Engl J Med. 2002;347(13):966–7. doi: 10.1056/NEJMp020096. [DOI] [PubMed] [Google Scholar]
- 14.Hall MJ, Williams SN, DeFrances CJ, Golosinskiy A. Inpatient care for septicemia or sepsis: a challenge for patients and hospitals. NCHS Data Brief. 2011;62:1–8. [PubMed] [Google Scholar]
- 15.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 16.Vincent JL. Clinical sepsis and septic shock–definition, diagnosis and management principles. Langenbecks Arch Surg. 2008;393(6):817–24. doi: 10.1007/s00423-008-0343-1. [DOI] [PubMed] [Google Scholar]
- 17.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 18.Hoebe K, Jiang Z, Georgel P, Tabeta K, Janssen E, Du X, et al. TLR signaling pathways: opportunities for activation and blockade in pursuit of therapy. Curr Pharm Des. 2006;12(32):4123–34. doi: 10.2174/138161206778743466. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9(8):535–42. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGettrick AF, O’Neill LA. Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr Opin Immunol. 2010;22(1):20–7. doi: 10.1016/j.coi.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17(6):666–72. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 24.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458(7237):430–7. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 25.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 26.Gay NJ, Gangloff M, O’Neill LA. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011;32(3):104–9. doi: 10.1016/j.it.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9(4):293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 28.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5(6):446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 29.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25(51):6685–705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 30.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–96. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 31.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 32.Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol. 2005;6(1):17–21. doi: 10.1038/ni1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–51. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5(10):975–9. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman ES, Smith RE, Renaud RC., Jr From the analyst’s couch: TLR-targeted therapeutics. Nat Rev Drug Discov. 2005;4(11):879–80. doi: 10.1038/nrd1880. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz DA, Cook DN. Polymorphisms of the Toll-like receptors and human disease. Clin Infect Dis. 2005;41(Suppl 7)(29):S403–7. doi: 10.1086/431985. [DOI] [PubMed] [Google Scholar]
- 37.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9(10):692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 38.Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, et al. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol. 2009;10(4):437–43. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 40.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. 2009;106(29):12061–6. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (Oxford) 2010;49(12):2281–9. doi: 10.1093/rheumatology/keq226. [DOI] [PubMed] [Google Scholar]
- 42.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature. 2008;452(7184):234–8. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 43.Saitoh S, Miyake K. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol Rev. 2009;227(1):32–43. doi: 10.1111/j.1600-065X.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 44.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 45.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–12. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 46.Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010;54(4):563–72. doi: 10.1002/pbc.22357. [DOI] [PubMed] [Google Scholar]
- 47.Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–6. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 48.Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res. 2009;22(3):268–82. doi: 10.1111/j.1755-148X.2009.00558.x. [DOI] [PubMed] [Google Scholar]
- 49.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206(9):1883–97. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakano S, Morimoto S, Suzuki S, Watanabe T, Amano H, Takasaki Y. Upregulation of the endoplasmic reticulum transmembrane protein UNC93B in the B cells of patients with active systemic lupus erythematosus. Rheumatology. 2010;49:876–81. doi: 10.1093/rheumatology/keq001. [DOI] [PubMed] [Google Scholar]
- 51.Alper S, Laws R, Lackford B, Boyd WA, Dunlap P, Freedman JH, et al. Identification of innate immunity genes and pathways using a comparative genomics approach. Proc Natl Acad Sci USA. 2008;105(19):7016–21. doi: 10.1073/pnas.0802405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burke B, Lewis CE. The macrophage. 2. Oxford: Oxford University Press; 2002. [Google Scholar]
- 53.Alper S. Model systems to the rescue: the relationship between aging and innate immunity. Commun Integr Biol. 2010;3(5):409–14. doi: 10.4161/cib.3.5.12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ewbank JJ. Signaling in the immune response. Worm-Book, editor. The C. elegans research community. 2006 doi: 10.1895/wormbook.1.83.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- 55.Irazoqui J, Ausubel F. 99th Dahlem conference on infection, inflammation, and chronic inflammatory disorders: Caenorhabditis elegans as a model to study tissues involved in host immunity and microbial pathogenesis. Clin Exp Immunol. 2010;160:48–57. doi: 10.1111/j.1365-2249.2010.04122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang IV, Alper S, Lackford B, Rutledge H, Warg LA, Burch LH, et al. Novel regulators of the systemic response to lipopolysaccharide. Am J Respir Cell Mol Biol. 2011;45(2):393–402. doi: 10.1165/rcmb.2010-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang IV, Jiang W, Rutledge HR, Lackford B, Warg LA, De Arras L, et al. Identification of novel innate immune genes by transcriptional profiling of macrophages stimulated with TLR ligands. Mol Immunol. 2011;48(15–16):1886–95. doi: 10.1016/j.molimm.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang IV, Wade CM, Kang HM, Alper S, Rutledge H, Lackford B, et al. Identification of novel genes that mediate innate immunity using inbred mice. Genetics. 2009;183(4):1535–44. doi: 10.1534/genetics.109.107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Arras L, Yang IV, Lackford B, Riches DW, Prekeris R, Freedman JH, et al. Spatiotemporal inhibition of innate immunity signaling by the Tbc1d23 RAB-GAP. J Immunol. 2012;188(6):2905–13. doi: 10.4049/jimmunol.1102595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–25. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 61.Barr F, Lambright DG. Rab GEFs and GAPs. Curr Opin Cell Biol. 2010;22(4):461–70. doi: 10.1016/j.ceb.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM. Illuminating the functional and structural repertoire of human TBC/ RABGAPs. Nat Rev Mol Cell Biol. 2012;13(2):67–73. doi: 10.1038/nrm3267. [DOI] [PubMed] [Google Scholar]
- 63.Fukuda M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci Rep. 2011;31(3):159–68. doi: 10.1042/BSR20100112. [DOI] [PubMed] [Google Scholar]
- 64.Wang D, Lou J, Ouyang C, Chen W, Liu Y, Liu X, et al. Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc Natl Acad Sci USA. 2010;107:13806–11. doi: 10.1073/pnas.1009428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Husebye H, Aune M, Stenvik J, Samstad E, Skjeldal F, Halaas O, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. 2010;33:583–96. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Chen T, Han C, He D, Liu H, An H, et al. Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood. 2007;110(3):962–71. doi: 10.1182/blood-2007-01-066027. [DOI] [PubMed] [Google Scholar]
- 67.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23(2):178–83. doi: 10.1016/j.coi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harris SJ, Parry RV, Westwick J, Ward SG. Phosphoinositide lipid phosphatases: natural regulators of phosphoinositide 3-kinase signaling in T lymphocytes. J Biol Chem. 2008;283(5):2465–9. doi: 10.1074/jbc.R700044200. [DOI] [PubMed] [Google Scholar]
- 69.Kashiwada M, Lu P, Rothman PB. PIP3 pathway in regulatory T cells and autoimmunity. Immunol Res. 2007;39(1–3):194–224. doi: 10.1007/s12026-007-0075-2. [DOI] [PubMed] [Google Scholar]
- 70.Oak JS, Fruman DA. Role of phosphoinositide 3-kinase signaling in autoimmunity. Autoimmunity. 2007;40(6):433–41. doi: 10.1080/08916930701464780. [DOI] [PubMed] [Google Scholar]
- 71.So L, Fruman DA. PI3 K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465–81. doi: 10.1042/BJ20112092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang TT, Li H, Cheung SM, Costantini JL, Hou S, Al-Alwan M, et al. Phosphoinositide 3-kinase-regulated adapters in lymphocyte activation. Immunol Rev. 2009;232(1):255–72. doi: 10.1111/j.1600-065X.2009.00838.x. [DOI] [PubMed] [Google Scholar]
- 73.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11(5):411–8. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 75.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38(5):1194–203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeng L, Liu YP, Sha H, Chen H, Qi L, Smith JA. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J Immunol. 2010;185(4):2324–30. doi: 10.4049/jimmunol.0903052. [DOI] [PMC free article] [PubMed] [Google Scholar]