

Abstract
The mammalian adenosine monophosphate–activated protein kinase (AMPK) is a serine-threonine kinase protein complex that is a central regulator of cellular energy homeostasis. However, the mechanisms by which AMPK mediates cellular responses to metabolic stress remain unclear. We found that AMPK activates transcription through direct association with chromatin and phosphorylation of histone H2B at serine 36. AMPK recruitment and H2B Ser36 phosphorylation colocalized within genes activated by AMPK-dependent pathways, both in promoters and in transcribed regions. Ectopic expression of H2B in which Ser36 was substituted by alanine reduced transcription and RNA polymerase II association to AMPK-dependent genes, and lowered cell survival in response to stress. Our results place AMPK-dependent H2B Ser36 phosphorylation in a direct transcriptional and chromatin regulatory pathway leading to cellular adaptation to stress.
Signaling pathways often involve cascades of protein phosphorylation that terminate in regulation of nuclear transcription. However, the central kinases in these pathways are generally not thought to directly modulate transcription. One such signaling kinase, adenosine monophosphate–activated protein kinase (AMPK) (1), is activated by high substrate adenosine monophosphate levels under conditions of energetic stress, such as nutrient deprivation or hypoxia. AMPK activation initiates a program of metabolic adaptation to preserve cellular energy (adenosine triphosphate conservation) and maintain cellular viability (2).
We investigated possible direct transcriptional mechanisms in the AMPK pathway. First, we examined whether AMPK broadly responds to cellular stress. Mouse embryonic fibroblasts (MEFs) were stressed by ultraviolet (UV) irradiation, γ irradiation, the topoisomerase inhibitor camptothecin, the DNA intercalating agent adriamycin, the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine, or hydrogen peroxide. AMPK was activated [Thr172 phosphorylation in the AMPK activation loop and phosphorylation of substrate ACCα (acetyl coenzyme A–carboxylase-α)] by all agents (Fig. 1A). Liver kinase B1 (LKB1) is one of three mammalian upstream AMPK kinases and activates AMPK upon bioenergetic stress (3). LKB1 also activated AMPK in response to either genotoxic (UV irradiation) or metabolic stress [the glycolytic inhibitor 2-deoxyglucose (2-DG)] (fig. S1).
Fig. 1.
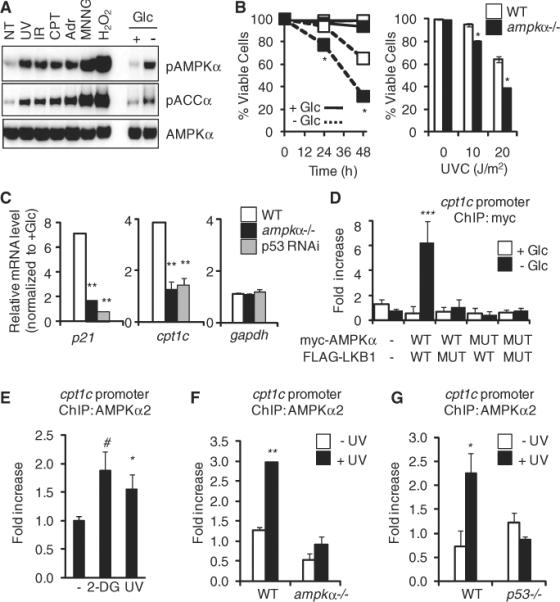
AMPK is required for stress-dependent transcription and localizes to stress-responsive genes. (A) Western blot of AMPK activation (AMPKα pThr172) and ACCα phosphorylation (ACCα pSer79) in MEFs in response to UV light, γ irradiation (IR), camptothecin (CPT), adriamycin (Adr), N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), or hydrogen peroxide (H2O2). Exposure times were 6 hours for UV, IR, CPT, and Adr and 30 min for MNNG and H2O2. NT, no treatment; Glc, glucose. (B) Viability of wild-type (WT) and ampkα−/− MEFs in response to indicated stresses. Left panel: glucose (Glc) withdrawal; right panel, UV irradiation (UVC). (C) Expression (qPCR) of p21, cpt1c, and gapdh mRNA in wild-type, AMPKα-deficient, or p53 RNAi–expressing MEFs after glucose withdrawal, relative to untreated cells. (D) ChIP of WT or dominant negative (MUT) myc-AMPK after glucose withdrawal in lkb1−/− MEFs cotransfected with either WT or catalytically inactive (MUT) FLAG-LKB1, both able to be immunoprecipitated [see (A), fig. S7, and (5)]. Data represent means ± SEM for n = 3. (E to G) Endogenous AMPKα2 ChIP in untreated MEFs (−), 2-DG (15 min), or UV (6 hours) (E); WT or ampkα−/− MEFs (F); and WT or p53−/− MEFs (G). Data represent means ± SEM for n = 3. #P < 0.06, *P < 0.05, **P < 0.01, ***P < 0.03.
We next used wild-type or AMPKα1- and AMPKα2-deficient MEFs (ampkα−/−) to examine whether AMPK influences cellular survival during stress (4). The ampkα−/− MEFs displayed decreased survival in the presence of metabolic stressors (Fig. 1B and fig. S2), UV irradiation (Fig. 1B), or hydrogen peroxide (fig. S2), indicating that AMPK promotes cell survival in response to various metabolic and genotoxic stresses.
Our previous work implicated AMPK and LKB1 in tumor suppressor p53–dependent metabolic and genotoxic stress responses (2, 5). We examined the involvement of AMPK in transcriptional regulation of p53-responsive genes. ampkα−/− MEFs showed decreased induction of p21, a well-characterized p53 target gene, in response to both glucose withdrawal and UV irradiation (Fig. 1C and fig. S3). The reduction in glucose-regulated gene expression was similar in magnitude to knockdown of p53 (Fig. 1C). Other p53-dependent genes, including reprimo, cyclinG, and cpt1c, showed reduced expression in ampkα−/− cells deprived of glucose, whereas expression of a control gene, gapdh, was unaffected (Fig. 1C and fig. S4). A similar defect in stress-dependent transcription was observed in human cancer cells (HCT116 colon carcinoma) (fig. S5). The transcriptional defect observed in UV-treated cells occurred downstream of the DNA-damage checkpoint, as phosphorylation of Chk1, Rad17, and p53 in both lkb1−/− and ampkα−/− cell lines was similar to that of wild-type controls (fig. S6). Thus, in cell culture, the LKB1-AMPK pathway responds to a wide variety of metabolic and genotoxic stresses and is required for maximal stress-induced transcription of multiple p53-dependent genes that promote survival.
These and previous results (6) implicate AMPK in gene activation, although the underlying mechanisms have remained elusive. We used chromatin immunoprecipitation (ChIP) and quantitative polymerase chain reaction (qPCR) to investigate whether AMPK plays a direct role at p53-regulated genes in MEFs. We found that LKB1 coimmunoprecipitated with AMPK in response to UV irradiation, and this interaction was dependent on AMPK kinase activity (fig. S7). ChIP showed ectopic myc-AMPK associated with the cpt1c and p21 gene promoters in response to glucose withdrawal and UV treatment (Fig. 1D and fig. S8). Ectopic kinase-deficient myc-AMPKα or FLAG-tagged LKB1 abrogated the binding of AMPK to chromatin (Fig. 1D and fig. S8). FLAG-LKB1 similarly associated with the p21 promoter (fig. S9). Binding of AMPK or LKB1 to promoters in unstressed cells was low (Fig. 1D and figs. S8 and S9). AMPK was also detected at the p21 promoter in human HCT116 cells after UV treatment (fig. S10). This AMPK activation appears to be specific for survival in response to stress, because AMPK was not detected at the PUMA promoter, a p53-dependent, pro-apoptotic gene (fig. S11). The immunoprecipitation and ChIP results suggest a functional, kinase-dependent interaction and promoter association of LKB1 and AMPK.
ChIP of endogenous AMPK, using an AMPKα2 antibody that immunoprecipitated the protein (fig. S12), showed increased AMPKα2 binding at p53 sites upstream of cpt1c in cells treated with 2-DG and UV light (Fig. 1E; at the p21 promoter, fig. S13). The binding was reduced in both ampkα−/− cells (Fig. 1F) and p53−/− cells (Fig. 1G). Thus, in response to cellular stress, LKB1 and AMPK physically localize to chromatin in an interdependent and p53-dependent fashion to regulate gene transcription.
Posttranslational modifications of core histones, including phosphorylation, function to regulate transcription (7, 8). To examine histones as substrates of AMPK, we epitope-tagged the predominantly nuclear isoform AMPKα2 (9) and immunoprecipitated wild-type AMPKα2 or a catalytic mutant (Asp157 → Ala) from transfected ampkα−/− MEFs after low glucose treatment to activate AMPK. The wild-type AMPKα2 complex specifically phosphorylated histone H2B above the nonspecific background activity of all four histones observed in the catalytic mutant and mock immunoprecipitations (Fig. 2A). AMPKα2 from untreated cells exhibited low activity on H2B relative to AMPKα2 from stressed cells (Fig. 2B and fig. S15). H2A was also phosphorylated in these assays; however, phospho-H2A was detected in the mock immunoprecipitation and did not respond to stress, which suggested that the activity is nonspecific. These data indicate that activated AMPKα2 specifically phosphorylates H2B.
Fig. 2.
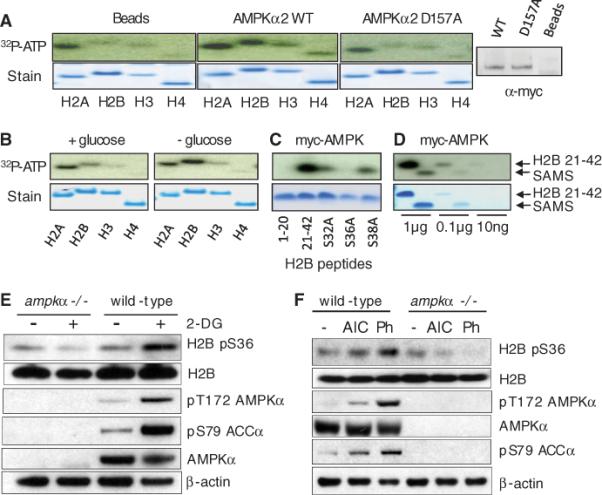
H2B is an AMPK target. (A to D) In vitro phosphorylation by myc-AMPK immunoprecipitated from ampkα−/− MEFs of recombinant human histones by beads alone, WT myc-AMPK, or catalytic mutant (D157A) myc-AMPK (right: myc Western blot) (A); recombinant human histones using myc-AMPK from glucose-treated cells (+glucose) or untreated cells (−glucose) (B); H2B peptides (C); and H2B 21–42 and SAMS peptide (D). (E) Western blots of wild-type or ampkα−/− MEFs treated with 2-DG (25 mM for 10 min) or untreated. (F) Western blots of MEFs treated for 1 hour with AICAR (AIC, 2 mM) or phenformin (Ph, 3 mM) or untreated.
Analysis of histone H2B revealed a potential AMPK target motif (10, 11) at Ser36 in the N terminus, similar to the AMPK substrates endothelial nitric oxide synthase (eNOS) and phosphofructokinase 2 (PFK2) (H2B, Arg-Ser-Arg-Lys-Glu-Ser; eNOS, Arg-Ile-Arg-Thr-Gln-Ser; PFK2, Arg-Met-Arg-Arg-Asn-Ser), with conserved Arg residues at P–5 and P–3 relative to the phosphoacceptor Ser36 (fig. S14) (12). We therefore compared peptides corresponding to residues 1 to 20 or 21 to 42 of H2B; only the H2B 21–42 peptide was phosphorylated by immunoprecipitated AMPKα2. Moreover, an H2B 21–42 peptide harboring a Ser36 → Ala (S36A) substitution was not phosphorylated, whereas S32A and S38A peptides were phosphorylated (Fig. 2C). In addition, myc-AMPKα2 phosphorylated the H2B 21–42 peptide more efficiently than did SAMS (His-Met-Arg-Ser-Ala-Met-Ser-Gly-Leu-His-Leu-Val-Lys-Arg-Arg) peptide, a canonical AMPK target sequence (Fig. 2D) (13). Thus, H2B Ser36 is a robust in vitro AMPK substrate.
We next examined whether AMPK is in volved in the regulation of H2B Ser36 phosphorylation in vivo. We generated an antibody against an H2B pS36 peptide and extensively characterized the affinity-purified antibody in vitro and in vivo (fig. S16). H2B pS36 occurs in cells treated with 2-DG, which induced metabolic stress shown by increased phosphorylation of AMPKα and ACCα (Fig. 2E), whereas other histone marks, including H3, K9me3 H3, and pS10 H3, did not significantly change (fig. S17). In ampkα−/− MEFs, H2B pS36 did not increase after 2-DG treatment (Fig. 2E). We detected similar induction of H2B pS36 in cells with two well-characterized AMPK activators, aminoimidazole carboxamide ribonucleotide (AICAR) and phenformin (Fig. 2F). In addition, H2B pS36 was not detected in lkb1−/− MEFs but increased after reintroduction of LKB1, and 2-DG stimulated H2B pS36 only when LKB1 was present (fig. S18). These results suggest that H2B Ser36 phosphorylation is downstream of the LKB1-AMPK signaling pathway in vivo in response to a broad range of metabolic stresses, and that AMPK is likely the direct kinase in this pathway. This global increase of H2B pS36 may not be limited to p53 target genes (Fig. 1G), because H2B pS36 levels were not reduced in p53−/− cells (fig. S19).
We found that transfected myc-AMPK and FLAG-H2B associate in 293T cells in response to the AMPK agonist AICAR (Fig. 3A). This interaction was ablated by an H2B S36A mutation (Fig. 3A), suggesting that Ser36 is critical for the interaction of AMPK and H2B. We next performed ChIP experiments in wild type and ampkα−/− MEFs to determine whether H2B pS36 is associated with AMPK-dependent genes, specifically at the p53 binding site in the cpt1c promoter. The H2B pS36 ChIP signal increased in response to both 2-DG and UV treatment, and the ChIP signal was reduced to background levels in AMPKα-deficient cells (Fig. 3B).
fig. 3.
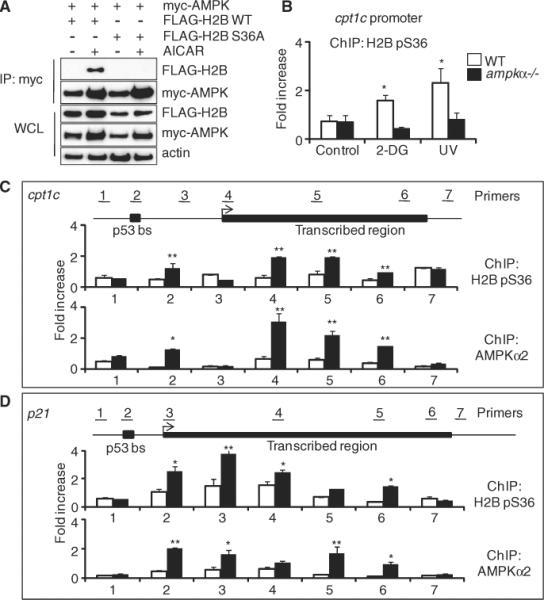
AMPK and H2B pS36 are localized to chromatin in response to stress. (A) Western blots showing myc-AMPK immunoprecipitation from WT H2B– or H2B S36A–expressing cells after AICAR treatment (1 mM, 24 hours). WCL, whole-cell lysate. (B) ChIP in WT or ampkα−/− MEFs. Data represent means ± SEM for n = 3. (C and D) ChIP along cpt1c gene (C) and p21 gene (D) with glucose (white bars) or without glucose (black bars). Data represent means ± SEM for n = 3. *P < 0.01, **P < 0.05.
To compare the locations of endogenous AMPK and H2B pS36, we performed ChIP at cpt1c under normal and glucose-free conditions, using primers along the promoter, transcribed region, and upstream or downstream nontranscribed regions (Fig. 3C, primer sets 1 to 7). Upon stress, an increase in H2B pS36 occurred at the p53 binding site (p53bs, primer set 2), a larger increase at the transcription start site (TSS, primer set 4), and only background levels upstream (primer set 1) and between (primer set 3) these sites, and downstream of the gene (primer set 7) (Fig. 3C). Notably, H2B pS36 associated throughout the transcribed region of cpt1c (primer sets 5 and 6) (Fig. 3C). We then used the AMPKα2 antibody and discovered a positive correlation between locations of endogenous AMPK and H2B pS36, again with localization throughout the transcribed region (Fig. 3C). The p21 gene showed a similar positive correlation between H2B pS36 and endogenous AMPK localization at the p53 binding sites and along the transcribed region, but not around the gene and only in response to low glucose (Fig. 3D). As a control, we tested the gapdh gene, which showed no difference in ChIP signal between the stressed and unstressed cells (fig. S20). Similar results were observed at the cpt1c gene in response to UV treatment (fig. S21). Ectopic myc-AMPK also localized along the p21 transcribed gene in response to low glucose and required AMPK catalytic activity (fig. S22). This correspondence in the location of AMPK and H2B pS36 suggests that the role of AMPK in transcriptional regulation is linked to the phosphorylation of H2B along transcribed regions of target genes.
We generated clonal MEF cell lines with stable ectopic expression of FLAG-H2B or mutant FLAG–H2B S36A, and, as shown above, only FLAG-H2B was detected by the H2B pS36 antibody (fig. S16). The level of FLAG-tagged wild-type or mutant histone H2B was lower than that of endogenous H2B as measured by Western blot (fig. S23); however, both ectopic wild-type and H2B S36A similarly incorporated into chromatin, as determined by ChIP and qPCR at the promoter and transcribed region of the cpt1c gene (fig. S24) and by coimmunoprecipitation of endogenous H3 and H2B with FLAG-H2B as shown by Western blot (fig. S25). We isolated several equivalently expressing FLAG-H2B– and FLAG–H2B S36A–expressing clonal lines (fig. S26). The cell lines treated with 2-DG showed comparable levels of AMPK and induction of AMPK Thr172 and pACCα Ser79 phosphorylation (fig. S27), suggesting that signaling to AMPK is unaffected by the S36A mutation. In response to glucose withdrawal, elevated transcription of the AMPK target genes p21, cpt1c, reprimo, and cyclinG was detected in wild-type FLAG-H2B–expressing cells; however, FLAG–H2B S36A–expressing cells showed significantly reduced induction of these genes (Fig. 4A). Expression of the control genes gapdh and hadh was unaffected by expression of the S36A mutant (fig. S28). Thus, although expression of FLAG-H2B is low relative to endogenous H2B (figs. S23 and S25), we observed a significant and specific transcriptional defect in the cells ectopically expressing S36A mutant H2B.
Fig. 4.
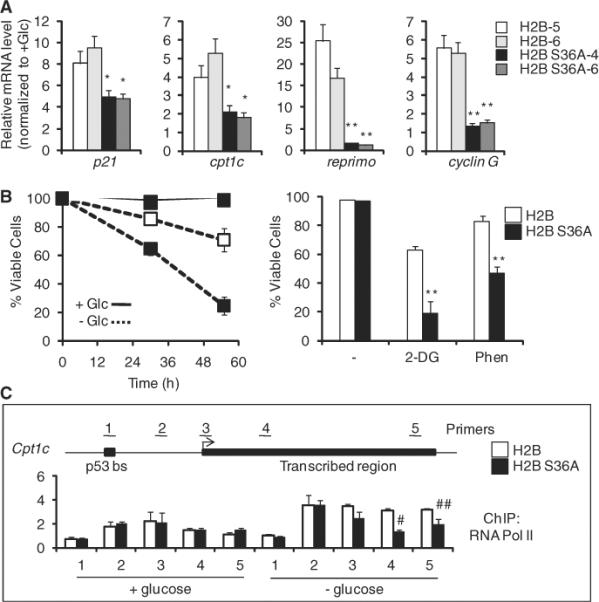
H2B Ser36 is essential for transcription and survival in response to metabolic stress. Data for MEF cell lines expressing wild-type H2B or H2B S36A are shown. (A) Relative expression (qPCR) of indicated genes after glucose withdrawal in individual MEF clones (H2B, clones 5 and 6; H2B S36A, clones 4 and 6). (B) Viability of H2B (open squares) or H2B S36A (solid squares) MEFs in no glucose, 2-DG (10 mM), or phenformin (3 mM). (C) ChIP of RNA Pol II across the cpt1c gene. Data represent means ± SEM for n = 3. *P < 0.05, **P < 0.01, #P < 0.03, ##P < 0.08.
We next tested the viability of these cells in response to various metabolic stresses including glucose withdrawal, 2-DG, and phenformin. The level of cell death induced by these stresses was significantly greater in the presence of ectopic H2B S36A, relative to cells expressing ectopic wild-type H2B (Fig. 4B).
Finally, we examined whether RNA polymerase II (Pol II) occupancy at genes is altered by ectopic expression of H2B S36A. ChIP analysis showed that Pol II was present through the cpt1c gene, and Pol II increased in glucose withdrawal (Fig. 4C). In FLAG–H2B S36A cells, the Pol II ChIP signal was reduced along the transcribed region of the cpt1c gene specifically under conditions of glucose withdrawal (Fig. 4C). These data point to AMPK-mediated phosphorylation of H2B at Ser36 as a critical transcriptional regulatory step in the cellular adaptation response to stress, to optimize cell survival.
Our results show that AMPK directly associates with chromatin to regulate transcriptional programs required to survive a wide variety of metabolic and environmental stresses. Mammalian AMPK is a histone kinase that preferentially phosphorylates histone H2B at Ser36. Phosphorylation of this site in vivo is acutely dependent on the LKB1-AMPK energy-sensing pathway, and through their direct action at genes.
AMPK and H2B pS36 appear to regulate transcription by direct interaction between the kinase and the H2B substrate. AMPK appears to be recruited to specific promoters by transcription factors such as p53. AMPK may similarly influence other stress-induced transcriptional activators, including Foxo3a, peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α), and transducer of regulated CREB activity 2 (TORC2) (2, 14–16). AMPK colocalizes with and phosphorylates H2B at p53 binding sites and along the transcribed cpt1c and p21 genes. This pattern may be due to activator recruitment of the enzyme, followed by enzyme transit across transcribed regions, as seen for the SAGA-Gcn5-Ubp8 complex and in the acetylation of H3 with deubiquitination of H2B (17, 18). Moreover, histone methylases associate with RNA Pol II to modify H3 across transcribed regions (19). Our findings represent evidence for histone phosphorylation across transcribed regions, which suggests that AMPK, via H2B Ser36 phosphorylation, may play a role in transcriptional elongation.
Ectopic expression of the H2B S36A mutant, even in the presence of higher levels of endogenous wild-type H2B, significantly reduces transcription of AMPK target genes and stress survival. Hence, the modification may function processively to facilitate transcription elongation. Indeed, H2B Ser36 phosphorylation promotes RNA Pol II association to transcribed regions. Ectopic expression of mutant histone genes may be of broad utility to unravel functions of other histone modifications in mammalian cells.
As part of the central energy-sensing pathway in eukaryotes, AMPK functions to maintain cellular energy homeostasis in the face of metabolic perturbations. Our data suggest that modification of H2B by AMPK may be a general stress-response pathway to tune specific transcriptional responses, regulate cellular metabolism, and promote cell survival.
Supplementary Material
Acknowledgments
Supported by NIH grants CA078831 (S.L.B.) and CA105463 (C.B.T.), NIH Training Program in Basic Cancer Research grant CA09171 (D.B.), Canadian Institutes of Health Research grant MOP-93799 (R.G.J.), the McGill Integrated Cancer Research Training Program (B.F.), and the Leukemia and Lymphoma Society, NIH, National Cancer Institute, and American Association for Cancer Research (AACR)/Stand Up to Cancer (C.B.T.). C.B.T. reports the filing of patent applications exploiting the use of AMPK activators in cancer therapy. He is also a member of the board of directors of Merck, the scientific advisory board of Agios Pharmaceuticals, and the medical advisory board of the Howard Hughes Medical Institute.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/science.1191241/DC1
References and Notes
- 1.Hardie DG. Nat. Rev. Mol. Cell Biol. 2007;8:774. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 2.Jones RG, et al. Mol. Cell. 2005;18:283. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG. Curr. Opin. Cell Biol. 2005;17:167. doi: 10.1016/j.ceb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Laderoute KR, et al. Mol. Cell. Biol. 2006;26:5336. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng PY, Berger SL. Cancer Res. 2006;66:10701. doi: 10.1158/0008-5472.CAN-06-0999. [DOI] [PubMed] [Google Scholar]
- 6.McGee SL, Hargreaves M. Front. Biosci. 2008;13:3022. doi: 10.2741/2907. [DOI] [PubMed] [Google Scholar]
- 7.Berger SL. Nature. 2007;447:407. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 8.Ivaldi MS, Karam CS, Corces VG. Genes Dev. 2007;21:2818. doi: 10.1101/gad.1604007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salt CJ, et al. Biochem. J. 1998;334:177. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gwinn DM, et al. Mol. Cell. 2008;30:214. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weekes J, Ball KL, Caudwell FB, Hardie DG. FEBS Lett. 1993;334:335. doi: 10.1016/0014-5793(93)80706-z. [DOI] [PubMed] [Google Scholar]
- 12.Towler MC, Hardie DG. Circ. Res. 2007;100:328. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Salt IP, Davies SP. Methods Mol. Biol. 2000;99:63. doi: 10.1385/1-59259-054-3:63. [DOI] [PubMed] [Google Scholar]
- 14.Shaw RJ, et al. Science. 2005;310:1642. [Google Scholar]
- 15.Greer EL, et al. J. Biol. Chem. 2007;282:30107. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 16.Jäger S, Handschin C, -Pierre J. St., Spiegelman BM. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12017. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Govind CK, Zhang F, Qiu H, Hofmeyer K, Hinnebusch AG. Mol. Cell. 2007;25:31. doi: 10.1016/j.molcel.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Wyce A, et al. Mol. Cell. 2007;27:275. doi: 10.1016/j.molcel.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 19.Shilatifard A. Annu. Rev. Biochem. 2006;75:243. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.