

Abstract
How do regulatory switches achieve high sensitivity within the noisy cellular milieu? Loewer et al. (2010) now use single-cell microscopy to demonstrate that alternative posttranslational modifications allow the tumor suppressor p53 to differentiate between benign breaks in DNA during the cell cycle and deleterious damage caused by mutagens.
The tumor suppressor p53 is one of the key sentinels in the cell for monitoring the integrity of genomic DNA. When deleterious mutations occur, p53 launches a DNA repair pathway and arrests the cell cycle to protect daughter cells from inheriting potentially mutagenic DNA (Vousden and Prives, 2009). Exquisitely sensitive to DNA damage, p53 can respond to even one or two breaks in nuclear DNA, but it apparently ignores harmless breaks that naturally form as DNA is opened during the replication phase of the cell cycle. Thus, a central question has been how p53 maintains its high sensitivity to mutagenic damage while simultaneously overlooking benign breaks during normal cell division. In this issue of Cell, Loewer et al. (2010) use an elegant single-cell microscopy approach to address this fundamental question. They find that the activity of p53 is surprisingly uncoupled from its cellular concentration; instead, its activity depends on the interplay of inhibitory and activating posttranslational modifications.
The classical model for activation of p53 is based primarily on experiments with large cell populations. These studies concluded that the levels of p53 protein in the cell are kept low under normal cell-cycle conditions. DNA damage triggers an initial boost in p53 concentration in the cell by activating the p53 promoter and stabilizing existing p53 protein (Kruse and Gu, 2009). If the DNA damage persists, p53 then can kick off a cell-cycle arrest response in part by stimulating gene expression of specific targets, including p21. Eventually, the activity of p53 is attenuated as it switches on another key target gene, mdm2. The Mdm2 protein represses transcriptional activation of p53 while simultaneously lowering p53 protein levels through ubiquitin-mediated degradation (Vousden and Prives, 2009; Kruse and Gu, 2009).
In their new study, Loewer et al. (2010) now uncover a new twist in the mechanism of p53 activation. By examining individual cells with time-lapse microscopy, they find that the levels of p53 protein actually pulse up and down during the normal cell cycle. Furthermore, the bursts in p53 concentration require kinases, such as the ataxia telangiectasia mutated (ATM) kinase, which phosphorylates and stabilizes p53 (Vousden and Prives, 2009; Kruse and Gu 2009). These oscillations in p53 during normal growth were not detected by earlier studies because they average out over a population of cells. Interestingly, Loewer et al. find that these “normal” pulses, which are probably triggered by transient DNA damage during DNA replication, do not lead to activation of p21 or cell-cycle arrest. In contrast, bursts of p53 triggered by extrinsic mutagens, such as radiation and drugs, do activate p21 and halt cell division. Remarkably, the intensity and duration of these p53 pulses were similar under both conditions. How then does p53 differentiate between benign breaks in DNA and potentially dangerous ones?
Loewer and colleagues find that the critical signal controlling the activity of p53 is an intricate balance of alternative posttranslational modifications of p53. Recent studies have found that, like histone proteins, p53 is the target of myriad posttranslational modifications at numerous lysine (K) residues, primarily at its carboxyl terminus region (Figure 1) (Vousden and Prives, 2009; Kruse and Gu 2009). As with many histone proteins, acetylation activates p53, whereas methylation can either activate (at K372) or repress (at K370, K373, and K382) this transcription factor (Huang and Berger, 2008; Huang et al., 2010). Interestingly, several of these alternative modifications occur on the same or adjacent lysine residues in the carboxyl terminus (Figure 1). In addition, these lysines can also be ubiquitinated to target p53 for degradation.
Figure 1. Posttranslational Modifications Regulate p53 Activity.
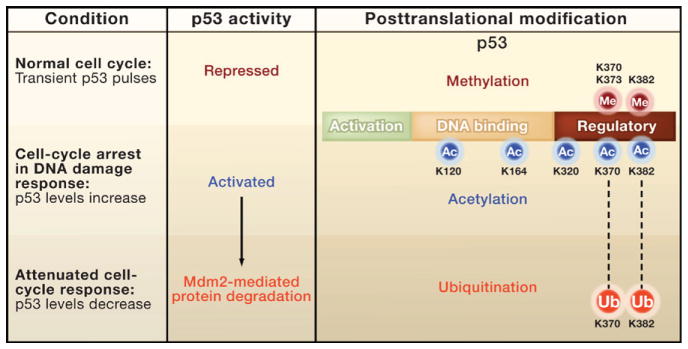
The tumor suppressor p53 is a modular protein that contains (from the amino to the carboxyl terminus) two tandem activation domains (green), a DNA binding domain (tan), and an oligomerization domain surrounded by regulatory domains (red). Levels of p53 increase transiently during the normal cell cycle (Loewer et al., 2010). However, the activity of p53 during these bursts is kept in check by methylation (red hexagons) of lysine residues (K370, K373, and K382) in the carboxyl terminus region. In contrast, dimethylation of K370 and monomethylation of K372 activate p53 (Huang and Berger, 2008). In response to DNA damage, sequential regulatory steps occur. The p53 protein is phosphorylated (not shown) and acetylated at multiple sites (K120, K164, K320, K370, and K382). The level of p53 increases in the cell, triggering factors that repair the DNA and arrest the cell cycle. For cell division to resume, the stress response mediated by p53 needs to be attenuated by deacetylation (not shown) and ubiquitin-mediated degradation of p53.
The importance and significance of these lysine modifications in p53’s carboxyl terminus have been controversial. Several studies with transgenic mice found that mutating a subset of these lysines had only modest effects on the activity of p53 (Toledo and Wahl, 2006). In contrast, a subsequent study in cell culture found that p53 function was dramatically reduced when all acetylated sites were mutated (Tang et al., 2008). However, studies with mice engineered to express this acetylation-deficient form of p53 have not been reported yet.
Adding to the complexity of the story, lysine methylation, which occurs at many of the same residues as acetylation (Figure 1), appears to repress p53 activity (Vousden and Prives, 2009; Huang and Berger, 2008). This is confusing because the context of repression has not been clear; does methylation maintain low basal activity of p53 during the normal cell cycle, or does it attenuate the activity of p53 after a stress response? One potential explanation for the conflicting results of the functional studies is that these lysines may be alternatively acetylated for activation, methylated for repression, and ubiquitinated for degradation. Thus, the opposing actions of these modifications may mask the effects of removing the lysine residues from p53. In other words, substitution of the lysines with other residues leads to the simultaneous loss of activating and repressing modifications and thus possible mutual suppression in vivo.
The single-cell approach used by Loewer and colleagues supports this latter hypothesis. They find that only cells experiencing true DNA mutagenesis possess acetylated p53 (i.e., the activated form of p53) and induce the transcription of p21. The application of deacetylase inhibitors during the normal cell cycle boosted the levels of acetylated p53 and increased expression of p21. Moreover, reducing the expression of a specific methyltransferase (SET8) stimulated the induction of p21 during the normal cell cycle. These results indicate that repressive methylations on p53 keep it in check as it pulses during cell division; when actual DNA damage occurs, acetylation replaces the methylation to trigger p53 transcriptional activity.
Although this new study provides an elegant framework for understanding how the balance between methylation and acetylation of p53 may regulate its activity, many questions emerge from these results. For example, does methylation of lysine residues in the DNA binding domain of p53 (at K120 and K164; Figure 1) also block acetylation and activation of p53 (Vousden and Prives, 2009)? In addition, there is evidence that deacetylases and demethylases also regulate p53 (Kruse and Gu 2009; Huang et al., 2007), and it is important to understand how these different classes of enzymes target p53, especially in terms of their role in cancer and tumorigenesis.
Further, it will be interesting to learn how ubiquitination at these same residues is integrated into the scheme that regulates p53. One reasonable overall scenario is that methylation represses p53 until irreconcilable breaks in the DNA occur outside of the normal cell cycle. Then activation of p53 occurs by a series of steps: acetylation blocks the repressive methylation, leading to an increase in the p53 protein and eventually to cell-cycle arrest (Figure 1). When the cell “wants” to resume growth, ubiquitination of these same lysines may attenuate this loop by stimulating degradation of p53.
Finally, placing the results of this study into the context of p53 research over the last decade also begs the question, “why is regulation of p53 so complex”? Is it because p53 is of profound importance to normal cell physiology, necessitating intricate regulation that involves at least three alternative forms of post-translational modifications on lysine? Or, is p53 simply studied more intensively than other transcription factors, and thus more is known about its regulation? This is a fascinating question, especially given that hundreds of non-histone proteins are acetylated. Moreover, in the coming decade, researchers will probably identify an equal number of proteins that are methylated. Will opposing modifications on lysines be a general theme for regulating these factors, too? Only time and hard work will provide the answer.
References
- Huang J, Berger SL. Curr Opin Genet Dev. 2008;18:152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Huang J, Dorsey J, Chuikov S, Pérez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. J Biol Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Batchelor E, Gaglia G, Lahav G. Cell. 2010 doi: 10.1016/j.cell.2010.05.031. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]