

Abstract
Fully understanding the influence of blood proteins on the assembly structure and dynamics within nanoparticles is difficult because of the complexity of the system and the difficulty in probing the diverse elements and milieus involved. Here we show the use of site-specific labeling with spin probes and fluorophores combined with electron paramagnetic resonance (EPR) spectroscopy and fluorescence resonance energy transfer (FRET) measurements to provide insights into the molecular architecture and dynamics within nanoparticles. These tools are especially useful for determining nanoparticle stability in the context of blood proteins and lipoproteins, and have allowed us to quantitatively analyze the dynamic changes in assembly structure, local stability and cargo diffusion of a class of novel telodendrimer-based micellar nanoparticles. When combined with human plasma and individual plasma components, we find that non-crosslinked nanoparticles immediately lose their original assembly structure and release their payload upon interaction with lipoproteins. In contrast, serum albumins and immunoglobulin gamma have moderate affects on the integrity of the nanoparticles. Disulfide crosslinked nanoparticles show minimal interaction with lipoproteins, and can better retain their assembly structure and payload in vitro and in vivo. We further demonstrate how the enhanced stability and release property of disulfide crosslinked nanoparticles can be reversed in reductive conditions. These findings identify factors are crucial to the performance of nanomedicines and provide design modes to control their interplay with blood factors.
Keywords: Protein-nanoparticle interaction, site-specific labeling, EPR, FRET, structure and dynamics, micellar nanoparticle, lipoprotein particle, drug delivery
Nanoparticles have emerged as a major class of vehicles to deliver conventional anticancer drugs.1–4 Nanoparticle drug delivery systems offer several distinct advantages, such as controlled release and prolonged circulation time, as well as passive and active tumor targeting.1–4 After entering into blood circulation, nanoparticles immediately confront blood proteins and lipoprotein particles. Understanding the interactions of nanoparticles with these factors is therefore crucial to the design of safer and more efficacious nanomedicines.5–7 Recent studies focused on the “nanoparticle-protein coronas” exemplify the extrinsic surface property that nanoparticles gained upon interaction with blood factors.5–11 However, blood proteins may also strongly affect the internal components of nanoparticles, promoting the early disintegration or aggregation of the nanoparticles and premature drug release before reaching the tumor target.7, 12–14
Despite extensive reports on nanoparticle syntheses for drug delivery, few publications address the multiple dynamic processes occurring inside nanoparticles in the presence of blood proteins and lipoproteins. This is likely due to the limitations of current analytical techniques. Dynamic light scattering (DLS) has been used to monitor the dynamic changes in particle size of nanoparticle in the presence of blood proteins.4, 12 However, this technique is limited by its sensitivity and interference by lipoprotein particles; and is generally not suitable for analyzing heterogeneous biological samples. Fluorescence based assays, such as fluorescence resonance energy transfer (FRET) and fluorescence-quenching,12, 15–18 have been developed to study the stability of nanoparticles. FRET is a distance dependent physical process by which excitation energy is absorbed by a molecular fluorophore (the donor) and then transferred to a nearby fluorophore (the acceptor). It is a highly sensitive technique for investigating a variety of biological phenomena that produce changes in molecular proximity.19, 20 Despite these efforts, a clear picture in molecular scale of what happens within nanoparticles during interaction with blood proteins is still absent, which severely hampers the rational design of nanoparticles.
Electron paramagnetic resonance (EPR) spectroscopy is a powerful tool for investigating dynamic phenomena and the microenvironment of colloidal systems.21 The EPR spectrum of the spin-labels provides discrete information on the local order, by reflecting the dynamics occurring on the nanosecond timescale. Since nanosecond reorientations are not assessable by nuclear magnetic resonance (NMR) due to short relaxation times of the samples,22 EPR provides a unique capability to probe the order within nanoparticles in a highly sensitive manner. Spatial information is also available with EPR, as the spectrum is also affected by dipolar coupling between nearby spin probes (< 2 nm, with longer distances possible using pulsed methods).21–24
We have reported a novel class of micellar nanoparticles assembled by polyethylene glycol (PEG)-block- dendritic oligomer of cholic acids (CA) copolymers (called telodendrimers). This nanocarrier system is chemically well-defined, size tuneable, easy to be functionalized in a site-specific manner, and capable to package a diverse array of drugs.16, 25–29 Furthermore, we have demonstrated that paclitaxel or doxorubicin formulated in this nanocarrier system are therapeutically more effective than their corresponding free drugs.16, 25–29 Very recently, we reported the development of more stable nanoparticles that are crosslinked via disulfide bonds to minimize the premature release of drugs while in circulation.28 To better understand the assembly, structure and dynamics of these crosslinked and non-crosslinked nanoparticles, we have designed a class of site-specific nitroxide spin label reporter systems and a series of position-specific FRET-based fluorescence reporter systems. The spin label reporter systems allow us to use EPR spectroscopy to quantitatively analyze the motions of the hydrophilic shell and hydrophobic core of nanoparticles in human plasma and individual plasma components. We also demonstrate the promise of time-resolved FRET approach for in vitro and in vivo probing the dynamic changes in local proximities within nanoparticles.
RESULTS
Site-specific spin label reporter system
To evaluate the potential of site-specific spin labels to report on the dynamic structure and local mobility of nanoparticles assembled from amphiphilic telodendrimers, we attached 2,2,6,6-tetramethylpiperidinyloxy (TEMPO), a nitroxide spin label to two specific sites on the reported PEG5k-CA8 telodendrimers. As shown in Scheme 1 and Scheme S1, the spin label of TEMPO1-PEG5k-CA8 is localized at the distal end of the hydrophilic PEG chain while that of TEMPO2-PEG5k-CA8 is attached to a lysine proximal to the oligocholic acid cluster. These telodendrimers can self-assemble in aqueous solution to form spin labelled non-crosslinked micellar nanoparticles with a typical core-shell structure, named as S-NCMN1 and S-NCMN2, respectively (Scheme 1). The spin labels of S-NCMN1 are located on the surface of the nanoparticles, reflecting the movements of PEG chain while that of S-NCMN2 are located closely at core-shell interface of the nanoparticles, indicating the dynamic changes of the oligocholic acid core of the nanoparticles. We also introduced TEMPO to the lysine proximal to the oligocholic acids of PEG5k-Cys4-CA8, a cysteine containing telodendrimers28 to prepare spin labelled disulfide crosslinked nanoparticles (S-DCMN2) (Scheme 1 and Scheme S1). The structure of the spin labeled telodendrimers was confirmed by 1H-NMR (Fig. S1a). The properties of the resulting nanoparticeles were characterized by DLS particle sizer, transmission electron microscopy (TEM) and EPR spectroscopy. The three types of spin labeled nanoparticles are all around 21 nm in diameter in phosphate buffered saline (PBS) with narrow size distribution (Fig. S2), which are similar to the parent nanoparticles without spin labels,26, 28 indicating the spin labels have negligible influence on the particle size of the nanoparticles.
Scheme 1.

Schematic illustration of the spin labelled non-crosslinked nanoparticles by the self-assembly of (a) the PEG5k-CA8 telodendrimer with a spin label attached to the end of the hydrophilic PEG chain and (b) the PEG5k-CA8 telodendrimer with a spin label attached to the lysine side chain at the junction between the linear PEG chain and the dendritic core. (c) Schematic illustration of the spin labelled disulfide crosslinked nanoparticles (S-DCMN2) by the self-assembly of the cysteine containing PEG5k-Cys4-CA8 telodendrimer with a spin label attached to the lysine side chain at the junction between the linear PEG chain and the dendritic core followed by oxidation to form disulfide crosslinks.
The EPR spectra of nanoparticles in PBS containing spin labels attached at two different locations of the non-cross linked nanoparticle are shown in Fig. 1. Consistent with our prediction for the ultra-structure of the nanoparticle assembly in solution, the EPR spectrum of S-NCMN1 (Fig. 1a, black line) is isotropic triplet revealing a typical fast motion of the nitroxide spin labels. The EPR spectrum of S-NCMN2 is considerably broader than the spectrum of S-NCMN1 under identical instrument condition and sample concentration (Fig. 1b, black line). This indicates the TEMPO molecules in S-NCMN2 are limited in their ability to reorient in time, consistent with their localization near a clustered hydrophobic core. The simple line shapes displayed in the spectra are indicative of single component. Thus, the correlation time for the spin label can be estimated from the line height ratios in a straightforward manner (see Equation 1 in Methods).30 The calculated rotational correlation times (τ) for S-NCMN1 and S-NCMN2 are 0.9×10−10 s and 4.4×10−10 s, respectively. The shorter τ reflects the faster motion of the label located in a more disordered environment, such as the terminal PEG position of S-NCMN1. Fig. 1 also shows the spectrum of spin labels located at core-shell interface of a nanoparticle comprised of cysteine containing telodendrimers28 that have been oxidized to form disulfide cross-linked nanoparticles (S-DCMN2) (Fig. 1c, black line). While the spectrum of cross-linked S-DCMN2 is similar to that of S-NCMN2, it also contains features of exchange between spins in very close proximity (<0.5 nm). Thus in addition to a more constrained environment, the spin-spin interaction indicates a tighter packing of the spin labels within the nanoparticles. Therefore, the TEMPO moieties in S-NCMN2 and S-DCMN2 display broader EPR spectra than that of S-NCMN1 due to their low mobility and spin-spin interaction. These results are supported by NMR characterizations in deuterium oxide (D2O) that also indicate the motions of the spin probes of S-NCMN2 and S-DCMN2 are in more restricted environments than that of S-NCMN1 (Fig. S1b).
Figure 1.

Representative EPR spectra of the S-NCMN1 (a), S-NCMN2 (b) and S-DCMN2 (c) in the presence of 1x PBS and 2.5 mg/mL SDS for 1 min, respectively. TEM images of S-NCMN1 (d), S-NCMN2 (e) and S-DCMN2 (f) in the presence of 2.5 mg/mL SDS for 1 min (scale bar: 100 nm).
We investigated the EPR spectra of the nanoparticles in the presence of sodium dodecyl sulfate (SDS), a strong ionic detergent, which has been reported to be able to completely break the down assembly structure of micellar nanoparticles.28, 31 As confirmed by TEM and DLS, the non-crosslinked nanoparticles (S-NCMN1 and S-NCMN2) are completely lost in SDS (Fig. 1d, 1e and Fig. S2). As the spin labels at the end of PEG are able to move freely regardless of the integrity of the nanoparticles, SDS disruption of particles containing S-NCMN1 results in only a nominal change in the EPR spectrum (Fig. 1a). In contrast, the EPR spectrum of S-NCMN2 becomes much sharper in SDS compared to PBS (Fig. 1b). The magnitude of change following SDS treatment reveals that much of the broadening in the S-NCMN2 is due to magnetic coupling of spins in close proximity. Upon disassembly of the nanoparticle, there is both increased motional freedom along with a greater spatial distribution of the spin probes, resulting in the dramatic spectral change observed. In contrast, the spectrum (Fig. 1c) of cross-linked S-DCMN2 particles28 are resistant to detergent disruption, as no significant change is observed following SDS treatment (Fig. 1f and Fig. S2). EPR signal changes can also be quantitatively analyzed by using the percentage of intensity increase (I1−I0)/I0, where I0 is the intensity of the highest peak in the EPR spectrum of the sample in PBS while I1 is the intensity of the corresponding peak in a different media under identical instrument condition and sample concentration. The (I1−I0)/I0 values in SDS for S-NCMN1, S-NCMN2 and S-DCMN2 in SDS are calculated to be 7%, 202%, and 48%, respectively.
EPR study of the nanoparticle-protein interactions
These nanoparticles are very stable in PBS,16, 25–29 therefore, there are minimal changes in the EPR spectra over time in PBS (Fig. S3a and S3b). The lipid assemblies normally present in human plasma may interact with nanoparticles, resulting in the disruption of their structural stability and cargo release. Thus it is critical that we have a method for real-time probing the dynamic structure of nanoparticles in human plasma. To this end, we have used EPR to quantitatively investigate the dynamic structural changes in the hydrophilic shell and hydrophobic core of a nanoparticle in human plasma and in the presence of individual plasma components. After incubation in human plasma, S-NCMN2 immediately loses its assembly order as reflected in the sharp spectrum, resulting in a signal of increased intensity (Fig. S4b) compared to that in PBS. The rotational correlation time decreases to 2.1×10−10 s (Fig. 2d), indicating the increased mobility of the spin labels. The EPR spectra and the (I1−I0)/I0 value of S-NCMN2 are similar to those in SDS (Fig. S4b and Fig. 2e) at initial time point, which is correlated with the disassembly of the oligocholic acid core. The subsequent changes in EPR signal and rotational correlation time are not obvious (Fig. S4b and Fig. 2d, 2e). In contrast, the EPR signal of S-NCMN1 increases slightly over time, indicating there are limited changes in the order of PEG chain (Fig. S4a and Fig. 2d, 2e). Such measurements will be highly valuable in assessing the local stability within different modules of the nanoparticle as well as determining the effectiveness of stabilization strategies and in nascent, modified and loaded nanoparticles.
Figure 2.

Representative time-resolved EPR spectra S-NCMN1 (a), S-NCMN2 (b) and S-DCMN2 (c) in the presence of CM (1.0 mg/mL), HDL (2.0 mg/mL), LDL (2.0 mg/mL), PBS and VLDL (1.0 mg/mL). The percentage changes in rotational correlation time ((τ1 − τ0)/τ0) of the three types of spin labelled nanoparticles in human plasma at different time points (d), where τ0 is the rotational correlation time of the sample in PBS while τ1 is the rotational correlation time of the corresponding peak in a different media under identical instruments conditions, values reported are the mean diameter ± SD for duplicate samples; The percentage of EPR intensity changes ((I1−I0)/I0) of the three types of spin labelled nanoparticles in human plasma at different time points (e), where I0 is the intensity of the highest peak in the EPR spectrum of the sample in PBS while I1 is the intensity of the corresponding peak in a different media under identical instruments conditions, values reported are the mean diameter ± SD for duplicate samples; The ((τ1 − τ0)/τ0) value of the three types of spin labelled nanoparticles in PBS and HSA at different time points (f).
Next we tested the ability of the spin label reporter system to investigate how the individual plasma components influence the dynamic structure of nanoparticles. The concentrations of the plasma components (e.g. albumins and lipoprotein particles) used in the following studies are close to their physiological level.32 We first selected serum albumins, the most abundant proteins in plasma (about 55% of total plasma proteins). We found the correlation time and (I1−I0)/I0 values of S-NCMN1 remained unchanged over time in the presence of human serum albumin (HSA) (Fig. 2f and Fig. S4d, S6), indicating these nanoparticles do not absorb serum albumins on the PEG surface. In contrast, there is a decrease in the EPR signals and (I1−I0)/I0 values of S-NCMN2 over time (Fig. S4e, S6) along with a significant increase of τ (Fig. 2f) in the presence of HSA. This is likely due to the insertion of small albumin particles (around 5 nm) into the nanoparticles (21 nm) restricting the motions of the spin labels located at core-shell interface. It should be mentioned that albumin does not cause disassembly of the nanoparticles over time. The EPR spectra of S-NCMN2 become immediately sharper, and thereby increasing dramatically in intensity upon incubation with three major lipoprotein particles: chylomicrons (CM, >100 nm), low density lipoprotein (LDL, 18–28 nm), and very low density lipoprotein (VLDL, 30–80 nm) (Fig. 2b, and Fig. S5c, S5e). Lipoprotein particles are amphiphilic assemblies that contain both apolipoproteins and lipids for fats delivery. Lipoprotein particles are able to absorb contents (assembly units and payloads) from nanoparticles.7, 12–14 Due to the similar amphiphilic nature of lipoprotein particles and micellar nanoparticles, they are also likely to exchange contents with each other.7, 12–14 These interactions will cause the disassembly of the nanoparticles. The EPR data also reveal that the interaction of nanoparticles with lipoprotein particles is concentration dependent. Higher concentrations of lipoprotein particles have a greater effect on the assembly structure of the micellar nanoparticles (Fig. S7). We also find that high density lipoprotein (HDL), a type of densest lipoprotein with smaller size (5–15 nm),7 only has moderate effect on the assembly structure of the nanoparticles (Fig. 2b, and Fig. S5c, S5e). This is probably due to the highly packed assembly structure of HDL, limiting the absorption of contents from nanoparticles.
Further, we tested the performance of the spin label reporter system to investigate whether the nanoparticles with intra-micellar disulfide crosslink could better retain the assembly structure in the presence of blood proteins. The spin labels of the S-DCMN2 are located at the core-shell interface of the nanoparticles which is at the same location as S-NCMN2. As shown in Fig. 2c, 2d, 2e and Fig. S4c, S5d, S5f, the EPR spectra and the correlation time of S-DCMN2 samples in human plasma and lipoproteins (e.g. LDL, HDL, VLDL and CM) only change slightly over this same period as S-NCMN2. The results indicate that the disulfide crosslinked nanoparticles can better retain their assembly structure in the presence of blood proteins. Similar to S-NCMN2, the EPR signal of S-DCMN2 also decreases slightly along with an increased τ in the presence of HSA and BSA, indicating serum albumins are able to cross the PEG shell and insert into S-DCMN2 affecting the movements of spin labels at core-shell interface (Fig. 2f, S4f, S5b and S6).
Position-specific FRET reporter systems
We further constructed three position-specific FRET-based fluorescence reporter systems to monitor the dynamic changes in local proximity within a nanoparticle upon interaction with blood proteins. The FRET pair of the first system (Fig. 3a) comprised of a green carbocyanine dye DiO (donor) physically encapsulated in the core of nanoparticles as the hydrophobic drug surrogate to track the payloads and a red-orange dye rhodamine B (acceptor) covalently conjugated to the telodendrimer units to track the nanocarriers. The FRET pair of the second system (Fig. 3b) comprised of DiO (donor) and a red-orange carbocyanine dye DiI (acceptor) both physically encapsulated in the core of nanoparticles as the hydrophobic drug surrogates. The FRET pair of the third system (Fig. 3c) comprised of a green dye FITC (donor) and rhodamine B (acceptor) both covalently conjugated to the telodendrimer units of the nanoparticles. The FRET signal of the first, second and third FRET-based reporter system reflects the proximity between the telodendrimer unit and drug surrogate, different drug surrogates and different telodendrimer units within a nanoparticle, respectively. We also constructed the first FRET reporter system (DiO and rhodamine B pair) on the disulfide crosslinked nanoparticles for comparison.
Figure 3.
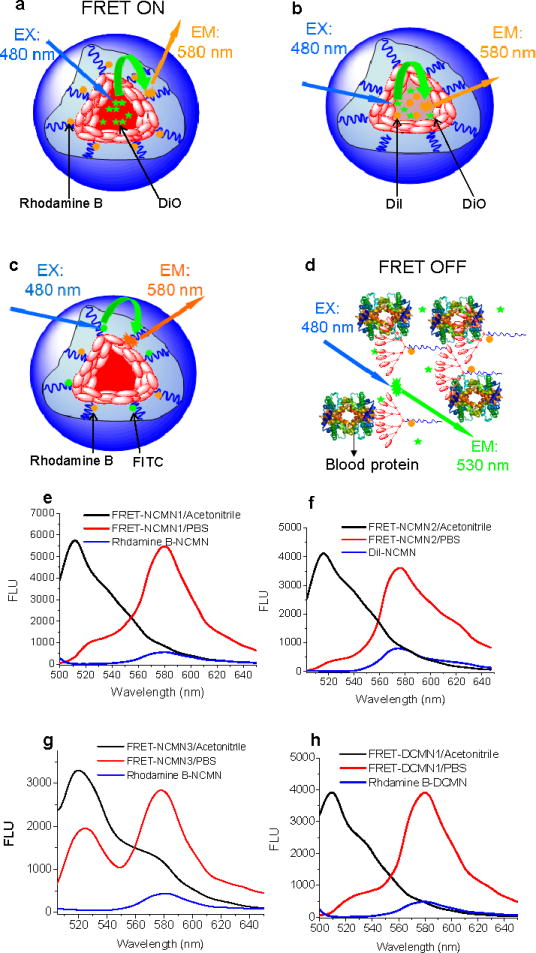
Schematic illustration of non-crosslinked FRET-based nanoparticles FRET-NCMN1 (DiO and rhodamine B pair) (a), FRET-NCMN2 (DiO and DiI pair) (b), FRET-NCMN3 (FITC and rhodamine B pair) in PBS (c) and FRET-NCMN1 (DiO and rhodamine B pair) in human plasma (d); representative fluorescence spectra of FRET-NCMN1 (DiO loading: 2.5%, rhodamine B conjugated PEG5k-CA8: 5.0 mg) in PBS (red line) and in acetonitrile (black line), NCMN with rhodamine B alone (rhodamine B conjugated PEG5k-CA8: 5.0 mg) (blue line) with 480 nm excitation (e); representative fluorescence spectra of FRET-NCMN2 (DiO loading: 2.5%, DiI loading: 2.5%) in PBS (red line) and in acetonitrile (black line), NCMN with DiI alone (DiI loading: 2.5%) (blue line) with 480 nm excitation (f); representative fluorescence spectra of FRET-NCMN3 (FITC conjugated PEG5k-CA8: 5.0 mg, rhodamine B conjugated PEG5k-CA8: 5.0 mg) in PBS (red line) and in acetonitrile (black line), NCMN with rhodamine B alone (rhodamine B conjugated PEG5k-CA8: 5.0 mg) with 480 nm excitation (g); representative fluorescence spectra of FRET-DCMN1 (DiO loading: 2.5%, rhodamine B conjugated PEG5k-Cys4-CA8: 5.0 mg) in PBS (red line) and in acetonitrile (black line), DCMN with rhodamine B alone (rhodamine B conjugated PEG5k-Cys4-CA8: 5.0 mg) (blue line) with 480 nm excitation (h).
The particle sizes of these FRET-based nanoparticles in PBS are in the range of 20–25 nm (Fig. S8), which are similar to their parent nanoparticles. When these FRET based nanoparticles retain their assembly structure in PBS, the drug surrogates and telodendrimer units are packed closely within nanoparticles about 20–25 nm in size. The distances between all the donor and acceptor pairs are within the FRET range allowing efficient energy transfer from donor to the acceptor upon excitation of donor at around 480 nm (Fig. 3). The apparent FRET efficiency reflects the distance between the FRET pair and is calculated as Eapp = IA/(IA + ID), where IA and ID represent acceptor and donor intensities, respectively.33 The apparent FRET efficiency for the FRET based non-crosslinked nanoparticles FRET-NCMN1 (DiO and rhodamine B pair), FRET-NCMN2 (DiO and DiI pair), FRET-NCMN3 (FITC and rhodamine B pair) and disulfide crosslinked nanoparticles, FRET-DCMN1 (DiO and rhodamine B pair) diluted 20 times with PBS were measured to be 79.1%, 81.0%, 63.7% and 84.2%, respectively. When the nanoparticles were diluted 20 times via acetonitrile, a dramatic increase of donor emission was observed along with a significant reduction of acceptor signal (Fig. 3e, 3f, 3g, 3h). The ratios of FRET-NCMN1, FRET-NCMN2, FRET-NCMN3 and FRET-DCMN1 decrease to 13.3%, 15.8%, 25.9% and 10.2%, respectively. We can further estimate the distance between the donor and acceptor based on the reported equation Eapp = (1 + (R/R0)6)−1, where R is the inter-dye distance, and R0 is the Förster radius at which E = 0.5.33 For example, the estimated distance between the FRET pair (FITC and rhodamine B) is 4.5 nm for FRET-NCMN3 in PBS and 7.2 nm for FRET-NCMN3 in acetonitrile, respectively, based on an estimated R0 of 5.0 nm.34 The loss of FRET signal is due to the solvation of telodendrimer units and payload when these nanoparticles are dissolved and the distance between the donor and acceptor pairs cannot be retained within the FRET range. There is negligible FRET signal of the nanoparticles labeled with acceptors alone using the donor excitation at around 480 nm in comparison with the corresponding FRET nanoparticles (Fig. 3e, 3f, 3g, 3h). Therefore, we are able to probe local proximity changes within nanoparticles in real time by monitoring the dynamic change of FRET efficiency.17, 28
In vitro and in vivo FRET studies
We tested the ability of these FRET reporter systems to report on the structure and dynamics of the micellar nanoparticles in vitro and in vivo. These nanoparticles are able to retain their assembly structure and have a slow cargo diffusion rate in PBS.26, 28 Thus, there is little change in FRET signal over time in PBS (Fig 4c). However, a dramatic decrease in FRET signal for FRET-NCMN1 and FRET-NCMN3 occurs within 30 min in the presence of human plasma (Fig. 4a, 4c). The FRET ratio decreases to 33.8% and 29.5% at 1.5 h, respectively. As indicated in the above EPR studies, human plasma can disrupt the non-crosslinked nanoparticles, resulting in the complete release of payload. Thus, there is a significant increase of the proximities between the telodendrimer unit and drug surrogate as well as that between different telodendrimer units. However, the FRET signal of FRET-NCMN2 changes slowly over 24 hrs in the present of human plasma (Fig 4c). The physically loaded DiO and DiI FRET pairs are the same class of hydrophobic carbocyanine tracers for membrane labeling. DiO and DiI may bind to the same protein or lipid membrane after the release from nanoparticles, which may still give FRET signal.12 To confirm this assumption, an equal molar ratio of DiI and DiO were dissolved directly in human plasma. The calculated apparent FRET efficiency is approximately 65.3%, which is similar to that of FRET-NCMN2 in human plasma at 24 h (67.4%). The FRET signal of FRET-NCMN1 was then monitored in the presence of single plasma components. We observed that all the four major groups of lipoprotein particles (CM, HDL, LDL and VLDL) cause a rapid decrease of FRET efficiency of FRET-NCMN1 while serum albumin (HSA) and immunoglobulin gamma (IgG, the most abundant antibody in blood) have minimal affect on the FRET efficiency (Fig 4e).
Figure 4.

Representative fluorescence emission spectra of non-crosslinked FRET based nanoparticles (FRET-NCMN1, DiO and rhodamine B pair) (a) and disulfide crosslinked FRET nanoparticles (FRET-DCMN1, DiO and rhodamine B pair) (b) in the presence of human plasma, respectively. The final concentrations of the nanoparticles were 0.1 mg/mL; the time-resolved apparent FRET efficiency (Eapp) change of FRET-NCMN1 (DiO and rhodamine B pair), FRET-NCMN2 (DiO and DiI pair), FRET-NCMN3 (FITC and rhodamine B pair), FRET-DCMN1 (DiO and rhodamine B pair) in human plasma and PBS (c), the apparent FRET efficiency is calculated as Eapp = IA/(IA + ID), where IA and ID represent acceptor and donor intensities, respectively; the time-resolved Eapp change of FRET-DCMN1 (DiO and rhodamine B pair) in the presence of human plasma and reducing agents (glutathione (GSH) and N-acetylcysteine (NAC)) (d); the time-resolved Eapp change of FRET-NCMN1 (DiO and rhodamine B pair) (e) and FRET-DCMN1 (DiO and rhodamine B pair) (f) in the presence of HSA (50 mg/mL), LDL (2.0 mg/mL), HDL (2.0 mg/mL), VLDL (1.0 mg/mL), CM (1.0 mg/mL) and IgG (10 mg/mL). Excitation: 480 nm. Values reported are the mean diameter ± SD for triplicate samples.
We then applied the FRET-based reporter system to study the structure and dynamics, as well as the redox response of disulfide crosslinked nanoparticles in biological-relevant media. As shown in Fig. 4b and 4c, there is minimal change in the FRET signal for FRET-DCMN1 up to 30 min in the presence of human plasma. The subsequent FRET efficiency decrease of FRET-DCMN1 is more slowly than that of FRET-NCMN1 using the same FRET pair. We also observed that the FRET ratio of FRET-DCMN1 decreases more slowly in the presence of HDL, LDL and VLDL than that of FRET-NCMN1 in the same media (Fig. 4h). Consistent with the observations in EPR studies, the disulfide crosslinked nanoparticles can better retain their assembly structure and slow down the payload release in the presence of blood proteins, therefore minimizing the drop of FRET signal. Additionally, we also used the FRET based reporter system to study the response of FRET-DCMN1 to reducing agents by detecting the FRET signal change over time in human plasma. In the presence of human plasma and reducing agents (glutathione (GSH) and N-acetylcysteine (NAC)), the FRET signal of FRET-DCMN1 drops faster over time compared to that without reducing agents (Fig 4d).
Furthermore, we presented data on the ability of the FRET reporter system to probe the dynamic assembly structure and cargo release of the nanoparticles in vivo. FRET-NCMN1 and FRET-DCMN1 were injected into nude mice via tail vein and the apparent FRET efficiency was monitored at pre-determined time points. The FRET signal of blood background is very low. The FRET ratio of FRET-NCMN1 drops immediately to 45.3% post-injection and then decreases to 20.5% after 18 min (Fig. 5a). FRET-DCMN1 exhibits a significantly slower decrease of FRET ratio compared to FRET-NCMN1 at the same nanoparticle concentration (Fig. 5a and Fig. S10), indicating the disulfide crosslinking greatly enhances the stability of the nanoparticles in vivo, by preventing the dissociation of nanoparticles and the premature payload release. In the blood elimination study, we demonstrated that rhodamine B labeled disulfide crosslinked nanoparticles have significantly longer circulation times than that of non-crosslinked nanoparticles (Fig. 5b). This result is consistent with the observations in the in vivo FRET studies.
Figure 5.

(a) The time-resolved apparent FRET efficiency (Eapp) change in blood of nude mice (n=3) over time after intravenous injection of 100 μL FRET-NCMN1 (DiO and rhodamine B pair) and FRET-DCMN1 (DiO and rhodamine B pair) (2.0 mg/mL). Excitation: 480 nm. (b) The fluorescence signal changes of rhodamine B conjugated NCMN and DCMN in the blood collected at different time points after intravenous injection in the nude mice (n=3). Excitation: 540 nm. Values reported are the mean diameter ± SD for triplicate samples.
DISCUSSION
By targeting spin labels within the specific sites within the nanoparticles, we have used EPR to define the dynamics, polarity and stability of each module within a novel micellar telodendrimer assembly. Furthermore, the high sensitivity of EPR (1–20 NM spin concentration) and low sample volume requirements (~4 NL) make this approach highly convenient for investigation with biological components.22 The site-specific nitroxide spin labeling technique from this study is generally applicable to many other nanoparticle drug delivery systems. The spin labels can be conveniently and specifically conjugated to many nanoparticles with amino groups to probe multiple locations within their structure.
One limitation of the commonly used FRET techniques is the low signal-to-noise ratio.33 In the three FRET pairs used in this study, there are decent overlaps between emission spectra of donors and excitation spectra of acceptors while the overlaps between the excitation spectra of donors and emission spectra acceptors are negligible, which significantly increases the signal-to-noise ratio. Furthermore, the acceptors and donors are well-positioned within nanoparticles around 20 nm in size, allowing efficient energy transfer from the donors to the acceptors. We were able to conveniently carry out our time-resolved FRET assay in 96-well plates, indicating it is a high-throughput method. The FRET-based fluorescence labeling technique described in this study can also be easily applied to many other nanoparticle drug delivery systems.
We demonstrated the ability of using the combination of site-specific labeling techniques (spin labeling and FRET-based fluorescence label) and two highly sensitive, quantitative and time-resolved analytical techniques (EPR and FRET) to probe the multiple dynamic processes occurring in nanoparticles during interactions with blood proteins and lipoprotein particles. As reported extensively, PEG is able to form a palisade avoiding protein adsorption of nanoparticles and subsequent nonspecific uptake by the reticuloendothelial system (RES) after intravenous injection.5, 27, 35 However, our data clearly demonstrated that lipoprotein particles could interact with the non-crosslinked nanoparticles comprised of a PEG shell and disrupt their assembly structure rapidly. Our results also demonstrated that disulfide crosslinked nanoparticles can better retain their assembly structure and payload in vitro and in vivo and are ready to disintegrate and release the payload under reductive conditions. We have previously demonstrated that paclitaxel loaded disulfide crosslinked nanoparticles have longer blood circulation time and are more efficacious than the non-crosslinked and free drug formulations in xenograft mice tumor models.28 The anti-tumor effect of the disulfide crosslinked formulation can be further enhanced by triggering the release of drug on-demand by the administration of the FDA approved reducing agent, N-acetyl cysteine, after the nanoparticles have reached the tumor site.28 This indicates that the EPR and FRET data obtained in this study are directly relevant to the treatment outcomes of the nanoparticles.
CONCLUSION
In summary, we have demonstrated the advantages of using EPR and FRET combined with the site-specific labelling technique to reveal structural and dynamic changes within nanoparticles during interaction with blood proteins and in vivo. Blood proteins and lipoproteins may affect the stability of nanoparticles and could disrupt their assembly structure rapidly. These interactions highly depend on the type and concentration of blood proteins. Our findings also revealed that one can minimize the interactions of nanoparticles with blood proteins via introducing disulfide crosslinkages into the nanoparticles and activate these interactions by using reducing agents to cleave the disulfide bonds subsequently. The incorporation of site-directed probes in the nanoparticle design can facilitate the characterization of these micellar-based drug delivery systems in blood, plasma and body fluids. This will ultimately result in better design nanomedicines with better pharmacodynamic and pharmacokinetic properties, and therefore therapeutically more efficacious.
EXPERIMENTAL SECTION
EPR studies
The synthesis of spin labelled telodendrimers and the resulting micellar nanoparticles were described in Supplementary Information (Supplementary Methods). The spin labelled nanoparticle solution was mixed with 20 times of PBS, human plasma, HSA, BSA, LDL, HDL, VLDL and CM solutions. The final concentrations of the nanoparticle were kept at 1.0 mg/mL and the blood proteins were kept close to their physiological concentrations ranges respectively (HSA: 50 mg/mL, BSA: 50 mg/mL, LDL: 2.0 mg/mL, HDL: 2.0 mg/mL, VLDL: 1.0 mg/mL, CM: 1.0 mg/mL)32. Approximately 4 microliters of the solution mixture was loaded into a micro capillary tube with immediate EPR measurement on a JEOL FA-100 X-band EPR spectrometer fitted with a loop-gap resonator. A free TEMPO solution in the same buffer was used as a control. Standard measurement parameters are: scan time= 2 min, power= 4 mW, scan width=100 G, and modulation width optimized to the line width of the spectrum (on the order of 0.5 to 2 G). To quantify the magnetic dipolar interaction between nearby (<2 nm) spins, deconvolution of Peak broadening will be performed as described previously.21 After each measurement, the sample was taken out and incubated at physiological body temperature (37 °C). The rotational correlation time, τ(s), which is the time taken for the axis of the nitroxyl group to rotate one radian, is highly sensitive to the mobility of the TEMPO radicals, and is calculated using the following equation:
| (Equation 1) |
Here, k is a constant determined from the principal values of A and the g factor; the value of 6.6 × 10−10/Gauss is used. As shown in Fig. S11, ΔH0 is the line width (in gauss) of the zero transition. I0 and I−1 are the peak intensity in the EPR spectra as marked in Fig. S11.
In vitro FRET studies
1) PEG5k-CA8 (15 mg), rhodamine B conjugated PEG5k-CA8 (5 mg) and DiO (0.5 mg); 2) PEG5k-CA8 (20 mg), DiO (0.5 mg)) and DiI (0.5 mg), 3) PEG5k-CA8 (10 mg), FITC conjugated PEG5k-CA8 (5 mg) and rhodamine B conjugated PEG5k-CA8 (5 mg) were used to prepare the three types of non-crosslinked FRET micellar nanoparticles (FRET-NCMN) as described in Supplementary Information (Supplementary Methods, Section 1.4). PEG5k-Cys4-CA8 (15 mg), rhodamine B conjugated PEG5k-Cys4-CA8 (5 mg), and DiO (0.5 mg) were used to prepare crosslinked FRET micellar nanoparticles (FRET-DCMN1). Micellar nanoparticles with DiO, rhodamine B, DiI or FITC alone at the same dye contents were also prepared for comparison. The particle size of these micellar nanoparticles was measured by DLS. The absorbance and fluorescence spectra of these micelles diluted by PBS were characterized by fluorescence spectrometry (SpectraMax M2, Molecular Devices. 5 μL of the FRET based nanoparticle solutions (20 mg/mL) were diluted 20 times by PBS or mixed with 95 μL whole human plasma, HSA (50 mg/mL), BSA (50 mg/mL), HDL (2.0 mg/mL), LDL (2.0 mg/mL), VLDL (1.0 mg/mL), chylomicrons (1.0 mg/mL) and IgG (10 mg/mL)17 solutions and incubated in an incubator at 37 °C with a stirring speed of 100 rpm. The final concentration of the total micellar nanoparticles was kept at 0.1 mg/mL. The fluorescence spectra of these nanoparticles were measured at pre-determined time points. In some experiments, reducing agents (glutathione (GSH, 20 mM) and N-acetylcysteine (NAC, 20 mM)) were added into the nanoparticle solutions to study the stimli-response of the disulfide crosslinked nanoparticles to reducing agents.
In vivo FRET studies
100 μL of FRET micellar nanoparticle solution (2.0 mg/mL) was injected into nude mice via tail vein to investigate their in vivo stability by monitoring FRET efficiency. 50 μL blood was collected at different time points post-injection to measure the fluorescence intensity.28
Supplementary Material
Acknowledgments
The authors thank Dr. Lorenzo Berti for his helpful suggestions, Drs. Nell Suby and Ivy Kekessie for their editorial help, andfinancial support from NIH/NCI R01CA115483 (to K.S.L.), NIH/NIBIB R01EB012569 (to K.S.L.), Prostate Cancer Foundation Creative Award (to K.S.L.), NIH/NCI R01CA140449 (to J.L.), and DoD PCRP Postdoctoral Training Award (W81XWH-12-1-0087 to Y.L.) for financial support.
Footnotes
Conflict of Interest: K.S. Lam is the founding scientist of LamnoTherapeutics which plan to develop the nanotherapeutic described in the manuscript. J. Luo and K.S. Lam are the inventors of pending patent on telodendrimers.
Supporting Information Available: Supplementary methods, synthesis and characterizations of nanoparticles, stability of nanoparticles and additional EPR and FRET data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, et al. Accumulation of Sub-100 nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 2.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable Long-Circulating Polymeric Nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Lee H, Allen C. Formulation of Drugs in Block Copolymer Micelles: Drug Loading and Release. Curr Pharma Des. 2006;12:4685–4701. doi: 10.2174/138161206779026263. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Pan S, Zhang W, Du Z. Novel Thermo-sensitive Core-shell Nanoparticles for Targeted Paclitaxel Delivery. Nanotechnology. 2009;20:065104. doi: 10.1088/0957-4484/20/6/065104. [DOI] [PubMed] [Google Scholar]
- 5.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding Biophysicochemical Interactions at the Nano-bio Interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 6.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Protein-nanoparticle Interactions: Opportunities and Challenges. Chem Rev. 2011;111:5610–5637. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 7.Cedervall T, Lynch I, Foy M, Berggard T, Donnelly SC, Cagney G, Linse S, Dawson KA. Detailed Identification of Plasma Proteins Adsorbed on Copolymer Nanoparticles. Angew Chem Int Ed. 2007;46:5754–5756. doi: 10.1002/anie.200700465. [DOI] [PubMed] [Google Scholar]
- 8.Casals E, Pfaller T, Duschl A, Oostingh GJ, Puntes V. Time Evolution of the Nanoparticle Protein Corona. ACS Nano. 2010;4:3623–3632. doi: 10.1021/nn901372t. [DOI] [PubMed] [Google Scholar]
- 9.Lundqvist M, Stigler J, Cedervall T, Berggard T, Flanagan MB, Lynch I, Elia G, Dawson K. The Evolution of the Protein Corona around Nanoparticles: A Test Study. ACS Nano. 2011;5:7503–7509. doi: 10.1021/nn202458g. [DOI] [PubMed] [Google Scholar]
- 10.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, Dawson KA, Linse S. Understanding the Nanoparticle-protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. P Natl Acad Sci USA. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. P Natl Acad Sci USA. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Owen SC, Shoichet MS. Stability of Self-Assembled Polymeric Micelles in Serum. Macromolecules. 2011;44:6002–6008. doi: 10.1021/ma200675w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the Barriers in Micellar Drug Delivery: Loading Efficiency, in vivo Stability, and Micelle-cell Interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 14.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv Drug Deliv Rev. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savic R, Azzam T, Eisenberg A, Maysinger D. Assessment of the Integrity of Poly(caprolactone)-b-poly(ethylene oxide) Micelles under Biological Conditions: A Fluorogenic-based Approach. Langmuir. 2006;22:3570–3578. doi: 10.1021/la0531998. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Xiao W, Xiao K, Berti L, Luo J, Tseng HP, Fung G, Lam KS. Well-Defined, Reversible Boronate Crosslinked Nanocarriers for Targeted Drug Delivery in Response to Acidic pH Values and cis-Diols. Angew Chem Int Ed. 2012;51:2864–2869. doi: 10.1002/anie.201107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng JX. Fast Release of Lipophilic Agents from Circulating PEG-PDLLA Micelles Revealed by in vivo Forster Resonance Energy Transfer Imaging. Langmuir. 2008;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- 18.Park K, Chen HT, Kim SW, Li L, Wang SY, Cheng JX. Release of Hydrophobic Molecules from Polymer Micelles into Cell Membranes Revealed by Forster Resonance Energy Transfer Imaging. P Natl Acad Sci USA. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jares-Erijman EA, Jovin TM. FRET Imaging. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 20.Sapsford KE, Berti L, Medintz IL. Materials for Fluorescence Resonance Energy Transfer Analysis: beyond Traditional Donor-acceptor Combinations. Angew Chem Int Ed Engl. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 21.Hess JF, Voss JC, FitzGerald PG. Real-time Observation of Coiled-coil Domains and Subunit Assembly in Intermediate Filaments. J Biol Chem. 2002;277:35516–35522. doi: 10.1074/jbc.M206500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lurie DJ, Mader K. Monitoring Drug Delivery Processes by EPR and Related Techniques--principles and Applications. Adv Drug Deliv Rev. 2005;57:1171–1190. doi: 10.1016/j.addr.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Walter ED, Sebby KB, Usselman RJ, Singel DJ, Cloninger MJ. Characterization of Heterogeneously Functionalized Dendrimers by Mass Spectrometry and EPR Spectroscopy. J Phys Chem B. 2005;109:21532–21538. doi: 10.1021/jp0515683. [DOI] [PubMed] [Google Scholar]
- 24.Klajnert B, Cangiotti M, Calici S, Majoral JP, Caminade AM, Cladera J, Bryszewska M, Ottaviani MF. EPR Study of the Interactions between Dendrimers and Peptides Involved in Alzheimer’s and Prion Diseases. Macromol Biosci. 2007;7:1065–1074. doi: 10.1002/mabi.200700049. [DOI] [PubMed] [Google Scholar]
- 25.Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, Xing L, Holland Cheng R, Liu GY, Lam KS. Well-defined, Size-tunable, Multifunctional Micelles for Efficient Paclitaxel Delivery for Cancer Treatment. Bioconjug Chem. 2010;21:1216–1224. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao K, Luo J, Fowler WL, Li Y, Lee JS, Xing L, Cheng RH, Wang L, Lam KS. A Self-Assembling Nanoparticle for Paclitaxel Delivery in Ovarian Cancer. Biomaterials. 2009;30:6006–6016. doi: 10.1016/j.biomaterials.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. The Effect of Surface Charge on in vivo Biodistribution of PEG-oligocholic Acid Based Micellar Nanoparticles. Biomaterials. 2011;32:3435–3446. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, Kato J, Dong TA, Lam KS. Well-defined, Reversible Disulfide Cross-linked Micelles for On-demand Paclitaxel Delivery. Biomaterials. 2011;32:6633–6645. doi: 10.1016/j.biomaterials.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao K, Luo J, Li Y, Lee JS, Fung G, Lam KS. PEG-oligocholic Acid Telodendrimer Micelles for the Targeted Delivery of Doxorubicin to B-cell Lymphoma. J Control Release. 2011;155:272–281. doi: 10.1016/j.jconrel.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beghein N, Rouxhet L, Dinguizli M, Brewster ME, Arien A, Preat V, Habib JL, Gallez B. Characterization of Self-assembling Copolymers in Aqueous Solutions Using Electron Paramagnetic Resonance and Fluorescence Spectroscopy. J Control Release. 2007;117:196–203. doi: 10.1016/j.jconrel.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH, Lee SC. Disulfide-cross-linked PEG-poly(amino acid)s Copolymer Micelles for Glutathione-mediated Intracellular Drug Delivery. Chem Commun. 2008;48:6570–6572. doi: 10.1039/b815918a. [DOI] [PubMed] [Google Scholar]
- 32.Rebbaa A, Portoukalian J. Distribution of Exogenously Added Gangliosides in Serum Proteins Depends on the Relative Affinity of Albumin and Lipoproteins. J Lipid Res. 1995;36:564–572. [PubMed] [Google Scholar]
- 33.Roy R, Hohng S, Ha T. A Practical Guide to Single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masuko M, Ohuchi S, Sode K, Ohtani H, Shimadzu A. Fluorescence Resonance Energy Transfer from Pyrene to Perylene Labels for Nucleic Acid Hybridization Assays under Homogeneous Solution Conditions. Nucleic Acids Res. 2000;28:E34. doi: 10.1093/nar/28.8.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology Applications in Cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.