

Abstract
Objectives
Class C β-lactamases are prevalent among Enterobacteriaceae; however, these enzymes are resistant to inactivation by commercially available β-lactamase inhibitors. In order to find novel scaffolds to inhibit class C β-lactamases, the comparative efficacy of monocyclic β-lactam antibiotics (aztreonam and the siderophore monosulfactam BAL30072), the bridged monobactam β-lactamase inhibitor BAL29880, and carbapenems (imipenem, meropenem, doripenem and ertapenem) were tested in kinetic assays against FOX-4, a plasmid-mediated class C β-lactamase (pmAmpC).
Methods
The FOX-4 β-lactamase was purified. Steady-state kinetics, electrospray ionization mass spectrometry (ESI-MS) and ultraviolet difference (UVD) spectroscopy were conducted using the β-lactam scaffolds described.
Results
The Ki values for the monocyclic β-lactams against FOX-4 β-lactamase were 0.04 ± 0.01 μM (aztreonam) and 0.66 ± 0.03 μM (BAL30072), and the Ki value for the bridged monobactam BAL29880 was 8.9 ± 0.5 μM. For carbapenems, the Ki values ranged from 0.27 ± 0.05 μM (ertapenem) to 2.3 ± 0.3 μM (imipenem). ESI-MS demonstrated the formation of stable covalent adducts when the monocyclic β-lactams and carbapenems were reacted with FOX-4 β-lactamase. UVD spectroscopy suggested the appearance of different chromophoric intermediates.
Conclusions
Monocyclic β-lactam and carbapenem antibiotics are effective mechanism-based inhibitors of FOX-4 β-lactamase, a clinically important pmAmpC, and provide stimulus for the development of new inhibitors to inactivate plasmidic and chromosomal class C β-lactamases.
Keywords: bridged monobactams, monosulfactam, β-lactamase inhibitors, carbapenems
Introduction
The prevalence and dissemination of plasmid-mediated class C β-lactamases (pmAmpCs) is a critical challenge in the treatment of infections caused by Gram-negative bacteria.1,2 Clinically important pmAmpCs include the CMY, MIR, MOX, LAT, FOX, DHA, ACT, ACC and CFE β-lactamase families.2 The FOX family of class C cephalosporinases is distinctly different in amino acid sequence when compared with other class C β-lactamases.3 All eight variants of the FOX family of β-lactamases that have been described thus far show substrate specificity for cephalosporins, including cephamycins.2,4–9 Studies have indicated that blaFOX is widely disseminated in the USA, is present on IncA/C and pMG252 plasmids, and is often associated with the presence of plasmid-mediated quinolone resistance determinants, qnr.8,10–19 FOX β-lactamases have also been found outside the USA – in Europe, Central and South America, and India.4–7,20,21 Significantly, the presence of FOX β-lactamases can complicate the laboratory detection of extended-spectrum β-lactamases by yielding false-positive or false-negative results.19
In this study, FOX-4 was selected from the FOX family as a model β-lactamase because the clinical impact of this class of pmAmpC is notable. blaFOX-4 was identified in an Escherichia coli clinical strain isolated from a patient with a urinary tract infection in Spain.7 FOX-4 possesses broad substrate specificity for β-lactam hydrolysis, especially cephamycins, the biochemical basis of which remains to be explored. The kinetic inhibitory profiles of monocyclic β-lactams and carbapenems against FOX-4 were investigated in this analysis to determine whether these compounds have broad inhibitory profiles against plasmid-encoded cephamycinases.
To date, studies of the inhibition of class C enzymes by β-lactamase inhibitors have focused on a select or restricted number of enzymes (e.g. AmpC of E. coli, and ADC-7, CMY-2 and AmpC of Pseudomonas aeruginosa).22–28 These studies do not fully define the mechanistic basis of AmpC inhibition. To illustrate, Bauvois et al.29 showed that the affinity constants of pmAmpCs were significantly different compared with chromosomal AmpCs and that enzyme production in the periplasmic space is variable. These uncertainties merit more careful study.
Class C cephalosporinases are usually resistant to the action of commercially available β-lactamase inhibitors (i.e. clavulanic acid, sulbactam and tazobactam). The search for new compounds, including β-lactamase-stable antibiotics and inhibitors of these enzymes, therefore assumes paramount importance.30 Many experimental β-lactamase inactivators have been described and at least three are in clinical development (i.e. avibactam, MK-7655 and RPX7009).31,32 However, some years will pass before any reach clinical use.
As an antimicrobial agent, aztreonam effectively inhibits class C β-lactamases by acting as a poor substrate because its C4 methyl substituent prevents rotation that would otherwise expose the ester bond of the acyl adduct to hydrolytic attack.29,33 The bridged monobactams were designed to exploit this principle,34 and BAL29880, which does not have intrinsic antimicrobial properties, has been shown to be an effective class C β-lactamase inhibitor.24,35 The siderophore monosulfactam BAL30072 (bearing a side chain that can enhance its entry to cells through bacterial iron uptake systems) is effective against many cephalosporin- and carbapenem-resistant strains.24,36,37 The potential of BAL30072 as a broad-based inhibitor of class C β-lactamases has not been fully evaluated.
Carbapenems such as imipenem were shown to act as class C β-lactamase inhibitors,2,24,38,39 and were studied as inhibitors of the P. aeruginosa and E. coli class C β-lactamases as well as of CMY-2 and ADC-7.22–24,40 These molecules form stable acyl–enzyme adducts in which the 6α-hydroxyethyl side chain is believed to force the electrophilic acyl centre to rotate away from the point of hydrolytic attack.39 The high apparent affinity of carbapenems for class C β-lactamases makes them attractive candidates for further development as drugs with dual properties (i.e. as antibiotics and β-lactamase inhibitors).38,39
Materials and methods
Antibiotics
Ampicillin was obtained from Sigma (St Louis, MO, USA). Aztreonam was purchased from Elan (Dublin, Ireland). Nitrocefin (NCF) was supplied by BD Biosciences (San Jose, CA, USA). Doripenem was obtained from Janssen-Cilag (Neuss, Germany), ertapenem and imipenem were purchased from Merck (Whitehouse Station, NJ, USA) and meropenem was procured from AstraZeneca Pharmaceuticals (Wilmington, DE, USA). BAL29880 and BAL30072 were prepared in the laboratories of Basilea Pharmaceutica International Ltd (Basel, Switzerland). The chemical structures of the monobactams and carbapenems tested are shown in Figure 1.
Figure 1.

Chemical structures of the β-lactamase inhibitors used in this study.
Plasmids and strains
The cloning of the blaFOX-4 gene into pGEX-6p-1 and pBGS18 was previously reported.3 The pGEX-6p-1 clone in E. coli BL21 (DE3) was used for protein expression. The pBGS18 clone expressed in E. coli TG1 was used for antimicrobial susceptibility testing.
Protein expression and purification
The expression and purification of the FOX-4 β-lactamase was previously described.3
Kinetic parameters
The purified enzyme was used in all the studies reported here. Kinetic experiments were carried out with an Agilent 8453 diode array spectrophotometer at room temperature in 10 mM PBS buffer at pH 7.4. Vmax and Km were determined from initial steady-state velocities for NCF, Δɛ482 = 17 400 M−1 cm−1. These parameters were calculated using iterative non-linear least-squares fit of the data to the Henri–Michaelis–Menten equation using EnzfitterTM (Biosoft Corporation) according to Equation 1:
| (1) |
Here, v is observed velocity, Vmax is maximum velocity, [S] is substrate concentration and Km is the Michaelis constant.
The simplified scheme in Figure 2 depicts the interactions of FOX-4 with inhibitors chosen for this study. Ki values correspond to a relative affinity or k−1/k1 of the inhibitor for the enzyme (for inhibitors that acylate slowly) or to the Michaelis constant or (k−1+k2)/k1 of the inhibitor for the enzyme (for inhibitors that acylate more rapidly). Ki was determined by direct competition assays using 200 μM NCF. Inverse initial steady-state velocities (1/v0) were plotted against the inhibitor concentration ([I]) to obtain a straight line. The v0 measured after mixing is represented by Equation 2:
| (2) |
Figure 2.
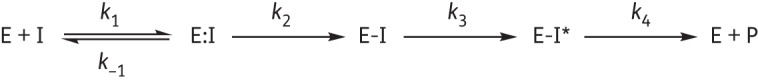
Interactions of FOX-4 (E) with inhibitors (I) are depicted by this reaction scheme. The formation of the Michaelis complex E:I is represented by the dissociation constant K = k−1/k1. k2 is the first-order rate constant for acylation or formation of E-I. The appearance of chromophores (E-I*), as suggested by UVD spectroscopy, is represented by the rate constant k3. Finally, product formation (E + P) as revealed by tn >1 for the majority of the compounds tested is denoted by the rate constant k4.
The plots were linear and provided y-intercept and slope values used for Ki determination. Ki was determined by dividing the value for the y-intercept by the slope of the line and then corrected by taking into account the NCF affinity (Equation 3):
| (3) |
Here, [S] is the concentration of NCF (200 μM) used in the assay and Km is the Michaelis constant determined for NCF (74 μM).
Progress curves with a fixed concentration of enzyme and NCF, and increasing inhibitor concentration, were used to determine the kinact and KI values, as previously described.41 kinact values indicate the number of molecules of enzyme that are inactivated per second; a higher value equates to a more rapid inactivation of the enzyme by the inhibitor. The KI value corresponds to the concentration of inhibitor required to reach ½ kinact. Progress curves were fit to Equation 4 to obtain kobs values using iterative non-linear least-squares fit of the data employing Origin 7.5 software.
| (4) |
Here, A is absorbance, v0 (expressed in variation of absorbance per unit time) is initial velocity, vf is final velocity and t is time. kobs was plotted versus the inhibitor concentrations of the experiment and fit to determine kinact according to Equation 5. The KI value obtained was corrected using Equation 3.
| (5) |
Turnover numbers (tn) or partition ratios (kcat/kinact) are defined herein as the ratio of inhibitor concentration to enzyme concentration necessary to decrease the enzyme activity by 95%. tn corresponds to the number of inhibitor molecules an enzyme turns over before the enzyme is inhibited; thus, the lower the number the more effective the inhibitor for the time period measured. The tn values were determined after 15 min and 24 h of incubation with increasing concentrations of the inhibitor. Incubations were done in a final volume of 300 μL, and 25 μL of this reaction mixture was added to a 1 mL final volume to determine the residual enzyme activity using 200 μM NCF.
Ultraviolet difference (UVD) spectroscopy and formation of spectroscopic intermediates
To monitor the appearance of chromophore intermediates, UVD spectroscopy was conducted using a molar ratio of 1000 : 1 (inhibitor/enzyme) from λ = 200–350 nm as previously described.42 Measurements were made every 12 s for 60 min.
Electrospray ionization mass spectrometry (ESI-MS)
β-Lactamase analysis was carried out on an Applied Biosystems (Foster City, CA, USA) QSTAR®-Elite quadrupole time-of-flight (Q-TOF) mass spectrometer equipped with a TurboIon spray™ source and an integrated syringe pump. Mass calibration was performed in the positive mode with renin solution, using the validated instrument protocol. Enzyme-to-inhibitor ratios were based on the amount of inhibitor required to inactivate >95% of the enzyme after 15 min or the tn at 15 min. The reaction mixtures of protein and inhibitor were desalted using C18 ZipTips (Millipore, Bedford, MA, USA) according to the protocol recommended by the manufacturer. The protein was eluted from the ZipTips in 50 μL of acetonitrile/H2O/formic acid (50 : 49 : 1) before analysis. The protein eluate was then infused into the mass spectrometer at a flow rate of 5 μL/min. Mass spectra were acquired using Analyst QS v2.0 software with 80 MCA cycles and an accumulation time of 1.5 s. The mass spectra were deconvoluted by BioAnalyst™ 2.0 software using the Bayesian protein reconstruction algorithm.
Antimicrobial susceptibility testing
Agar dilution was used to determine the MICs of ampicillin, aztreonam, BAL29880, BAL30072, doripenem, ertapenem, imipenem and meropenem alone and ampicillin in combination with aztreonam, BAL29880 and BAL30072. For combination experiments, the concentrations of BAL29880, BAL30072 and aztreonam were fixed at 4 mg/L.
Results
Inhibition kinetics
Kinetic parameters of inhibition for the monocyclic β-lactams and carbapenems are summarized in Table 1. Aztreonam possessed the lowest Ki and KI values, at 0.04 ± 0.01 μM and 0.010 ± 0.001 μM, respectively, while BAL29880 demonstrated the highest Ki and KI values, at 9 ± 1 μM and 41 ± 4 μM, respectively. The inactivation efficiencies (i.e. kinact/KI) were the lowest for BAL29880 and imipenem at 0.010 ± 0.001 μM−1 s−1 and 0.04 ± 0.01 μM−1 s−1, respectively, and highest for ertapenem and aztreonam at 1.1 ± 0.2 μM−1 s−1 and 5 ± 1 μM−1 s−1, respectively. BAL30072 displayed an inactivation efficiency similar to those of meropenem and doripenem (0.29 ± 0.03, 0.62 ± 0.08 and 0.14 ± 0.02 μM−1 s−1, respectively). The partition ratio (kcat/kinact) for aztreonam and BAL29880 was 1 at 24 h. For the other compounds tested, ≥40 molecules of inhibitor were required to decrease FOX-4 activity by 95% after 24 h; thus, these compounds appear to be slowly hydrolysed by FOX-4. Fewer molecules were required to inhibit FOX-4 activity after 15 min for most inhibitors, except for BAL29880, which may be due to its large Ki and KI values for FOX-4.
Table 1.
Steady-state kinetic inhibition parameters for FOX-4 with monocyclic β-lactams and carbapenems
| Inhibitor | Ki (μM) | KI (μM) | kinact (s−1) | kinact/KI (μM−1 s−1) | tn (15 min) | tn (24 h) |
|---|---|---|---|---|---|---|
| Aztreonam | 0.04 ± 0.01 | 0.010 ± 0.001 | 0.05 ± 0.01 | 5 ± 1 | 1 | 1 |
| BAL29880 | 9 ± 1 | 41 ± 4 | 0.34 ± 0.03 | 0.010 ± 0.001 | 20 | 1 |
| BAL30072 | 0.7 ± 0.1 | 0.35 ± 0.01 | 0.10 ± 0.01 | 0.29 ± 0.03 | 70 | 80 |
| Doripenem | 1.1 ± 0.1 | 1.4 ± 0.2 | 0.19 ± 0.02 | 0.14 ± 0.02 | 50 | 80 |
| Ertapenem | 0.27 ± 0.05 | 0.09 ± 0.02 | 0.10 ± 0.01 | 1.1 ± 0.2 | 3 | 60 |
| Imipenem | 2.3 ± 0.3 | 10 ± 1 | 0.37 ± 0.04 | 0.04 ± 0.01 | 80 | 100 |
| Meropenem | 0.28 ± 0.04 | 0.13 ± 0.01 | 0.08 ± 0.01 | 0.62 ± 0.08 | 15 | 40 |
UVD spectroscopy
UVD spectroscopy was used to detect the presence of intermediates (chromophores) formed during the inactivation process. A clear peak in the difference spectrum of the reaction mixture of FOX-4 and BAL29880 occurred at λ = 255 nm (Figure 3a). A0 at λ = 255 nm increased rapidly to a stable level (representing formation of the acyl–enzyme adduct). Surprisingly, the formation of chromophores was distinctly different when aztreonam and BAL30072 were reacted with FOX-4. The difference spectrum of these reaction mixtures showed the formation of chromophores at λ = 215 nm and λ = 290 nm with aztreonam, and at λ = 235 nm and λ = 290 nm with BAL30072 (Figure 3b and c). The UVD spectra revealed that the chemistry of BAL29880 upon interaction with FOX-4 is different from that of aztreonam and BAL30072, as different chromophores are formed.
Figure 3.

UVD spectroscopy of FOX-4 reacted with (a) BAL29880, (b) aztreonam and (c) BAL30072. Absorbance (Abs) values in arbitrary units (a.u.) were measured from wavelengths of 200–350 nm at 12 s (solid line), 15 min (dotted line), 30 min (dashed line), 45 min (smaller dashed line) and 60 min (dashed-dotted line) timepoints.
ESI-MS
ESI-MS was employed to gain insight into the nature of the stable covalent intermediates formed in the inactivation of the FOX-4 β-lactamase. The expected mass of FOX-4 was 39 230 ± 5 amu, as the recombinant enzyme consists of FOX-4 residues 24 to 382 plus eight extra residues (GPLGSPEF) at the amino terminus following cleavage of the fused N-terminal GST-tag expressed from the pGEX-6p-1 vector (Figure 4a and Table 2).
Figure 4.

Deconvoluted mass spectra of FOX-4 β-lactamase alone and with inhibitors in variable ratios (in parentheses). Mass increases (products of inactivation) are shown for (a) FOX-4 β-lactamase alone, (b) FOX-4 with aztreonam, (c) FOX-4 with BAL29880 and (d) FOX-4 with BAL30072. All measurements have an error of ±5 amu.
Table 2.
Mass spectrometry of FOX-4 alone and in combination with monocyclic β-lactams and carbapenems
| Theoretical mass amu (±5) | Experimental mass amu (±5) | Experimental mass Δ amu (±5) | |
|---|---|---|---|
| FOX-4 | 39 230 | 39 230 | |
| FOX-4 + aztreonam (435 Da) | 39 665 | 39 667 | 437 |
| 39 586 | 356 | ||
| FOX-4 + BAL29880 (412 Da) | 39 642 | 39 642 | 412 |
| FOX-4 + BAL30072 (518 Da) | 39 748 | 39 749 | 519 |
| FOX-4 + doripenem (420 Da) | 39 650 | 39 653 | 423 |
| 39 609 | 379 | ||
| FOX-4 + ertapenem (475 Da) | 39 705 | 39 707 | 477 |
| 39 663 | 433 | ||
| FOX-4 + meropenem (383 Da) | 39 613 | 39 614 | 384 |
| 39 565 | 335 | ||
| FOX-4 + imipenem (299 Da) | 39 529 | 39 533 | 303 |
| 39 489 | 259 |
FOX-4 inactivated by aztreonam produced the full adduct of aztreonam equivalent to Δ +437 ± 5 amu, as well as another smaller adduct of Δ+356 ± 5 amu (Figure 4b and Table 2). The smaller adduct is hypothesized to represent the complexed β-lactamase with elimination of the SO32− of aztreonam.24 Complete adducts were found for FOX-4 in combination with BAL29880 and BAL30072 (Δ +412 ± 5 amu and Δ +519 ± 5 amu, respectively) (Figure 4c and d, and Table 2).
Each combination of FOX-4 with a carbapenem demonstrated the full carbapenem-complexed β-lactamase, as well as an additional peak representing the acyl–enzyme complex with elimination of the C6 hydroxyethyl side chain of the carbapenem (Figure 5a–d and Table 2).23,24,43,44 For all tested combinations, FOX-4 enzyme alone (39 230 amu) was not identified, indicating complete inactivation of FOX-4. An unexpected mass of 39 560 ± 5 amu was seen in most cases; the nature of this species remains under investigation.
Figure 5.

Deconvoluted mass spectra of FOX-4 β-lactamase with inhibitors in variable ratios (in parentheses). Mass increases (products of inactivation) are shown for (a) FOX-4 β-lactamase with ertapenem, (b) FOX-4 with meropenem, (c) FOX-4 with doripenem and (d) FOX-4 with imipenem. All measurements have an error of ±5 amu.
Antimicrobial susceptibility assays
To determine the antibacterial properties of the compounds studied and relate these to the biochemical analyses, agar dilution MICs were determined for the monocyclic β-lactams and carbapenems, and the data are presented in Table 3. For E. coli TG1 with pBGS18 blaFOX-4, MICs were the lowest for BAL30072, doripenem, ertapenem, meropenem and ampicillin/aztreonam, at 0.06 mg/L, and highest for BAL29880, at >256 mg/L.
Table 3.
MICs of monocyclic β-lactams and carbapenems
| Antibiotic | MIC (mg/L) |
|
|---|---|---|
| E. coli TG1 with pBGS18 blaFOX-4 | E. coli TG1 with pBGS18 | |
| Ampicillin | 32 | 4 |
| Aztreonam | 4 | 0.06 |
| Ampicillin + aztreonam (4 mg/L) | 0.06 | 0.06 |
| Ampicillin + BAL29880 (4 mg/L) | 1 | 1 |
| BAL29880 | >256 | >256 |
| Ampicillin + BAL30072 (4 mg/L) | 0.06 | 0.06 |
| BAL30072 | 0.06 | 0.06 |
| Doripenem | 0.06 | 0.06 |
| Ertapenem | 0.06 | 0.06 |
| Imipenem | 0.25 | 0.25 |
| Meropenem | 0.06 | 0.06 |
Discussion
The clinically used β-lactamase inhibitors (clavulanic acid, sulbactam and tazobactam) have limited activity against class C β-lactamases; ampicillin and ceftazidime MICs for E. coli TG1 carrying blaFOX-4 are completely unaffected when combined with these inhibitors.3,30 There is therefore a growing need to identify novel compounds that are active against these enzymes. In this context, the aim of this study was to test different monocyclic β-lactams (aztreonam and a siderophore monosulfactam, BAL30072), a bridged monobactam (BAL29880) and carbapenems for their ability to inhibit the class C β-lactamase FOX-4.
Among the monocyclic β-lactams, aztreonam showed the highest inactivation efficiency (kinact/KI = 5 ± 1 μM−1 s−1), owing to a very low KI (0.01 ± 0.001 μM), followed by BAL30072 and then BAL29880. BAL29880 inactivates FOX-4 rapidly, with a high kinact value of 0.34 ± 0.03 s−1; however, BAL29880 is slow to bind and/or acylate the enzyme, as evidenced by high Ki, KI and tn values at 15 min. The tn at 24 h for aztreonam and BAL29880 were both 1, which shows that recovery was not detected after 24 h of incubation and that these inhibitors are not hydrolysed by FOX-4. These data are in accordance with previous results obtained with BAL29880 as an inhibitor of CMY-2.24 The kinact/KI values are identical for CMY-2 and FOX-4 at 0.01 μM−1 s−1. In bacteria, BAL30072 was the most effective monocyclic β-lactam, with an MIC of 0.06 mg/L; this may be owing to the enhanced ability BAL30072 to enter cells as a result of its siderophore side chain.
These observations were anticipated, as the suspected mechanism of inactivation by aztreonam and BAL29880 depends upon restricted rotation in the acyl adduct and blockade of the trajectory of the hydrolytic water molecule.33,34 It has been noted that the monosulfactams form less stable adducts with class C β-lactamases, probably because of the greater flexibility of the N-O-SO3− group compared with N-SO3−.45
Ertapenem was the best carbapenem inhibitor of FOX-4, with a kinact/KI value of 1.1 ± 0.2 μM−1 s−1, followed closely by meropenem (0.62 ± 0.08 μM−1 s−1); as with aztreonam, this is mostly due to low KI values (0.09 ± 0.02 μM and 0.13 ± 0.01 μM, respectively). Imipenem and doripenem demonstrated higher KI values and thus were less effective inhibitors by comparison. The kinact values were similar for all the carbapenems, indicating that they inactivate FOX-4 at similar rates. In bacteria, all the carbapenems were similarly effective against E. coli TG1 pBGS18 blaFOX-4, as evidenced by their MIC values of 0.06–0.25 mg/L. In clinical practice, the use of ertapenem is more limited than the other carbapenems because of its lack of activity against P. aeruginosa and Acinetobacter baumannii,46,47 but its in vivo t1/2 is greater than those of the other carbapenems.48
Conclusions
In summary, our testing of aztreonam, new monocyclic β-lactams and carbapenems against a pmAmpC, FOX-4, offers a better understanding of their inhibitory behaviour against enzymes with a substrate preference for cephamycins. The data we report here propose a rationale to use both these classes of compounds with dual properties for the treatment of infections caused by microorganisms possessing the blaFOX-4 gene; one exception is BAL29880, which must be partnered with a β-lactam for activity.38,39 Further spectroscopic and structural studies are warranted to decipher the nature of the intermediates suggested by the UVD studies. The novel chromophores are suspected to occur due to differences in the chemistry of the R1 side chains, which may be important for FOX-4 inactivation by these compounds.
Funding
G. B. was funded by the Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III-FEDER, Spanish Network for Research in Infectious Diseases (REIPI RD06/0008). This work was also funded by FIS PI12/00552, PS09/00687 and PS07/90 from Xunta de Galicia. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01 AI100560 and R01 AI063517 to R. A. B. This study was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program and the Geriatric Research Education and Clinical Center VISN 10 to R. A. B. and the Veterans Affairs Career Development Program to K. M. P.-W.
Transparency declarations
M. G. P. P. and E. D. are employees of Basilea Pharmaceutica International Ltd. They hold stock options (M. G. P. P. and E. D.) and stock (M. G. P. P.) in the company. All other authors: none to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Cejas D, Fernandez Canigia L, Quinteros M, et al. Plasmid-encoded AmpC (pAmpC) in Enterobacteriaceae: epidemiology of microorganisms and resistance markers. Rev Argent Microbiol. 2012;44:182–6. [PubMed] [Google Scholar]
- 2.Jacoby GA. AmpC β-lactamases. Clin Microbiol Rev. 2009;22:161–82. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallo S, Perez-Llarena FJ, Kerff F, et al. A tripeptide deletion in the R2 loop of the class C β-lactamase enzyme FOX-4 impairs cefoxitin hydrolysis and slightly increases susceptibility to β-lactamase inhibitors. J Antimicrob Chemother. 2010;65:1187–94. doi: 10.1093/jac/dkq115. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez Leiza M, Perez-Diaz JC, Ayala J, et al. Gene sequence and biochemical characterization of FOX-1 from Klebsiella pneumoniae, a new AmpC-type plasmid-mediated β-lactamase with two molecular variants. Antimicrob Agents Chemother. 1994;38:2150–7. doi: 10.1128/aac.38.9.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauernfeind A, Wagner S, Jungwirth R, et al. A novel class C β-lactamase (FOX-2) in Escherichia coli conferring resistance to cephamycins. Antimicrob Agents Chemother. 1997;41:2041–6. doi: 10.1128/aac.41.9.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marchese A, Arlet G, Schito GC, et al. Characterization of FOX-3, an AmpC-type plasmid-mediated β-lactamase from an Italian isolate of Klebsiella oxytoca. Antimicrob Agents Chemother. 1998;42:464–7. doi: 10.1128/aac.42.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bou G, Oliver A, Ojeda M, et al. Molecular characterization of FOX-4, a new AmpC-type plasmid-mediated β-lactamase from an Escherichia coli strain isolated in Spain. Antimicrob Agents Chemother. 2000;44:2549–53. doi: 10.1128/aac.44.9.2549-2553.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Queenan AM, Jenkins S, Bush K. Cloning and biochemical characterization of FOX-5, an AmpC-type plasmid-encoded β-lactamase from a New York City Klebsiella pneumoniae clinical isolate. Antimicrob Agents Chemother. 2001;45:3189–94. doi: 10.1128/AAC.45.11.3189-3194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miro E, Aguero J, Larrosa MN, et al. Prevalence and molecular epidemiology of acquired AmpC β-lactamases and carbapenemases in Enterobacteriaceae isolates from 35 hospitals in Spain. Eur J Clin Microbiol Infect Dis. 2013;32:253–9. doi: 10.1007/s10096-012-1737-0. [DOI] [PubMed] [Google Scholar]
- 10.Hazen TH, Robinson GL, Harris AD, et al. Genome sequence of Klebsiella oxytoca 11492-1, a nosocomial isolate possessing a FOX-5 AmpC β-lactamase. J Bacteriol. 2012;194:3028–9. doi: 10.1128/JB.00391-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coudron PE, Hanson ND, Climo MW. Occurrence of extended-spectrum and AmpC β-lactamases in bloodstream isolates of Klebsiella pneumoniae: isolates harbor plasmid-mediated FOX-5 and ACT-1 AmpC β-lactamases. J Clin Microbiol. 2003;41:772–7. doi: 10.1128/JCM.41.2.772-777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Martinez JM, Conejo MC, Martinez-Martinez L, et al. Evaluation of antimicrobial susceptibility of bacteria containing the qnr gene and FOX-5 β-lactamase by four automated systems. Clin Microbiol Infect. 2005;11:402–4. doi: 10.1111/j.1469-0691.2005.01097.x. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez M, Tran JH, Chow N, et al. Epidemiology of conjugative plasmid-mediated AmpC β-lactamases in the United States. Antimicrob Agents Chemother. 2004;48:533–7. doi: 10.1128/AAC.48.2.533-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby GA, Chow N, Waites KB. Prevalence of plasmid-mediated quinolone resistance. Antimicrob Agents Chemother. 2003;47:559–62. doi: 10.1128/AAC.47.2.559-562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang M, Sahm DF, Jacoby GA, et al. Emerging plasmid-mediated quinolone resistance associated with the qnr gene in Klebsiella pneumoniae clinical isolates in the United States. Antimicrob Agents Chemother. 2004;48:1295–9. doi: 10.1128/AAC.48.4.1295-1299.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshpande LM, Jones RN, Fritsche TR, et al. Occurrence of plasmidic AmpC type β-lactamase-mediated resistance in Escherichia coli: report from the SENTRY Antimicrobial Surveillance Program (North America, 2004) Int J Antimicrob Agents. 2006;28:578–81. doi: 10.1016/j.ijantimicag.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 17.Rhomberg PR, Deshpande LM, Kirby JT, et al. Activity of meropenem as serine carbapenemases evolve in US medical centers: monitoring report from the MYSTIC Program (2006) Diagn Microbiol Infect Dis. 2007;59:425–32. doi: 10.1016/j.diagmicrobio.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Castanheira M, Mendes RE, Rhomberg PR, et al. Rapid emergence of blaCTX-M among Enterobacteriaceae in U.S. medical centers: molecular evaluation from the MYSTIC Program (2007) . Microb Drug Resist. 2008;14:211–6. doi: 10.1089/mdr.2008.0827. [DOI] [PubMed] [Google Scholar]
- 19.Robberts FJ, Kohner PC, Patel R. Unreliable extended-spectrum β-lactamase detection in the presence of plasmid-mediated AmpC in Escherichia coli clinical isolates. J Clin Microbiol. 2009;47:358–61. doi: 10.1128/JCM.01687-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodford N, Reddy S, Fagan EJ, et al. Wide geographic spread of diverse acquired AmpC β-lactamases among Escherichia coli and Klebsiella spp. in the UK and Ireland. J Antimicrob Chemother. 2007;59:102–5. doi: 10.1093/jac/dkl456. [DOI] [PubMed] [Google Scholar]
- 21.Manoharan A, Sugumar M, Kumar A, et al. Phenotypic and molecular characterization of AmpC β-lactamases among Escherichia coli, Klebsiella spp. and Enterobacter spp. from five Indian medical centers . Indian J Med Res. 2012;135:359–64. [PMC free article] [PubMed] [Google Scholar]
- 22.Beadle BM, Shoichet BK. Structural basis for imipenem inhibition of class C β-lactamases. Antimicrob Agents Chemother. 2002;46:3978–80. doi: 10.1128/AAC.46.12.3978-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drawz SM, Babic M, Bethel CR, et al. Inhibition of the class C β-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry. 2010;49:329–40. doi: 10.1021/bi9015988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Endimiani A, Doi Y, Bethel CR, et al. Enhancing resistance to cephalosporins in class C β-lactamases: impact of Gly214Glu in CMY-2. Biochemistry. 2010;49:1014–23. doi: 10.1021/bi9015549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drawz SM, Taracila M, Caselli E, et al. Exploring sequence requirements for C3/C4 carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: insights into plasticity of the AmpC β-lactamase. Protein Sci. 2011;20:941–58. doi: 10.1002/pro.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papp-Wallace KM, Bethel CR, Gootz TD, et al. Inactivation of a class A and a class C β-lactamase by 6-β-(hydroxymethyl)penicillanic acid sulfone. Biochem Pharmacol. 2012;83:462–71. doi: 10.1016/j.bcp.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahiri SD, Mangani S, Durand-Reville T, et al. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother. 2013;57:2496–505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morandi F, Caselli E, Morandi S, et al. Nanomolar inhibitors of AmpC β-lactamase. J Am Chem Soc. 2003;125:685–95. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 29.Bauvois C, Ibuka AS, Celso A, et al. Kinetic properties of four plasmid-mediated AmpC β-lactamases. Antimicrob Agents Chemother. 2005;49:4240–6. doi: 10.1128/AAC.49.10.4240-4246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Llarena FJ, Bou G. β-Lactamase inhibitors: the story so far. Curr Med Chem. 2009;16:3740–65. doi: 10.2174/092986709789104957. [DOI] [PubMed] [Google Scholar]
- 31.Shlaes DM. New β-lactam–β-lactamase inhibitor combinations in clinical development. Ann N Y Acad Sci. 2013;1277:105–14. doi: 10.1111/nyas.12010. [DOI] [PubMed] [Google Scholar]
- 32.Livermore DM, Mushtaq S. Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J Antimicrob Chemother. 2013;68:1825–31. doi: 10.1093/jac/dkt118. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson AS, Bryant PK, Meroueh SO, et al. A dynamic structure for the acyl–enzyme species of the antibiotic aztreonam with the Citrobacter freundii β-lactamase revealed by infrared spectroscopy and molecular dynamics simulations. Biochemistry. 2003;42:1950–7. doi: 10.1021/bi0266941. [DOI] [PubMed] [Google Scholar]
- 34.Heinze-Krauss I, Angehrn P, Charnas RL, et al. Structure-based design of β-lactamase inhibitors. 1. Synthesis and evaluation of bridged monobactams. J Med Chem. 1998;41:3961–71. doi: 10.1021/jm980023c. [DOI] [PubMed] [Google Scholar]
- 35.Page MG, Dantier C, Desarbre E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant Gram-negative bacilli. Antimicrob Agents Chemother. 2010;54:2291–302. doi: 10.1128/AAC.01525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mushtaq S, Warner M, Livermore D. Activity of the siderophore monobactam BAL30072 against multiresistant non-fermenters. J Antimicrob Chemother. 2010;65:266–70. doi: 10.1093/jac/dkp425. [DOI] [PubMed] [Google Scholar]
- 37.Page MG, Heim J. New molecules from old classes: revisiting the development of β-lactams. IDrugs. 2009;12:561–5. [PubMed] [Google Scholar]
- 38.Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp-Wallace KM, Endimiani A, Taracila MA, et al. Carbapenems: past, present, and future. Antimicrob Agents Chemother. 2011;55:4943–60. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monks J, Waley SG. Imipenem as substrate and inhibitor of β-lactamases. Biochem J. 1988;253:323–8. doi: 10.1042/bj2530323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papp-Wallace KM, Bethel CR, Distler AM, et al. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob Agents Chemother. 2010;54:890–7. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pattanaik P, Bethel CR, Hujer AM, et al. Strategic design of an effective β-lactamase inhibitor: LN-1–255, a 6-alkylidene-2′-substituted penicillin sulfone. J Biol Chem. 2009;284:945–53. doi: 10.1074/jbc.M806833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tremblay LW, Fan F, Blanchard JS. Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems, ertapenem and doripenem. Biochemistry. 2010;49:3766–73. doi: 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hugonnet JE, Tremblay LW, Boshoff HI, et al. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–8. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hubschwerlen C, Angehrn P, Gubernator K, et al. Structure-based design of β-lactamase inhibitors. 2. Synthesis and evaluation of bridged sulfactams and oxamazins. J Med Chem. 1998;41:3972–5. doi: 10.1021/jm9800245. [DOI] [PubMed] [Google Scholar]
- 46.Martinez MJ, Garcia MI, Sanchez EG, et al. [Available carbapenems: properties and differences] Enferm Infecc Microbiol Clin. 2010;28(Suppl 2):53–64. doi: 10.1016/S0213-005X(10)70031-8. [DOI] [PubMed] [Google Scholar]
- 47.Wexler HM. In vitro activity of ertapenem: review of recent studies. J Antimicrob Chemother. 2004;53(Suppl 2):ii11–21. doi: 10.1093/jac/dkh204. [DOI] [PubMed] [Google Scholar]
- 48.Parakh A, Krishnamurthy S, Bhattacharya M. Ertapenem. Kathmandu University Med J. 2009;7:454–60. [PubMed] [Google Scholar]