

Abstract
Objectives
Trypanosoma brucei drug transporters include the TbAT1/P2 aminopurine transporter and the high-affinity pentamidine transporter (HAPT1), but the genetic identity of HAPT1 is unknown. We recently reported that loss of T. brucei aquaglyceroporin 2 (TbAQP2) caused melarsoprol/pentamidine cross-resistance (MPXR) in these parasites and the current study aims to delineate the mechanism by which this occurs.
Methods
The TbAQP2 loci of isogenic pairs of drug-susceptible and MPXR strains of T. brucei subspecies were sequenced. Drug susceptibility profiles of trypanosome strains were correlated with expression of mutated TbAQP2 alleles. Pentamidine transport was studied in T. brucei subspecies expressing TbAQP2 variants.
Results
All MPXR strains examined contained TbAQP2 deletions or rearrangements, regardless of whether the strains were originally adapted in vitro or in vivo to arsenicals or to pentamidine. The MPXR strains and AQP2 knockout strains had lost HAPT1 activity. Reintroduction of TbAQP2 in MPXR trypanosomes restored susceptibility to the drugs and reinstated HAPT1 activity, but did not change the activity of TbAT1/P2. Expression of TbAQP2 sensitized Leishmania mexicana promastigotes 40-fold to pentamidine and >1000-fold to melaminophenyl arsenicals and induced a high-affinity pentamidine transport activity indistinguishable from HAPT1 by Km and inhibitor profile. Grafting the TbAQP2 selectivity filter amino acid residues onto a chimeric allele of AQP2 and AQP3 partly restored susceptibility to pentamidine and an arsenical.
Conclusions
TbAQP2 mediates high-affinity uptake of pentamidine and melaminophenyl arsenicals in trypanosomes and TbAQP2 encodes the previously reported HAPT1 activity. This finding establishes TbAQP2 as an important drug transporter.
Keywords: drug transport, protozoan, parasite, resistance mutation, aquaporin
Introduction
The protozoan parasite Trypanosoma brucei is the aetiological agent of human African trypanosomiasis (HAT or sleeping sickness). The subspecies T. b. gambiense and T. b. rhodesiense are responsible for West African and East African sleeping sickness, respectively, and T. b. brucei is one of the pathogens that cause animal African trypanosomiasis, a wasting disease of livestock. Despite the recent introduction of nifurtimox/eflornithine combination therapy for the late, cerebral stage of HAT,1 there is an urgent need for new drugs, driven in part by resistance to the diamidines, phenanthridines and melaminophenyl arsenicals (MPAs) that have been the central pillars of African trypanosomiasis treatment for decades.2 An understanding of the mechanisms of resistance, and particularly of cross-resistance, is of great importance. Firstly, molecular markers are required to study the epidemiology of resistance, particularly as phenotypic assessment in primary clinical/veterinary isolates is impossible for many species of African trypanosome and there is an unresolved debate about the extent of treatment failure versus genuine resistance, especially with respect to melarsoprol.3 Secondly, in the absence of new drugs we need to make best use of the treatments available and, for this, insight into resistance mechanisms and levels of cross-resistance is essential. Importantly, new drug development must take into account the resistance mechanisms to the current drugs, in order to avoid cross-resistance.
Melarsoprol/pentamidine cross-resistance (MPXR) is a well-known phenomenon in HAT and was first noted by Rollo and Williamson in 1951;4 although its causes have never been completely resolved, it has long been clear this is linked to reduced drug accumulation.5–7 The first drug transporter identified in trypanosomes was the P2 adenosine/adenine transporter, which was initially implicated in melarsoprol uptake8 and subsequently also in diamidine transport;9–11 the gene was designated TbAT1.12 All protozoan nucleoside and nucleobase transporters identified to date have been of the equilibrative nucleoside transporter family.13 Although the evidence of diamidine and arsenical transport by TbAT1/P2 has become incontrovertible, it has become equally clear that this transporter mediates only part of the uptake and that this proportion is different for different diamidines in particular, as deletion of the TbAT1 gene led to a high level of resistance to the veterinary diamidine diminazene aceturate14 and the newer clinical candidates furamidine and CPD0801,15 but only to a relatively minor loss of susceptibility to MPAs and pentamidine.14,16 Two additional, adenosine-insensitive pentamidine transport activities were detected and functionally characterized in T. b. brucei: a high-affinity pentamidine transporter (HAPT1) and a low-affinity pentamidine transporter (LAPT1).17,18 HAPT1 was additionally found to be the secondary transporter for the arsenical drugs, with the loss of both the P2 and HAPT1 activities simultaneously leading to high-level MPXR.16,19 Despite the HAPT1 and LAPT1 activities having been first characterized over a decade ago,17 the genes encoding these transporters remained unknown.
Recently, we reported that the aquaglyceroporin TbAQP2 controls MPXR in T. b. brucei.20 Aquaporins (AQPs) are major intrinsic proteins (MIPs) that are present in virtually every organism and are commonly implicated in osmotic balance and, in the case of aquaglyceroporins, in the bidirectional flux of some small, usually uncharged solutes, such as glycerol and urea.21 AQPs have attracted increasing pharmacological interest because of their important roles in many human physiological and pathophysiological processes, including cancer, post-traumatic brain oedema, glaucoma and epilepsy.22,23 Further pharmacological interest in AQPs emerged when it became clear that these water channels can also mediate the uptake of a wider array of molecules, including some that are cytotoxic and display antimicrobial activity.24 Some AQPs, including Leishmania major AQP1, transport antimony and arsenic, most likely in the form of As(OH)3 and Sb(OH)3, which structurally resemble glycerol.25,26 This has attracted much attention, because pentavalent antimonials such as Glucantime and Pentostam, which are activated to a form of Sb(III), are a first-line treatment for leishmaniasis.
T. brucei members of the AQP family are classified functionally27,28 and phylogenetically29 as aquaglyceroporins. They are closely related to LmAQP1 and human aquaglyceroporins, including hAQP9, which reportedly allows the uptake of a wide variety of uncharged solutes, including carbamides, polyols, purines and pyrimidines.30 The three T. b. brucei AQPs appear to have very similar permeation patterns, mediating the uptake of glycerol, dihydroxyacetone, ribitol and urea.27 However, only TbAQP2 was implicated in MPXR, with the re-expression of TbAQP3 in an aqp2/aqp3 null line having no effect on drug susceptibility.20
Here, we report that loss of the wild-type TbAQP2 open reading frame (ORF) was observed in all MPXR strains (T. b. brucei, T. b. gambiense and T. b. rhodesiense), whether they were selected for resistance to MPAs or pentamidine, including strains selected in vivo and able to be transmitted by tsetse flies. Based on our detailed genetic, pharmacological and kinetic analysis, we conclude that TbAQP2 encodes the HAPT1 activity and that loss of AQP2 function is sufficient and likely required for high-level MPXR.
Materials and methods
Trypanosome strains and culture
Bloodstream-form T. b. brucei, strain Lister 427 (s427; MiTat 1.2/BS221), and its derivatives were maintained as previously described.16 Several derivative lines were used: tbat1−/−,14 B48,16 2T1,31 aqp2/aqp3 null strains32 and P1000 cells (this paper). Procyclic-form T. b. gambiense STIB 386 wild-type and Cymelarsan-resistant (386MR) lines, and T. b. brucei STIB 247 wild-type and Cymelarsan-resistant (247MR) lines were grown as described previously.33 The P1000 line was generated by further subculturing of bloodstream forms of the B48 line in incrementally increasing concentrations of pentamidine, starting at 75 nM, until the trypanosomes proliferated in 1 μM pentamidine. This process took almost a year of continuous in vitro adaptation (Figure S1a, available as Supplementary data at JAC Online), which was presumably genetic in nature as the resistance phenotype has proven to be completely stable even after storage in liquid nitrogen or transformation to procyclic cells. There was no apparent in vitro growth defect associated with the P1000 adaptations (Figure S1b, available as Supplementary data at JAC Online). The STIB 900 line is T. b. rhodesiense, originally isolated from a human patient in Tanzania, and was adapted in vitro for resistance to pentamidine (STIB 900-P) or melarsoprol (STIB 900-M).34
Leishmania strains and culture
Leishmania mexicana promastigotes of strain MNYC/BZ/62/M37935 were cultured in HOMEM medium (Invitrogen) supplemented with 10% fetal bovine serum at 25°C exactly as described for L. major promastigotes.36 Promastigotes were passed to fresh culture medium or used for analysis when in mid-log culture.
Expression of aquaglyceroporins in T. b. brucei cell lines
AQP2 was expressed in the B48 and P1000 lines by modification of the expression vector pHD133637 to give pHDK21. This plasmid was digested with NotI prior to transfection into trypanosomes. The pRPaiGFPx construct38 was modified to express either the AQP2 or AQP2-3 chimera genes and was digested with AscI prior to transfection. Primer sequences are given in Table S1 (available as Supplementary data at JAC Online). The AQP3 and AQP2-3 genes with their selectivity region altered to that of AQP220 were synthesized by GenScript (New Jersey, USA) for insertion into pRPaiGFPx,38 to give N-terminally tagged proteins. The constructs were digested with AscI prior to transfection. B48, P1000 or aqp2/aqp3 null strains were washed in Human T Cell Nucleofector Solution for transfection using the appropriate cassette with an Amaxa Nucleofector as described previously.39 Transfectants were grown and cloned out by limiting dilution in standard HMI-9/FBS containing the relevant antibiotic (5 μg/mL blasticidin for pHDK21 and 2 μg/mL hygromycin for pRPaAQP2/pRPaAQP2-3).
Genome sequencing of STIB 900 lines
Whole genome sequencing of the three T. b. rhodesiense lines STIB 900, STIB 900-M and STIB 900-P was carried out by 454 Life Sciences (Branford, CT, USA) on the Genome Sequencer FLX Titanium, performing two shotgun runs per line. All the high-quality reads were mapped to the reference genome T. b. brucei 92740 from EBI-EMBL (version October 2011) using SMALT (www.sanger.ac.uk/resources/software/smalt). Consensus sequence and variants were identified with SAMtools41 and inspected using Artemis.42
Sequencing of AQP2 and AQP3 genes in drug-resistant lines
The AQP2 and AQP3 genes were sequenced from the drug-resistant lines as well as from their respective wild-type lines. The genes were amplified from genomic DNA using a proofreading polymerase and ligated into the pGEM-T Easy subcloning vector and sequenced using standard procedures. The primers used for amplification of the various genes are given in Table S1 (available as Supplementary data at JAC Online).
Cellular localization of chimeric AQP2/3
Localization of green fluorescent protein (GFP)-coupled chimeric AQP2/3 (GFP-chAQP2/3) and western blot with anti-GFP antiserum were performed exactly as described for wild-type TbAQP2 and TbAQP3.20 Nuclear staining was performed with 4′,6-diamidino-2-phenylindole (DAPI).
Transport assays
Transport assays with procyclic-43 and bloodstream-form trypanosomes,44,45 and L. mexicana promastigotes,36,46 were performed as described previously. Cultures were harvested at the mid-log growth phase and washed into assay buffer (AB; 33 mM HEPES, 98 mM NaCl, 4.6 mM KCl, 0.55 mM CaCl2, 0.07 mM MgSO4, 5.8 mM NaH2PO4, 0.3 mM MgCl2, 23 mM NaHCO3 and 14 mM glucose, pH 7.3) at a final concentration of 108 cells/mL. Transport was initiated by the addition of 100 μL cells to 100 μL of a solution of the appropriate radiolabel in AB layered over oil [7 : 1 dibutylphthalate/mineral oil (v/v); Sigma–Aldrich, St Louis, MO, USA] and terminated by the addition of an ice-cold solution of 1 mL of unlabelled permeant and immediate centrifugation through the oil layer. Radioactivity in the cell pellet was determined by liquid scintillation counting and corrected for non-specific association of radiolabel with the cells as described previously.45 All experiments were performed in triplicate on at least three independent occasions and analysed using the appropriate linear and non-linear regression equations in GraphPad Prism 5.0. [3H]pentamidine (3.26 TBq/mmol; product TRQ40084, batch 1) was custom synthesized by Amersham using tritium gas, producing a general labelling of the pentamidine molecule.
Drug susceptibility assays
Drug susceptibilities of the bloodstream-form trypanosomes47 and L. mexicana promastigotes48 were determined using the Alamar blue assay exactly as described previously, measuring fluorescence in 96-well plates with a FLUOstar Optima (BMG Labtech, Durham, NC, USA) at wavelengths of 544 nm for excitation and 620 nm for emission. EC50 values were calculated by non-linear regression using an equation for a sigmoidal dose–response curve with variable slope (GraphPad Prism 5.0; GraphPad Software Inc., San Diego, CA, USA).
Heterologous expression of T. brucei AQPs in Leishmania promastigotes
TbAQP2 and TbAQP3 were amplified from genomic DNA by PCR using Phusion polymerase (Thermo Scientific) and subcloned into the pNUS vector for expression in Leishmania.49 The construct was verified by sequencing before transfection into L. mexicana promastigotes using an Amaxa Nucleofector (program U-33).
Results
Status of drug transporters and AQP2 in isogenic susceptible/resistant trypanosome pairs
The study by Baker et al.20 established a clear link between TbAQP2 and MPXR in the s427/B48 isogenic pair of T. b. brucei. The B48 line was produced from the TbAT1-KO line (derived from s427 by targeted deletion of TbAT1)14 followed by in vitro exposure to pentamidine.16 For TbAQP2 to be confirmed as a general genetic marker for MPXR in trypanosomes, however, it is essential that this link be upheld in further isogenic pairs showing MPXR, particularly where resistance has been induced (i) in vivo or (ii) to the arsenical component rather than pentamidine and (iii) in human-infective trypanosome subspecies. We thus widened our investigations to the strains described in Table 1, which lists a number of well-described isogenic strains with the desired characteristics. These include strains of both human-infective subspecies, T. b. gambiense and T. b. rhodesiense, in addition to the closely related animal parasite T. b. brucei. The strains were adapted by drug exposure in vivo or in vitro, by exposure to an MPA compound (melarsoprol or Cymelarsan, its water-soluble derivative) or pentamidine. Some were shown to be transmissible by tsetse flies and to mate in this vector.33 In all of these highly resistant strains, the TbAT1/P2 transport activity is known to be deleted, non-expressed or mutated (Table 1 and references therein) and it is believed that this explains part, but crucially not all, of the resistant phenotype.
Table 1.
Overview of trypanosome strains used in this study
| Strain | Subspecies | Susceptibilitya |
Adapted from | Resistance induction | Infectivity |
Transport activity |
References | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pentamidine | arsenical | rodents | tsetse | P2 | HAPT1 | LAPT1 | |||||
| Lister 427 | T. b. brucei | +++ | +++ | NA | NA | √ | √b | √ | √ | √ | 14,16,17 |
| 2T1 | T. b. brucei | +++ | +++ | Lister 427 | NA | unkn | unkn | √ | √ | √ | 31,20 |
| aqp2/aqp3 null | T. b. brucei | +/− | + | 2T1 | NA | unkn | unkn | √ | NP | √ | 20 |
| TbAT1-KO | T. b. brucei | ++ | ++ | Lister 427 | TGD | √ | unkn | NP | √ | √ | 14 |
| B48 | T. b. brucei | − | +/− | TbAT1-KO | in vitro, pentamidine | √ | unkn | NP | NP | √ | 16 |
| P1000 | T. b. brucei | −−− | +/− | B48 | in vitro, pentamidine | unkn | unkn | NP | NP | √ | — |
| STIB 247 | T. b. brucei | +++ | +++ | NA | NA | √ | √ | √ | √ | √ | 33,57,52 |
| STIB 247MR | T. b. brucei | +/− | − | STIB 247 | in vivo, Cymelarsan | √ | √ | NP | NP | √ | 33,57,52 |
| STIB 386 | T. b. gambiense | +++ | ++ | NA | NA | √ | √ | √ | √ | √ | 33,57,52 |
| STIB 386MR | T. b. gambiense | +/− | − | STIB 386 | in vivo, Cymelarsan | √ | √ | NP | NP | √ | 33,57,52 |
| STIB 900 | T. b. rhodesiense | +++ | +++ | NA | NA | √ | unkn | √ | unkn | unkn | 56 |
| STIB 900-M | T. b. rhodesiense | − | − | STIB 900 | in vitro, Cymelarsan | √ | unkn | NP | unkn | unkn | 56 |
| STIB 900-P | T. b. rhodesiense | − | +/− | STIB 900 | in vitro, pentamidine | √ | unkn | mutc | unkn | unkn | 56 |
√, present; NP, not present; NA, not applicable; unkn, unknown; TGD, targeted gene deletion of the TbAT1 gene creating a tbat1 null line; MR or -M, resistance induced to melaminophenyl arsenicals (melarsoprol and/or Cymelarsan); -P, resistance induced to pentamidine.
aSusceptibility to pentamidine or arsenical drugs is indicated on a relative scale as highly sensitive (+++) ranging to highly resistant to pentamidine or melaminophenyl arsenicals (−−−).
bThe clone used in this paper is not tsetse transmissible but other clones of s427 have been shown to infect tsetse flies.34
cBernhard et al.56 reported that STIB 900-P contained a wild-type TbAT1 gene; later analysis revealed that in fact the TbAT1 open reading frame contains one coding mutation (G1288C, leading to Gly430 → Arg).
The characterization of the isogenic pair s427/TbAT1-KO clearly showed loss of P2 activity to be associated with only a minor loss of susceptibility to pentamidine and MPAs, in addition to a much higher degree of resistance to diminazene.14,50 Thus, high-level MPXR is clearly a function of the loss of TbAT1 function in addition to other mutation(s).51 We investigated MPXR in the T. b. brucei 247 and T. b. gambiense 386 isogenic pairs, both generated by in vivo adaptation to Cymelarsan.33 Pentamidine cross-resistance for T. b. brucei 247 was first reported by Scott et al.52 with >50-fold pentamidine resistance in vivo for T. b. brucei 247MR relative to its parental line and we confirm here that procyclic 247MR were 74- and 755-fold resistant to pentamidine and Cymelarsan, respectively; these numbers were 90- and 83-fold for 386MR (Figure S2, available as Supplementary data at JAC Online). Neither strain displayed resistance to phenylarsine oxide (PAO). Assessment of the HAPT1 and LAPT1 transporters in both isogenic pairs confirmed that MPXR was associated with loss of HAPT1 activity: 247MR and 386MR did not express the activity whereas both parental lines did. Figure S3 (available as Supplementary data at JAC Online) illustrates this in detail for the 247 isogenic pair and Table 2 summarizes the characterization of HAPT1 and LAPT1 of the 247 and 386 isogenic lines.
Table 2.
Kinetic parameters of high-affinity and low-affinity pentamidine transport in 247 and 386 procyclics
| Pentamidine |
Propamidine | Pentamidine |
|||
|---|---|---|---|---|---|
| HAPT1 Km (μM) | HAPT1 Vmax (pmol/107 cells/s) | HAPT1 Ki (μM) | LAPT1 Km (μM) | LAPT1 Vmax (pmol/107 cells/s) | |
| STIB 247WT | 0.029 ± 0.001 | 0.008 ± 0.002 | 14 ± 2 | 49 ± 19 | 0.65 ± 0.17 |
| STIB 247MR | NP | 56 ± 19 | 0.41 ± 0.10 | ||
| STIB 386WT | 0.027 ± 0.004 | 0.007 ± 0.002 | 22 ± 6 | 46 ± 9 | 0.70 ± 0.15 |
| STIB 386MR | NP | 51 ± 2 | 1.2 ± 0.4 | ||
Uptake of [3H]pentamidine by suspensions of 107 procyclic trypanosomes was measured at 25 nM or 1 μM for the determination of parameters of high-affinity transport (HAPT1 mediated) or low-affinity transport (LAPT1 mediated), respectively. In the 247MR and 386MR strains, no high-affinity pentamidine transport was observed and transport rates were very low, with saturation only at very high concentrations of unlabelled pentamidine, consistent with uptake by LAPT1. NP, not present in these cells.
We thus analysed the AQP2-AQP3 locus of the 247 and 386 isogenic pairs and discovered that the AQP2 gene was completely absent from the 386MR line, whilst the AQP3 gene was identical to that in the 386 wild-type line. In the 247MR line, however, a chimeric gene of AQP2andAQP3 had been formed in place of both wild-type genes. This chimera is in-frame, producing a 939 bp ORF composed of the first 363 bp of AQP2 and the last 576 bp of AQP3, and is thus different from the chimera found in strain B48 (see below).
Detailed analysis of the AQP locus of the STIB 900 lines revealed that both the pentamidine- and melarsoprol-resistant lines had lost the AQP2 gene whilst retaining the AQP3 gene. The organization of the AQP2-AQP3 locus in all the various strains so far examined is shown in Figure S4 (available as Supplementary data at JAC Online). It appears that, in all cases, AQP2 is either lost completely or recombined into a chimeric gene that encodes most of the residues comprising the AQP3 selectivity filter.
Expression of AQP2 reverses high levels of pentamidine and melarsoprol resistance
We used the laboratory-generated cell line B4816 and its derivative P1000 to investigate whether expression of wild-type (WT) TbAQP2 can fully reverse the multidrug resistance phenotype of these clones. P1000 was developed by further adaptation of B48 to 1 μM pentamidine in vitro and thus its resistance phenotype is believed to be multifactorial; B48 itself was derived from the tbat1−/− strain and additionally lacks HAPT1 activity.16 Figure 1 shows the resistance profile of WT s427 and of B48 and P1000 transfected with the empty vector (EV) pHD1336 and with the same vector containing WT TbAQP2.37 The lipophilic arsenical PAO was used as a control as it has been shown to diffuse across the T. b. brucei plasma membrane,16 making its action independent of transporters and showing that the resistance phenotype is not to arsenic per se.
Figure 1.
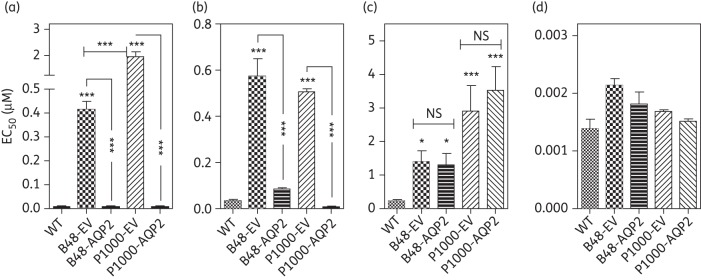
Expression of TbAQP2 in multidrug-resistant trypanosomes sensitizes to (a) pentamidine and (b) Cymelarsan but not (c) diminazene or (d) phenylarsine oxide. EC50 values were obtained using the Alamar blue assay and bars represent the mean and SEM of 3 to >10 independent determinations; data were analysed for significant differences from the wild-type control using one-way ANOVA/Tukey's test (GraphPad Prism 5.0). NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; unless otherwise indicated, relative to wild-type controls. EV, empty-vector control.
The EV controls B48 and P1000 were strongly resistant to the diamidines pentamidine and diminazene as well as to the MPA drug Cymelarsan, but not to PAO. Expression of TbAQP2 in B48 and P1000 completely reversed resistance to pentamidine (50- and 240-fold, respectively) and Cymelarsan (16.7- and 15.0-fold, respectively) in both resistant clones (Figure 1). However, the level of diminazene resistance in these lines was not affected, consistent with the lack of diminazene resistance in an aqp2 null line.20
A chimeric AQP2/3 gene in the AQP2 locus of B48 and P1000 is distributed over the cell surface and does not affect pentamidine and arsenic sensitivity
In B48, the AQP2 locus has been replaced by a chimeric in-frame fusion of AQP2 and AQP3 (chAQP2/3) whereas AQP3 has remained unchanged.20 An identical chAQP2/3 and AQP3 locus was found in P1000, showing that the higher level of pentamidine resistance in P1000 was not due to further changes to the AQP2-AQP3 locus.
The observation that expression of WT AQP2 reverses the B48 and P1000 MPXR phenotypes (Figure 1) suggests that chAQP2/3 coincides with the loss of AQP2 function. This was further investigated by expressing either WT AQP2 or chAQP2/3 in the aqp2 null line produced from this strain.20 Expression of WT AQP2 reversed the aqp2 null phenotype, whereas expression of chAQP2/3 had much less effect on sensitivity to any of the drugs tested (Figure 2). This confirms that the chimeric form does not function like AQP2, at least with respect to pentamidine susceptibility (P > 0.05), whether it still exercises an AQP-like activity or not. We did observe a small but significant (1.4-fold; P < 0.05) difference in Cymelarsan susceptibility between the aqp2 null clone and the same cells expressing chAQP2/3, although the effect was very much smaller than the expression of WT AQP2 (4.1-fold; P < 0.001). The (over)expression of WT AQP2 in an aqp2 null or aqp2/aqp3 double-null background appeared to make the cells slightly less susceptible to diminazene, the reverse of its effect on pentamidine and Cymelarsan sensitivity. There were no significant differences between any of the lines with respect to PAO. Expression of AQP2 in the WT trypanosomes did not elicit any change in drug susceptibility (Figure 2).
Figure 2.

Expression of TbAQP2 and chAQP2/3 in aqp2 null and aqp2/aqp3 null trypanosomes. EC50 values were determined for (a) pentamidine, (b) diminazene, (c) Cymelarsan and (d) phenylarsine oxide. All data are the mean of ≥10 independent determinations. See legend of Figure 1 for details. Significance was tested relative to the wild-type control unless otherwise indicated.
Apart from the primary sequence differences between TbAQP2 and TbAQP3, their cellular location is also different, with AQP2 restricted to the flagellar pocket whereas AQP3 is present throughout the plasma membrane.20 As this could have bearing on the MPXR phenotype, a fusion gene of chAQP2/3 N-terminally coupled to GFP was constructed and introduced into the aqp2/aqp3 null strain. Expression of the fusion protein was confirmed by western blotting (Figure 3a). The localization in trypanosomes was observed directly by fluorescence microscopy. It was found that the GFP-tagged chAQP2/3, unlike AQP2, was present on the plasma membrane (Figure 3b).
Figure 3.
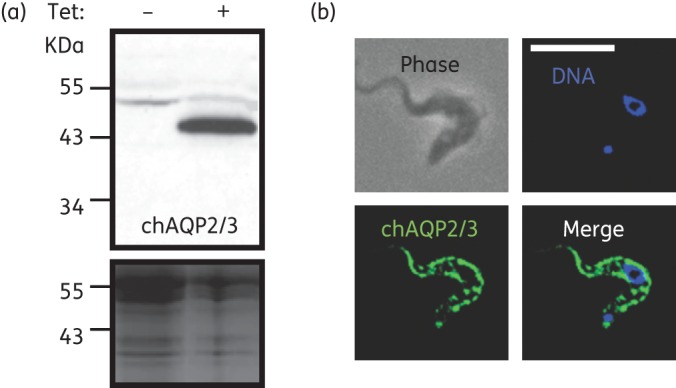
Localization of GFP-chAQP2/3 expressed in bloodstream-form aqp2/aqp3 null trypanosomes. (a) Western blot using anti-GFP antiserum as described previously.20 Blotting was performed after induction with 1 μg/mL tetracycline (+) or without induction as control (−). The Coomassie-stained panel shows relative loading. (b) GFP-chAQP2/3 was localized to the plasma membrane. Blue colour is DAPI staining of nucleus and kinetoplast. The scale bar represents 10 μm.
Expression of WT TbAQP2 correlates with the HAPT1 pentamidine transport activity
The strong phenotype of pentamidine resistance in aqp2 null lines (16.3-fold; n = 22; P < 0.001) prompted us to investigate the uptake of [3H]pentamidine in these cells and compare this with that in the parental 2T1 cells. We chose a [3H]pentamidine concentration of 30 nM in the presence of 1 mM adenosine; this concentration of adenosine fully blocks the P2 aminopurine transporter53 and the 30 nM label concentration produces a biphasic uptake that visualizes uptake through both high- and low-affinity pentamidine transporters (HAPT1 and LAPT1).16 Propamidine was used as a specific inhibitor of HAPT1, having no effect on LAPT1 activity.17 In four independent experiments in triplicate, [3H]pentamidine uptake was assessed, in parallel, in WT and in aqp2 null cells.
As expected in control trypanosomes, inhibition of the uptake of [3H]pentamidine with unlabelled pentamidine in the range 10 nM–1 mM produced a biphasic curve, of which only the first (high-affinity) phase was sensitive to inhibition by propamidine (IC50 = 29.9 ± 4.3 μM; n = 3). Plotting the pentamidine inhibition data to an equation for a biphasic sigmoidal curve (two-site inhibition; GraphPad Prism 5.0) yielded mean IC50 values that were entirely compatible with the HAPT1/LAPT1 system: 0.060 ± 0.017 and 29.9 ± 4.3 μM (n = 3), respectively (Figure 4). In the aqp2 null cells, [3H]pentamidine uptake was much lower, completely lacked the high-affinity inhibition phase with unlabelled pentamidine and was insensitive to propamidine (Figure 4). The mean IC50 value of 33.4 ± 9.5 μM was statistically identical to the lower-affinity phase of the WT 2T1 cells and with the published pentamidine Km for LAPT1 (56.2 ± 8.3 μM).17 We conclude that aqp2 null cells do not express HAPT1, but do express LAPT1.
Figure 4.
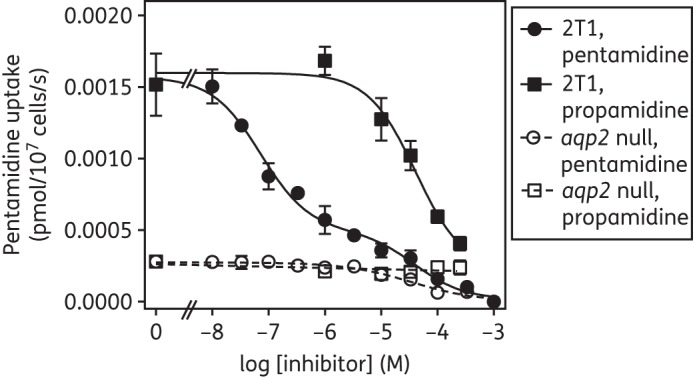
Uptake of 30 nM [3H]pentamidine by aqp2 null and wild-type control cells. Cells of T. b. brucei strain 2T1 (closed symbols) or the derived aqp2 null strain (open symbols) were incubated with [3H]pentamidine for 90 s in the presence or absence of various concentrations of unlabelled pentamidine (circles) or propamidine (squares). The incubation was stopped by the addition of 1 mL ice-cold 1 mM pentamidine and immediate centrifugation through oil. The experiment shown is representative of four identical but independent experiments, each performed in triplicate and showing virtually identical outcomes. Data points are mean ± SEM of triplicates.
We previously reported that the pentamidine-resistant clone B48 similarly lacks HAPT1 pentamidine transport activity as a result of in vitro resistance selection with pentamidine16 and that this clonal line lacks a WT AQP2 gene.20 We therefore tested whether introduction of wild-type TbAQP2, in addition to restoring pentamidine susceptibility (Figure 1), would reinstate HAPT1 activity. Figure 5 shows the rates of uptake at [3H]pentamidine concentrations that favour uptake by HAPT1 (50 nM; Figure 5a) or LAPT1 (1 μM; Figure 5b); an s427-based cell line from which the TbAT1/P2 transporter was removed by homologous recombination (TbAT1-KO)14 was used as the control, as this was the strain B48 was derived from.16 Consistent with earlier findings, uptake of 50 nM [3H]pentamidine was reduced 15.5-fold in B48 compared with in the TbAT1-KO control (P < 0.001), whereas uptake of 1 μM pentamidine was not significantly different. Expression of TbAQP2 in B48 increased uptake of 50 nM pentamidine 27.7-fold (P < 0.02; Figure 5a) to a level that appeared somewhat higher than that in TbAT1-KO, although that difference did not reach statistical significance. Uptake of 1 μM pentamidine was also increased upon introduction of TbAQP2, but by only 2.5-fold (P < 0.02; Figure 5b) and again was not significantly different from the level in TbAT1-KO. These data confirm that expression of TbAQP2 in B48 restores the pentamidine transport conditions of TbAT1-KO, completely reversing transport-related resistance.
Figure 5.

[3H]pentamidine uptake by various s427-derived trypanosome lines. (a) [3H]pentamidine concentration was 50 nM, reflecting predominantly HAPT1-mediated uptake. (b) [3H]pentamidine concentration was 1 μM, reflecting predominantly LAPT1 uptake. Rates were determined from the slopes of time courses over 10 min with timepoints at 0, 1, 3, 5, 7.5 and 10 min. Lines were linear, with none of the lines showing significant deviation from linearity in a runs test (GraphPad Prism 5.0). Slopes of control time courses in the presence of 1 mM unlabelled pentamidine were all non-significantly different from zero (F-test, GraphPad Prism 5.0), whereas uptake with unlabelled inhibitor was highly significantly different from zero (typically P < 0.0001). Results are the mean of three to four independent experiments, each performed in triplicate. (c, d) Uptake of [3H]pentamidine in bloodstream forms of B48/AQP2 was determined as described in the legend to Figure 4. The experiments shown are representative of three independently performed replicates. (c) Uptake of 30 nM [3H]pentamidine. (d) Uptake of 1 μM [3H]pentamidine. Solid squares, coincubation with various concentrations of unlabelled pentamidine; open circles, coincubation with propamidine. **P < 0.01; ***P < 0.001.
We next investigated whether the increased pentamidine uptake rates were indeed due to the expression of a HAPT1 activity in B48 cells re-expressing AQP2. Km and Vmax values of HAPT1 and LAPT1 were determined in B48/AQP2 (Figure 5c and d) and compared with previously obtained values for WT s427, B48 and TbAT1-KO (Table 3). No clear or significant differences were observed with the kinetic parameters of pentamidine transport in bloodstream forms of other s427-derived strains (WT s427, TbAT1-KO and B48), including P1000, for which the pentamidine Km and Vmax values (Figure S5; available as Supplementary data at JAC Online) were also added to Table 3. We conclude that expression of TbAQP2 reinstated a high-affinity pentamidine transport activity that is indistinguishable from the well-characterized HAPT1, whilst making no detectable change to the LAPT1 activity.
Table 3.
Kinetic parameters of high-affinity and low-affinity pentamidine transport in various strains of T. b. brucei bloodstream forms and in Leishmania mexicana expressing TbAQP2
| Strain | HAPT1 pentamidine Km (μM) | HAPT1 propamidine Ki (μM) | HAPT1 diminazene Ki (μM) | LAPT1 pentamidine Km (μM) |
|---|---|---|---|---|
| WT s427a | 0.036 ± 0.006 | 4.6 ± 0.7 | 63 ± 3 | 56 ± 8 |
| TbAT1-KOb | 0.029 ± 0.008 | 13.0 ± 3.0 | ND | 50 ± 17 |
| B48c | NP | 56 ± 7 | ||
| P1000 | NP | 99 ± 24 | ||
| B48/AQP2 | 0.046 ± 0.014 | 15.2 ± 1.6 | ND | 66 ± 1 |
| L. mexicana/TbAQP2 | 0.055 ± 0.004 | 8.1 ± 0.8 | 100 ± 21 | not present in these cells |
TbAQP2 displays HAPT1 activity when expressed in L. mexicana
Leishmania spp. are relatively insusceptible to pentamidine and tolerant to MPAs, as they lack high-affinity uptake systems for these drugs. In order to investigate whether T. b. brucei AQPs directly mediate uptake of these drugs, we expressed TbAQP2 and TbAQP3 in promastigotes of L. mexicana. Comparisons of control cells (transfected with EV) and cells expressing TbAQP2 showed 40-fold sensitization in two independent clones (Figure 6a) and a >1000-fold sensitization to MPAs (Figure 6b). There was no statistically significant effect on sensitivity to diminazene, relative to the EV control (Figure 6c), or to amphotericin B (Figure 6d); the latter was used as a control antileishmanial drug thought to bind to plasma membrane ergosterol, thereby forming pores.54 Expression of TbAQP3 in two independent clones led to minor (1.5–2.5-fold) sensitization to pentamidine.
Figure 6.
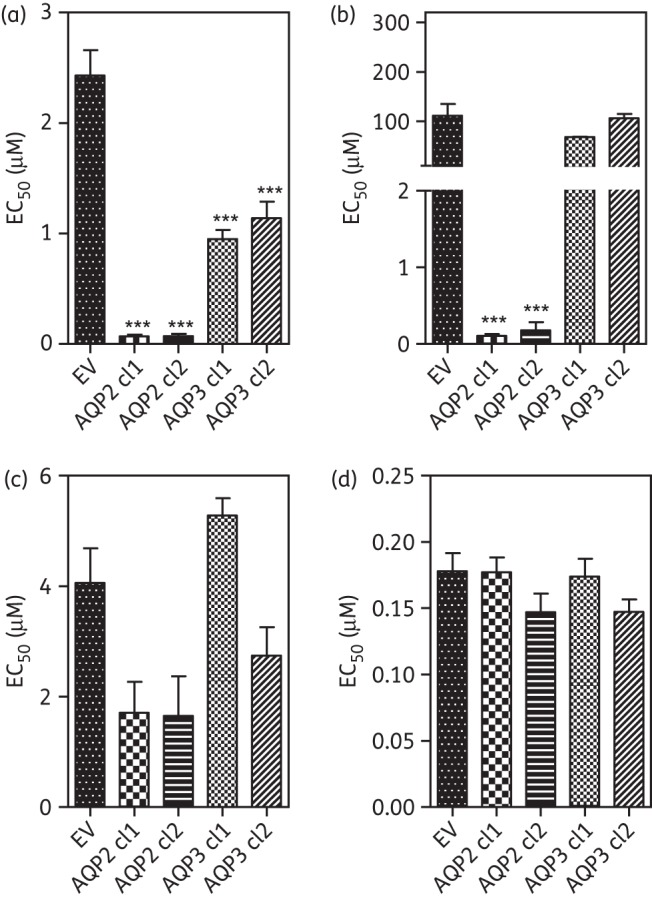
Effect of expression of T. brucei aquaporins on drug susceptibility of Leishmania mexicana. TbAQP2 and TbAQP3 were expressed in promastigotes using the pNUS vector. Two independent clones of each resulting cell line and the promastigotes transfected with the ‘empty’ pNUS vector were tested for sensitivity to (a) pentamidine, (b) Cymelarsan, (c) diminazene and (d) amphotericin B using the Alamar blue fluorimetric assay. Bars are means of three to eight independent determinations; error bars are SEM. ***P < 0.001 by one-way ANOVA with Tukey's correction (GraphPad Prism 5.0). EV, empty-vector control.
Consistent with this minor effect of AQP3 expression, uptake of 100 nM [3H]pentamidine in TbAQP3-transfected promastigotes was not significantly different from that in the control when measured over 5 min (Figure 7a and data not shown). In contrast, pentamidine uptake in TbAQP2-expressing cells was increased almost 15-fold (Figure 7a). Indeed, uptake of very low concentrations of [3H]pentamidine (50 nM) was measurable in TbAQP2-expressing promastigotes within 3 s, was linear with a rate of 0.032 ± 0.002 pmol/107 cells/s and completely saturable with excess unlabelled pentamidine (Figure 7b). Competition with unlabelled pentamidine, propamidine and diminazene was dose dependent (Figure 7c) and followed Michaelis–Menten kinetics (Figure 7d). Mean Km and Ki values for these inhibitors are listed in Table 3 and show a kinetic profile entirely consistent with the T. b. brucei HAPT1 activity, with an apparent Km value of 55 ± 4 nM (n = 5).
Figure 7.

Expression of T. b. brucei aquaporins in promastigotes of Leishmania mexicana. (a) Specific uptake of 100 nM [3H]pentamidine over 5 min in L. mexicana promastigotes transfected with empty pNUS vector (control), or promastigotes transfected with TbAQP2 or with TbAQP3. In each case, mediated uptake of 100 nM radiolabel was compared with total association of [3H]pentamidine with the cell pellet in the presence of a saturating concentration (1 mM) of unlabelled pentamidine. The data shown are the mean of triplicates ± SEM. ***P < 0.001 by one-way ANOVA, compared with all other datasets. (b) Time course of 50 nM [3H]pentamidine uptake, over 15 s, using L. mexicana promastigotes transformed with TbAQP2 in the presence and absence of 1 mM unlabelled pentamidine. Uptake at 50 nM pentamidine was linear (r2 = 0.98) and rapid (0.032 ± 0.002 pmol/107 cells/s, compared with 0.00026 ± 1.8 × 10−6 pmol/107 cells/s in the presence of 1 mM pentamidine). (c) Characterization of 20 nM [3H]pentamidine uptake in L. mexicana promastigotes expressing TbAQP2, in the presence of varying concentrations of unlabelled inhibitor. (d) Michaelis–Menten plot of 20 nM [3H]pentamidine uptake; conversion of pentamidine inhibition plot in (c).
TbAQP2 does not affect the activity of the P2 transporter
It has long been established that the P2 transport activity, encoded by TbAT1, is an important determinant of diamidine and arsenical sensitivity in T. brucei.8,12 It is thus important to establish whether the TbAQP2/HAPT1 activity has any effect on P2 activity. It has been previously reported that the expression of P2 can be assessed through susceptibility to some adenosine analogues.55 Indeed, we found that susceptibility to tubercidin (7-deazaadenosine) decreased 10-fold in the tbat1−/− strain (EC50 = 2.65 ± 0.23 μM) relative to s427 WT (0.23 ± 0.03 μM; P < 0.001; Student's t-test). Similarly, susceptibility to tubercidin, cordycepin (3′-deoxyadenosine) and 5′-deoxyadenosine increased >20-fold when TbAT1 was expressed in the tbat1−/− strain B48 (P < 0.001) (Figure S6; available as Supplementary data at JAC Online), confirming susceptibility to cytotoxic adenosine analogues as a sensitive indicator for TbAT1 expression levels. However, the susceptibility of 2T1 WT and aqp2 null cells to these same adenosine analogues was identical, as was the susceptibility of B48 + TbAQP2 versus control B48 cells (Figure S6, available as Supplementary data at JAC Online). Susceptibility to pentamidine, used as a positive control, was highly dependent on the expression of TbAQP2 and/or TbAT1. We conclude that TbAQP2 does not regulate P2 activity and is not itself a conduit for cytotoxic nucleosides.
Investigation of the TbAQP2 and TbAQP3 selectivity filters as the determinant for MPXR
The TbAQP2 region that was replaced with the corresponding TbAQP3 sequence in the recombination event that generated chAQP2/3 contains most of the selectivity filter that is believed to determine the distinct permeation profiles of AQPs.20 As the chimeric AQPs found in B48 and 247MR did not appear to have the TbAQP2 functionality with respect to pentamidine and melarsoprol susceptibility, we investigated whether MPXR is determined principally by the few amino acids of the selectivity filter. We used synthetic genes of chAQP2/3 and of TbAQP3 with a TbAQP2 selectivity filter (chAQP2/3sf2 and AQP3sf2, respectively; alignment in Figure S7, available as Supplementary data at JAC Online). These were expressed in the aqp2/aqp3 null cell line. Analysis of the drug susceptibility phenotype for the resultant lines showed that the transplantation of the amino acid residues of the AQP2 selectivity filter did not result in an AQP2 phenotype with respect to drug susceptibility; there was only a minor increase in susceptibility and only to pentamidine (EC50 values of 158 ± 2 and 131 ± 3 nM, P < 0.02, for aqp2/aqp3 null and AQP3sf2, respectively; Figure 8). In contrast, the cell line expressing the chAQP2/3sf2 construct was significantly more susceptible to Cymelarsan than the aqp2/aqp3 null background, reaching susceptibility to this drug halfway between the aqp2/aqp3 null and the same line expressing WT AQP2 (Figure 8). These findings indicate that the selectivity filter residues do play a role in arsenical drug sensitivity, but that the changes described here were insufficient to produce the same effect as WT AQP2 on pentamidine susceptibility.
Figure 8.

Expression of synthetic aquaporin constructs in bloodstream forms of the aqp2/aqp3 double-null line. EC50 values were determined for (a) pentamidine, (b) Cymelarsan, (c) diminazene and (d) phenylarsine oxide. Expression was induced by the addition of 1 μg/mL tetracycline 24 h before setting up the plates for the assay, followed by 48 h of incubation of the cells in the presence of a doubling dilution of the test compound followed by a further 24 h of incubation with the dye before fluorescence was determined. All data are the mean of at least three independent determinations; error bars are SEM. Statistical significance with aqp2/aqp3 null was determined using a one-way ANOVA with Tukey's correction (GraphPad Prism 5.0); **P < 0.02; ***P < 0.01. In the Cymelarsan panel (b), it is also indicated that Groups 4 and 5 are significantly different from Group 3.
Discussion
Although we recently reported that the absence of a wild-type AQP2 gene correlates with MPXR in T. b. brucei,20 many important questions remained. It remained unclear (i) whether this phenomenon is relevant for human-infective trypanosome subspecies, (ii) whether loss of AQP2 occurs as a result of in vivo drug exposure, (iii) whether loss of AQP2 alone is sufficient for high levels of pentamidine and melarsoprol resistance, (iv) by which mechanism AQP2 facilitated pentamidine and melarsoprol sensitivity and (v) whether AQP2 has any impact on TbAT1/P2 activity. We now provide answers to these questions.
Only TbAQP2 was implicated in MPXR, with the expression of TbAQP3 in an aqp2/aqp3 null line having no effect on drug susceptibility.20 This is most likely the result of differences in permeation, as all three TbAQPs are located on the cell surface, although AQP2 was found to be restricted to the flagellar pocket rather than dispersed over the plasma membrane.20,28 Interestingly, a chimeric AQP, made up from TbAQP2 and TbAQP3 (chAQP2/3) was present instead of wild-type TbAQP2 in the highly MPXR strain T. b. brucei B48, which retained a wild-type copy of TbAQP3 found in the parental, drug-susceptible strain.20 Here, we report that in other isogenic MPXR pairs, the adapted strain similarly showed a replacement of TbAQP2 with a different chimeric AQP2/3 fusion or an outright deletion of TbAQP2. This was the case whether the strain was originally adapted to pentamidine or to an MPA drug, whether the subspecies was T. b. brucei, T. b. gambiense or T. b. rhodesiense and for strains that were selected by either in vivo or in vitro drug pressure. It thus follows that for T. brucei subspecies, loss of TbAQP2 function represents an important component of acquiring high levels of resistance to MPAs and that this necessarily leads to pentamidine resistance as well, and vice versa. We also conclude that loss of AQP2 can be the result of in vivo drug pressure and that this does not significantly affect virulence and does not impede transmission by tsetse flies, as STIB 386MR and STIB 247MR produce similar parasitaemias in mice compared with the wild-type lines from which they were derived and were fly transmissible.33
One important question is whether loss of TbAQP2 alone is sufficient for MPXR in vivo. Certainly, the deletion of just TbAQP2 rendered a highly drug-susceptible T. b. brucei strain resistant to pentamidine (but not diminazene) and Cymelarsan (but not PAO) in vitro;20 this shows that the resistance is not to all aromatic diamidines or to arsenic per se. Similarly, the reintroduction of wild-type TbAQP2 into B48 or P1000 restored sensitivity to Cymelarsan and pentamidine (but not diminazene) to wild-type levels. Both strains retained their diminazene resistance, which is linked to the loss of the TbAT1/P2 transporter,14,50 which is absent in B48 and P1000 cells.16 Indeed, TbAT1 is absent or mutated in all the drug-adapted MPXR strains used here,56,57 opening up the possibility that both transport activities must be absent for high levels of MPXR. It has already been established that homozygous deletion of TbAT1 alone results in a relatively minor (2.5–3-fold) loss of sensitivity to pentamidine and MPAs;14 a similar level of MPA resistance was observed in the aqp2 null and aqp2/aqp3 null lines, which do retain a wild-type copy of TbAT1. Although the data presented here and by Baker et al.20 show that the deletion of TbAQP2 alone is sufficient to give a strong pentamidine resistance phenotype in vitro (17.5-fold; Figure 2), it is worth exploring this in detail. Firstly, TbAT1/P2 activity is very low in cultured T. b. brucei bloodstream forms, compared with expression in vivo,15 dramatically reducing the influence of this transporter on drug sensitivity. Secondly, the pHD1336 vector used to reintroduce TbAQP2 in B48 and P1000 gives a very robust expression level,37 well above that in the wild-type, which helps explain the complete reversal of MPA/pentamidine resistance even in the absence of TbAT1. Finally, the reintroduction of TbAT1 in B48 in the same vector also led to an almost complete reversal of resistance to pentamidine, Cymelarsan and diminazene, even in the continued absence of a wild-type TbAQP2.58
The conclusion from the above must be that loss of either TbAQP2 or TbAT1 activity can lead to some loss of MPA and pentamidine susceptibility, but that the high MPXR phenotype is the result of both being lost concomitantly. B48, which lacks both genes, is more resistant to pentamidine and MPAs than either aqp2 null or the TbAT1 knockout strain from which it was derived. Thus, the combined data strongly suggest that TbAQP2 is the previously described HAPT1 and transports MPAs and pentamidine. A number of observations reported in the present article strongly support this notion: (i) the T. b. brucei strains B48, P1000 and 247MR and T. b. gambiense 386MR have all lost HAPT1 activity and also lack a wild-type copy of TbAQP2; (ii) targeted deletion of TbAQP2 specifically removes the high-affinity pentamidine transport component, leaving the low-affinity transport activity unchanged; (iii) expression of TbAQP2 in B48 restores HAPT1 activity; (iv) expression of TbAQP2 in L. mexicana causes a massive sensitization to pentamidine and Cymelarsan; and (v) the introduction of a high-affinity pentamidine transporter that is indistinguishable from HAPT1 by Km value and inhibitor profile. Thus, the evidence overwhelmingly supports a model that TbAQP2 mediates the saturable uptake of pentamidine and MPAs, following standard Michaelis–Menten kinetics, in addition to its more conventional function as a water/glycerol channel.
As discussed by Baker et al.,20 TbAQP2 has a number of unusual residues in the motifs that are believed to be involved in selectivity: NPS/IVLL is replaced with the classical regions of NPA/IGYR in the chimera. To begin to unravel the structural determinants of AQP2 action, we synthesized two new genes, with the NPS/IVLL motif transplanted to either chAQP2/3 or TbAQP3, and expressed these constructs in an aqp2/aqp3 null strain. The resulting cell lines displayed slightly higher pentamidine sensitivity than the control and the cells expressing chAQP2/3sf2 showed a substantial sensitization to Cymelarsan. It follows that the usual selectivity filter of TbAQP2 does indeed contribute to its drug transport capacity, but is not the sole determinant.
In summary, we have shown that an aquaglyceroporin, TbAQP2, mediates the saturable uptake of some first-line trypanocides, Cymelarsan and pentamidine, compounds of substantially higher molecular weight than has so far been reported for any AQP. The mechanism by which this MIP transports these drugs with Michaelis–Menten kinetics remains to be investigated.
Funding
This work was funded by a grant to H. P. dK., M. P. B. and R. J. S. B. from the Medical Research Council (84733) and a grant to D. H. from The Wellcome Trust (093010/Z/10/Z) and by the Medical Research Council (MR/K000500/1). D. H. is also supported by a Wellcome Trust Senior Investigator Award (100320/Z/12/Z) and by the Medical Research Council (MR/K000500/1). A. A. E. was supported by a British Commonwealth scholarship. N. B. was supported by a Bloomsbury Colleges studentship. F. E. G. was supported by the Swiss National Science Foundation (31003A_135746), P. L. by the Emilia Guggenheim-Schnurr Foundation, the Freiwillige Akademische Gesellschaft Basel, and the Mathieu Foundation of the University of Basel.
Transparency declarations
None to declare.
Supplementary data
Table S1 and Figures S1–S7 are available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
Acknowledgements
We gratefully acknowledge a 10 Giga-base sequencing award from Roche. This work was supported by the Wellcome Trust. The Wellcome Trust Centre for Molecular Parasitology is supported by core funding from the Wellcome Trust (085349). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1.Franco JR, Simarro PP, Diarra A, et al. Monitoring the use of nifurtimox-eflornithine combination therapy (NECT) in the treatment of second stage gambiense human African trypanosomiasis. Res Rep Trop Med. 2012;3:93–101. doi: 10.2147/RRTM.S34399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delespaux V, De Koning HP. Drugs and drug resistance in African trypanosomiasis. Drug Resist Update. 2007;10:30–50. doi: 10.1016/j.drup.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Brun R, Schumacher R, Schmid C, et al. The phenomenon of treatment failures in human African trypanosomiasis. Trop Med Int Health. 2001;6:906–14. doi: 10.1046/j.1365-3156.2001.00775.x. [DOI] [PubMed] [Google Scholar]
- 4.Rollo IM, Williamson J. Acquired resistance to ‘melarsen’, tryparsamide and amidines in pathogenic trypanosomes after treatment with ‘melarsen’ alone. Nature. 1951;167:147–8. doi: 10.1038/167147a0. [DOI] [PubMed] [Google Scholar]
- 5.Damper D, Patton CL. Pentamidine transport and sensitivity in brucei-group trypanosomes. J Protozool. 1976;23:349–56. doi: 10.1111/j.1550-7408.1976.tb03787.x. [DOI] [PubMed] [Google Scholar]
- 6.Frommel TO, Balber AE. Flow cytofluorimetric analysis of drug accumulation by multidrug-resistant Trypanosoma brucei brucei and T. b. rhodesiense. Mol Biochem Parasitol. 1987;26:183–91. doi: 10.1016/0166-6851(87)90142-3. [DOI] [PubMed] [Google Scholar]
- 7.De Koning HP. Transporters in African trypanosomes: role in drug action and resistance. Int J Parasitol. 2001;31:512–22. doi: 10.1016/s0020-7519(01)00167-9. [DOI] [PubMed] [Google Scholar]
- 8.Carter NS, Fairlamb AH. Arsenical-resistant trypanosomes lack an unusual adenosine transporter. Nature. 1993;361:173–6. doi: 10.1038/361173a0. [DOI] [PubMed] [Google Scholar]
- 9.Carter NS, Berger BJ, Fairlamb AH. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J Biol Chem. 1995;270:28153–7. doi: 10.1074/jbc.270.47.28153. [DOI] [PubMed] [Google Scholar]
- 10.Barrett MP, Zhang ZQ, Denise H, et al. A diamidine-resistant Trypanosoma equiperdum clone contains a P2 purine transporter with reduced substrate affinity. Mol Biochem Parasitol. 1995;73:223–9. doi: 10.1016/0166-6851(95)00120-p. [DOI] [PubMed] [Google Scholar]
- 11.Carter NS, Barrett MP, De Koning HP. A drug resistance determinant from Trypanosoma brucei. Trends Microbiol. 1999;7:469–71. doi: 10.1016/s0966-842x(99)01643-1. [DOI] [PubMed] [Google Scholar]
- 12.Mäser P, Sutterlin C, Kralli A, et al. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science. 1999;285:242–4. doi: 10.1126/science.285.5425.242. [DOI] [PubMed] [Google Scholar]
- 13.De Koning HP, Bridges DJ, Burchmore R. Purine transporters of protozoa: from biology to therapy. FEMS Microbiol Rev. 2005;29:987–1020. doi: 10.1016/j.femsre.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Matovu E, Stewart M, Geiser F, et al. The mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot Cell. 2003;2:1003–8. doi: 10.1128/EC.2.5.1003-1008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward CP, Wong PE, Burchmore RJ, et al. Trypanocidal furamidine analogues: influence of pyridine nitrogens on trypanocidal activity, transport kinetics and resistance patterns. Antimicrob Agents Chemother. 2011;55:2352–61. doi: 10.1128/AAC.01551-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges D, Gould MK, Nerima B, et al. Loss of the high affinity pentamidine transporter is responsible for high levels of cross-resistance between arsenical and diamidine drugs in African trypanosomes. Mol Pharmacol. 2007;71:1098–108. doi: 10.1124/mol.106.031351. [DOI] [PubMed] [Google Scholar]
- 17.De Koning HP. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters. Implications for cross resistance with arsenicals. Mol Pharmacol. 2001;59:586–92. doi: 10.1124/mol.59.3.586. [DOI] [PubMed] [Google Scholar]
- 18.De Koning HP, Jarvis SM. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by the P2 adenosine transporter and at least one novel, unrelated transporter. Acta Trop. 2001;80:245–50. doi: 10.1016/s0001-706x(01)00177-2. [DOI] [PubMed] [Google Scholar]
- 19.Bray PG, Barrett MP, Ward SA, et al. Pentamidine uptake and resistance in pathogenic protozoa. Trends Parasitol. 2003;19:232–9. doi: 10.1016/s1471-4922(03)00069-2. [DOI] [PubMed] [Google Scholar]
- 20.Baker N, Glover L, Aguinaga A, et al. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc Natl Acad Sci USA. 2012;109:10996–1001. doi: 10.1073/pnas.1202885109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benga G. Water channel proteins (later called aquaporins) and relatives: past, present, and future. IUBMB Life. 2009;61:112–33. doi: 10.1002/iub.156. [DOI] [PubMed] [Google Scholar]
- 22.Verkman AS. Aquaporins in clinical medicine. Annu Rev Med. 2012;63:303–16. doi: 10.1146/annurev-med-043010-193843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber VJ, Tsujita M, Nakada T. Aquaporins in drug discovery and pharmacotherapy. Mol Aspects Med. 2012;33:691–703. doi: 10.1016/j.mam.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Wu B, Beitz E. Aquaporins with selectivity for unconventional permeants. Cell Mol Life Sci. 2007;64:2413–21. doi: 10.1007/s00018-007-7163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Shen J, Carbrey JM, et al. Arsenite transport by mammalian aquaglyceroporins AQP7 and AQP9. Proc Natl Acad Sci USA. 2002;99:6053–8. doi: 10.1073/pnas.092131899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukhopadhyay R, Mandal G, Atluri VS, et al. The role of alanine 163 in solute permeability of Leishmania major aquaglyceroporin LmAQP1. Mol Biochem Parasitol. 2011;175:83–90. doi: 10.1016/j.molbiopara.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uzcategui NL, Szallies A, Pavlovic-Djuranovic S, et al. Cloning, heterologous expression, and characterization of three aquaglyceroporins from Trypanosoma brucei. J Biol Chem. 2004;279:42669–76. doi: 10.1074/jbc.M404518200. [DOI] [PubMed] [Google Scholar]
- 28.Bassarak B, Uzcátegui NL, Schönfeld C, et al. Functional characterization of three aquaglyceroporins from Trypanosoma brucei in osmoregulation and glycerol transport. Cell Physiol Biochem. 2011;27:411–20. doi: 10.1159/000327968. [DOI] [PubMed] [Google Scholar]
- 29.Baker N, De Koning HP, Mäser P, et al. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol. 2013;29:110–8. doi: 10.1016/j.pt.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsukaguchi H, Weremowicz S, Morton CC, et al. Functional and molecular characterization of the human neutral solute channel aquaporin-9. Am J Physiol. 1999;277:F685–96. doi: 10.1152/ajprenal.1999.277.5.F685. [DOI] [PubMed] [Google Scholar]
- 31.Alsford S, Horn D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol Biochem Parasitol. 2008;161:76–9. doi: 10.1016/j.molbiopara.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alsford S, Eckert S, Baker N, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2012;482:232–6. doi: 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott AG, Tait A, Turner CMR. Characterisation of cloned lines of Trypanosoma brucei expressing stable resistance to MelCy and Suramin. Acta Trop. 1996;60:251–62. doi: 10.1016/0001-706x(96)00131-3. [DOI] [PubMed] [Google Scholar]
- 34.Peacock L, Ferris VR, Bailey M, et al. Fly transmission and mating of Trypanosoma brucei brucei strain 427. Mol Biochem Parasitol. 2008;160:100–6. doi: 10.1016/j.molbiopara.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Salabi MI, De Koning HP. Purine nucleobase transport in amastigotes of Leishmania mexicana: involvement in allopurinol uptake. Antimicrob Agents Chemother. 2005;49:3682–9. doi: 10.1128/AAC.49.9.3682-3689.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Salabi MI, Wallace LJM, De Koning HP. A Leishmania major nucleobase transporter responsible for allopurinol uptake is a functional homologue of the Trypanosoma brucei H2 transporter. Mol Pharmacol. 2003;63:814–20. doi: 10.1124/mol.63.4.814. [DOI] [PubMed] [Google Scholar]
- 37.Biebinger S, Wirtz LE, Lorenz P, et al. Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol Biochem Parasitol. 1997;85:99–112. doi: 10.1016/s0166-6851(96)02815-0. [DOI] [PubMed] [Google Scholar]
- 38.Alsford S, Kawahara T, Glover L, et al. Tagging a T. brucei RRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol Biochem Parasitol. 2005;144:142–8. doi: 10.1016/j.molbiopara.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burkard G, Fragoso CM, Roditi I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol. 2007;153:220–3. doi: 10.1016/j.molbiopara.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Berriman M, Ghedin E, Hertz-Fowler C, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–22. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carver T, Harris SR, Berriman M, et al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–9. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Salabi MI, Wallace LJM, Lüscher A, et al. Molecular interactions underlying the unusually high affinity of a novel Trypanosoma brucei nucleoside transporter. Mol Pharmacol. 2007;71:921–9. doi: 10.1124/mol.106.031559. [DOI] [PubMed] [Google Scholar]
- 44.Natto M, Wallace LJM, Candlish D, et al. Trypanosoma brucei: expression of multiple purine transporters prevents the development of allopurinol resistance. Exp Parasitol. 2005;109:80–6. doi: 10.1016/j.exppara.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Wallace LJM, Candlish D, De Koning HP. Different substrate recognition motifs of human and trypanosome nucleobase transporters: selective uptake of purine antimetabolites. J Biol Chem. 2002;277:26149–56. doi: 10.1074/jbc.M202835200. [DOI] [PubMed] [Google Scholar]
- 46.Papageorgiou IG, Yakob L, Al Salabi MI, et al. Identification of the first pyrimidine nucleobase transporter in Leishmania: similarities with the Trypanosoma brucei U1 transporter and antileishmanial activity of uracil analogues. Parasitology. 2005;130:275–83. doi: 10.1017/s0031182004006626. [DOI] [PubMed] [Google Scholar]
- 47.Gould MK, Vu XL, Seebeck T, et al. Propidium iodide-based methods for monitoring drug action in the kinetoplastidae: comparison with the Alamar blue assay. Anal Biochem. 2008;382:87–93. doi: 10.1016/j.ab.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 48.Ibrahim HM, Al-Salabi MI, El Sabbagh N, et al. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J Antimicrob Chemother. 2011;66:111–25. doi: 10.1093/jac/dkq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tetaud E, Lecuix I, Sheldrake T, et al. A new expression vector for Crithidia fasciculata and Leishmania. Mol Biochem Parasitol. 2002;120:195–204. doi: 10.1016/s0166-6851(02)00002-6. [DOI] [PubMed] [Google Scholar]
- 50.De Koning HP, Stewart M, Anderson L, et al. The trypanocide diminazene aceturate is accumulated predominantly through the TbAT1 purine transporter; additional insights in diamidine resistance in African trypanosomes. Antimicrob Agents Chemother. 2004;48:1515–9. doi: 10.1128/AAC.48.5.1515-1519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Koning HP. The ever-increasing complexities of arsenical-diamidine cross-resistance in African trypanosomes. Trends Parasitol. 2008;24:345–9. doi: 10.1016/j.pt.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 52.Scott AG, Tait A, Turner CMR. Trypanosoma brucei: lack of cross-resistance to melarsoprol in vitro by cymelarsan-resistant parasites. Exp Parasitol. 1997;86:181–90. doi: 10.1006/expr.1997.4167. [DOI] [PubMed] [Google Scholar]
- 53.De Koning HP, Jarvis SM. Adenosine transporters in bloodstream forms of T. b. brucei: substrate recognition motifs and affinity for trypanocidal drugs. Mol Pharmacol. 1999;56:1162–70. doi: 10.1124/mol.56.6.1162. [DOI] [PubMed] [Google Scholar]
- 54.Chattopadhyay A, Jafurulla M. A novel mechanism for an old drug: amphotericin B in the treatment of visceral leishmaniasis. Biochem Biophys Res Commun. 2011;416:7–12. doi: 10.1016/j.bbrc.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 55.Geiser F, Lüscher A, De Koning HP, et al. Molecular pharmacology of adenosine transport in Trypanosoma brucei: P1/P2 revisited. Mol Pharmacol. 2005;68:589–95. doi: 10.1124/mol.104.010298. [DOI] [PubMed] [Google Scholar]
- 56.Bernhard SC, Nerima B, Mäser P, et al. Melarsoprol- and pentamidine-resistant Trypanosoma brucei rhodesiense populations and their cross-resistance. Int J Parasitol. 2007;37:1443–8. doi: 10.1016/j.ijpara.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 57.Stewart ML, Burchmore RJS, Clucas C, et al. Multiple genetic mechanisms lead to the loss of functional TbAT1 expression in drug resistant trypanosomes. Eukaryot Cell. 2010;9:336–43. doi: 10.1128/EC.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munday JC, Rojas López KE, Eze AA, et al. Functional expression of TcoAT1 reveals it to be a P1-type nucleoside transporter with no capacity for diminazene uptake. Int J Parasitol Drugs Drug Resist. 2013;3:69–76. doi: 10.1016/j.ijpddr.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.