

Abstract
An important function of steroidogenic cytochromes P450 is the transformation of cholesterol to produce androgens, estrogens, and the cortico-steroids. The activities of cytochrome P450c17 (CYP17) are essential in sex hormone biosynthesis, with severe developmental defects being a consequence of deficiency or mutations. The first reaction catalyzed by this multifunctional P450 is the 17α-hydroxylation of pregnenolone (PREG) to 17 α -hydroxypregnenolone (17-OH PREG) and progesterone (PROG) to 17 α -hydroxyprogesterone (17-OH PROG). The hydroxylated products then are either used for production of corticoids or undergo a second CYP17 catalyzed-transformation, representing the first committed step of androgen formation. While the hydroxylation reactions are catalyzed by the well known Compound I intermediate, the lyase reaction is believed to involve nucleophilic attack of the earlier peroxo- intermediate on the C20-carbonyl. Herein resonance Raman (rR) spectroscopy reveals that substrate structure does not impact heme structure for this set of physiologically important substrates. On the other hand, rR spectra obtained here for the ferrous CO adducts with these four substrates show that substrates do interact differently with the Fe-C-O fragment, with large differences between the spectra obtained for the samples containing 17-OH PROG and 17-OH PREG, the latter providing evidence for the presence of two Fe-C-O conformers. Collectively, these results demonstrate that individual substrates can differentially impact the disposition of a heme-bound ligand, including dioxygen, altering the reactivity patterns in such a way as to promote preferred chemical conversions, thereby avoiding the profound functional consequences of unwanted side reactions.
Keywords: Resonance Raman, cytochrome P450, CYP17, nanodiscs
The cytochromes P450 are members of a family of heme containing monooxygenases, present in eukaryotic, prokaryotic, and archeal organisms that are able to catalyze a number of difficult chemical transformations. These enzymes facilitate a very wide range of physiologically important processes including metabolism of pharmaceuticals and other xenobiotics, and the production of essential endogenous biomolecules.1 In contrast to the xenobiotic metabolizing P450s which generally possess somewhat malleable distal-side pockets that can adjust to allow binding of a wide range of substrate structures, a given cytochrome P450 involved in the synthesis and modification of key endogenous biomolecules typically binds a very limited number of substrates and orchestrates multi-stage oxidative transformations on a single substrate by selectively enhancing different reactivity patterns at different stages.2
In vertebrates, one important function performed by a group of cytochromes P450 is the transformation of cholesterol to produce essential steroid hormones including androgens, estrogens and the cortico-steroids.2,3 One crucial enzyme involved in steroid hormone biosynthesis, cytochrome P450c17, also commonly known as CYP17, is expressed in steroidogenic tissues, such as gonads and adrenal glands.4,5 As outlined in Figure1, the primary reactions catalyzed by CYP17 are 17α-hydroxylation of pregnenolone (PREG) to 17-hydroxypregnenolone (17-OH PREG) and progesterone (PROG) to 17-hydroxyprogesterone (17-OH PROG). The 17-OH PREG and 17-OH PROG are further transformed in a 17,20-lyase reaction to dehydroepiandrosterone (DHEA) and androstenedione (AD), respectively, though it is emphasized that the catalytic efficiency of this transformation is 50-fold greater for 17-OH PREG than for 17-OH PROG.3,6 It is important to recognize that CYP17 reactivity represents a key branching point in human steroidogenesis as the hydroxylated products of initial reactions are either shunted towards production of corticoids or serve as a substrate for a second round of CYP17 catalyzed-transformation to generate androgens. While it is generally accepted that the hydroxylation reactions are catalyzed by the ferryl-cation radical of the heme prosthetic group (i.e., Compound I), emerging evidence suggests that the lyase reaction involves a nucleophilic attack of the peroxo- intermediate on the C20-carbonyl.
Figure 1.

Proposed pathway for biosynthesis of androstenedione and dehydroepiandrosterone from progesterone and pregnenolone.
Notable in Figure 1 are the relatively minor structural differences between the primary substrates of CYP17 that nevertheless exhibit structurally, kinetically, and mechanistically distinct interactions with this enzyme. Whether the role of substrate structure in defining these properties is passive, with the substrate “selecting” from possible alternate protein conformations, or active, inducing changes in the heme structure or protein-heme interactions, different A and D ring substituents can be viewed as a crucial factor in controlling the reactivity of CYP17. Relevant to this point is a groundbreaking recent report by DeVore and Scott of the crystal structure of CYP17 bound to a promising cancer drug.6 These authors also included results of molecular modeling procedures based on this structure suggesting that interactions of the 3β-alcohol or corresponding ketone fragments of PREG and PROG with active site H-bonding residues are among those important in defining orientation of the substrates with respect to the heme-site reactive center. While such studies provide useful insight, the authors also cautioned that the structure of the substrate obviously can impact the topology of the active site (i.e., the active role of substrate structure mentioned above) and stressed the need for application of other experimental techniques.6
While many spectroscopic techniques have been effectively applied to study heme proteins, resonance Raman (rR) has proven itself to be an especially powerful probe of these systems.7-9 Thus, high frequency marker modes respond to changes in oxidation- or spin-state of the central iron in well-established and documented ways, while low frequency modes report changes in protein interactions with the heme periphery.7-9 This is important because the presence of the propionic acid and potentially conjugated vinyl peripheral substituents have long been considered as possibly important structural determinants of heme reactivity whose influence may be sensitively manipulated by protein-heme interactions.10,11 Excitation within various ligand to metal and metal to ligand charge transfer transitions, or within the strong Soret band of the heme can lead to efficient enhancement of internal modes of Fe-N(histidine), Fe-S− (cysteine), or Fe-XY (XY = O2, NO or CO) fragments providing a very effective probe of the key linkages between the heme prosthetic group and these endogenous or exogenous ligands.7-9,12 While the power of rR spectroscopy to interrogate active site structure in heme proteins presents an especially effective approach to explore the complex mechanism of cytochromes P450, application of this method to the 57 human members of this superfamily has been impeded by their native membrane association. Fortunately, in contrast with the aggregated detergent solubilized preparations of the past, the recently developed Nanodisc system allows functional incorporation of these membrane proteins into a homogenous and monodisperse membrane environment. This native-like environment yields exceptionally well-behaved ligand binding properties, as evidenced by clean conversions of spin-state populations, and also enhances stability of their dioxygen adducts.13-16
In the present work a combination of Nanodisc and rR spectroscopic methods enable interrogation of the active site structure of CYP17 in its interaction with all four of the substrates shown in Figure 1 above. Specifically, following up on a recently reported preliminary study of the dioxygen adducts of this system17 which documented differential H-bonding interactions with the bound Fe-O-O fragment among the four substrate-bound dioxygen adducts, results are now expanded to include detailed studies of the ferric and CO-bound ferrous states of CYP17. Herein we provide insight on differential substrate-induced alterations in protein-heme interaction and differences in substrate interactions with the Fe-C-O fragment of the CO-ligated species; the latter species being the accepted paradigm for probing distal- and proximal-side effects on heme-bound exogenous ligands.7-9,12 The results obtained are consistent with those reported in our preliminary work17 and support the conclusion that the presence of the R-OH group in the two hydroxylated substrates alter the active site interactions relative to the two parent substrates, PROG and PREG. In addition, the previously noted differential H-bonding interactions of the 17-OH PROG and 17-OH PREG with the heme bound exogenous ligand, in this case the Fe(II)-C-O fragments, is clearly manifested in the corresponding rR spectra, such structural differences carrying with them important implications for crucial functional differences.
EXPERIMENTAL PROCEDURES
Sample preparation
Expression, Purification, and Nanodisc Incorporation of Human CYP17A1
Heterologous co-expression of full-length human CYP17A1 and the pGro7 GroEL/ES chaperone system (Takara Bio) was conducted under previously described conditions.18 Cytochrome P450 was subsequently purified to electrophoretic homogeneity utilizing a modification of the method devised by Imai et al.17-19 CYP17A1 was subsequently inserted into Nanodiscs containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) as published in Luthra et al.20
Resonance Raman measurements
1. Preparation of samples for rR measurements
Substrate stock solutions containing PROG, 17-OH PROG, PREG and 17-OH PREG in methanol were prepared at a concentration of 8-10 mM; the first three substrates were purchased from Sigma-Aldrich (Milwaukee, WI), while the 17-OH PREG was purchased from both Sigma-Aldrich and Steraloids (Newport, RI) All substrates were of the highest quality commercially available and special efforts were made to document the purity of 17-OH PREG (see Supporting Information). CYP17 Nanodisc (ND:CYP17) solutions in 100 μM potassium phosphate, pH 7.4 buffer containing 15% ultrapure glycerol were concentrated to ~100 μM and PROG- and PREG-bound samples were prepared by the addition of the substrate to a final concentration of 400 μM. The 17-OH PROG- and 17-OH PREG bound samples were similarly prepared by the addition of the appropriate substrate to 500 μM. The binding constants (Ks) of substrates to nanodisc incorporated CYP17 were estimated by application of the quadratic tight binding equation to isotherms acquired during substrate titrations with a CYP17 Nanodisc concentration of 1 μM. Ks values for PROG, 17-OH PROG, PREG, and 17-OH PREG were determined to be 90 nM, 1 μM, 65 nM, and 120 nM, respectively in 100 μM phosphate buffer at 25 °C. Electronic absorption spectroscopy was used to estimate HS state population, with results obtained being around 97 % HS for PROG-bound and PREG-bound samples while approximately 62 % HS for 17-OH PROG- and 17-OH PREG-bound samples. The values of HS content are in good agreement with those calculated from rR data (vide infra). Samples were subsequently flash frozen in liquid N2 before further use. It is important to point out that complete substrate binding, even in the case of substrates that cause only partial spin state conversion in the ferric state, can be confirmed by inspection of the rR spectra of ferrous CO forms (vide infra). As will be seen later, the absence of even residual stretching modes characteristic for substrate-free ferrous CO form (at 471 and 485 cm−1) in the spectra of ferrous-CO substrate-bound samples clearly indicates that these substrates are bound: the spectrum of the 17-OH PROG bound sample shows only a single Fe-CO stretching mode at 491 cm−1 and there is no evidence for residual substrate-free Fe-CO stretching modes, even though this particular substrate has the lowest affinity.
The carbonmonoxy ferrous adducts of ND:CYP17 samples were prepared as follows. A 100 μL volume of ~ 100 μM ferric ND:CYP17 sample in a septum-sealed 5 mm NMR tube (WG-5 Economy, Wilmad) was degassed, using needles, by application of vacuum and filling with argon gas. After the final gas evacuation, the CO gas was introduced to the NMR tube and the ferric sample was reduced by addition of ~2-5 molar equivalents (~2 μL) of sodium dithionite dissolved in freshly degassed 100 mM potassium phosphate buffer, pH 7.4; specifically, the concentration of sodium dithionite was 5.0-12.5 mM, as determined spectrophotometrically using a molar absorption coefficient, ε315 = 8050 M−1cm−1.21 While the rR spectra of ferrous form of the ND:CYP17 were not measured, the ferrous samples were examined by UV-Vis before the CO gas was added to form the ferrous-CO adducts. The visible region of all ferrous samples exhibited spectra characteristic for 5cHS state (the position of a single Q band at around 540 nm), indicating that there is no ligation of substrates with ferrous state. The formation of ferrous CO adducts was confirmed by absorption spectroscopy in the Q band region; i.e., the UV-Vis spectra of these samples exhibit a single Q band at around 552 nm.22 The samples contained no P420 impurities as judged by the lack of bands near 540 nm and 570 nm characteristic for ferrous CO adducts of P420.23
2. Resonance Raman measurements
The samples of ferric CYP17 samples were measured using the 406.7 nm and 356.7 nm excitation lines from a Kr+ laser (Coherent Innova Sabre Ion Laser) and the Fe(II)-CO adducts were excited by a 441.6 nm line provided by a He-Cd laser (IK Series He-Cd laser, Kimmon Koha CO., LTD.). The rR spectra of all samples were measured using a Spex 1269 spectrometer equipped with Spec-10 LN-cooled detector (Princeton Instruments). The slit width was set at 100 μm and the 1200 g/mm grating was used; with this grating, the resultant spectral dispersion is 0.46 cm−1/pixel. The laser power at the ferric sample was adjusted to ~ 10 mW while for ferrous CO adducts it was kept at ~1 mW to minimize photodissociation. Moreover, to avoid laser-induced heating and protein degradation the samples were contained in spinning NMR tubes (5 mm outside diameter, WG-5 ECONOMY, Wilmad). The 180° backscattering geometry was used for all measurements and the laser beam was focused onto the sample using a cylindrical lens.24 All samples were measured at room temperature. Spectra were calibrated with fenchone (Sigma-Aldrich, WI), toluene-2H6 and acetone-2H6 (Cambridge Isotope Laboratories, Inc., MA) and processed with Grams/32 AI software (Galactic Industries, Salem, NH).
RESULTS AND DISCUSSION
A. Effect of substrate structure on the ferric form of ND:CYP17
1. High frequency resonance Raman spectra
The spectrum of substrate-free ND:CYP17 (Figure S1, trace A) exhibits features characteristic of the ferric state, with the ν4 oxidation state marker occurring at 1372 cm−1; furthermore, the so-called spin state marker bands, including the ν3 at 1502 cm−1, the ν2 at 1581 cm−1 and the ν10 at1636 cm−1, are indicative of a pure low spin 6-coordinated state (LS6c) (Figure 2), presumably reflecting coordination by a water molecule associated with a distal pocket cluster of water molecules.25,26 The two overlapped features observed at 1621 cm−1 and 1629 cm−1 are confidently assigned to the ν(C=C) stretching modes of the two peripheral vinyl groups with respective dispositions in and out of the planes of the pyrrole rings to which they are bound, these assignments being consistent with previously published data.27-32
Figure 2.

The high frequency RR data of ferric ND:CYP17 in substrate-free state (A) and with the following substrates: PROG (B), 17-OH PROG (C), PREG (D) and 17-OH PREG (E). The spectra were normalized to the glycerol mode observed at 1468 cm−1.
Binding of substrates by cytochromes P450 is generally accompanied by change of the spin and coordination states of the heme iron, from LS6c to a high spin, five-coordinate state (HS5c), a conversion that results in a large increase of the heme reduction potential.33,34 As can be seen in the spectrum of PROG-bound CYP17 (Figure 2, trace B), the spin state markers are observed at 1486 cm−1 (ν3), 1565 cm−1 (ν2) and 1627 cm−1 (ν10), the last being overlapped with vinyl v(C=C) mode(s); these heme-core mode frequencies are indeed indicative of a pure HS5c state.14,25-32 On the other hand, the binding of 17-OH PROG (Figure 2, trace C) leads to only a partial spin state conversion, as seen by the presence of bands characteristic for both LS6c and HS5c states, noting that the frequencies of vinyl stretching modes in the spectra of substrate-bound samples are difficult to establish because of their overlap with the HS ν10 mode; i.e., the ν10 mode for HS5c is expected to occur near 1623 cm−1.25,26,31 Quite similar spectral patterns are observed for the PREG and 17-OH PREG substrates, as can be seen in traces E and F of Figure 2, where only partial spin state conversion is seen for the hydroxylated substrate. Such partial spin state conversions upon binding of certain substrates, such as the 17-OH PROG and 17-OH PREG samples above, has been seen previously for other cytochrome P450 enzymes and is most often attributed to incomplete expulsion of the distal pocket water cluster and coordination of a residual water molecule.1,2 On the other hand, in the present situation it is noted that the two substrates producing only partial spin-state conversion both have 17α-hydroxy substituents positioned near the heme binding site for exogenous ligands; i.e., it is also reasonable to suggest that these R-OH fragments might interact directly with the heme iron. Alternatively, the presence of this R-OH group within the heme pocket might enhance retention of one or more residual water molecules. Inasmuch as the components in the rR spectra that are associated with the LS state don’t significantly differ among traces A,C and F (i.e., 1636, 1581 and 1502 cm−1), there is no support for arguing that the coordination environment is different for the substrate-free (SF) and the samples bound with the hydroxylated substrates. On the other hand, the high degree of specificity associated with the hydroxylation reactions and the relatively crowded distal side pocket revealed in the crystal structure6 tends to favor the explanation involving direct interaction between the R-OH and heme iron. In either case it is reasonable to suggest that the retention of a significant amount of LC6c form for the 17-OH PROG and 17-OH PREG substrates is directly attributable to the juxtaposition of the 17-OH fragment to the heme iron. Additionally, it is also important to note that the HS states formed with all four substrates apparently have virtually identical heme structures; this conclusion is supported by the fact that the rR spectra of the HS fractions of the samples containing the hydroxylated substrates (i.e., traces D and G) are virtually identical to those obtained for samples containing PROG and PREG (traces B and E).
It is possible to estimate the spin state populations for these 5 samples using previously determined relative rR scattering cross-sections of the ν3 modes for the LS6c and HS5c forms of cytochrome P450cam (CYP101) as described by Mak et al., where an internal standard was employed to obtain the IHS/ILS, which was determined to be 1.24.35 There it is also shown that application to rR data sets for several cytochromes P450 gave good agreement with the HS/LS ratios that had been previously determined by other methods such as deconvolution of electronic absorption spectra. Using this procedure, the substrate-free enzyme is estimated to be 95% LS, whereas the HS populations of the PROG- and PREG-bound samples are calculated to be 94% and 95%, respectively. On the other hand, the HS components for the 17-OH PROG- and 17-OH PREG-bound samples are calculated to be 59% and 60%, respectively, also in satisfying agreement with the values determined in the present work by deconvolution of the electronic absorption spectra (not shown).
2. Low frequency resonance Raman spectra
The rR spectra in the low frequency region provide useful information not only on the binding of endogenous and exogenous heme axial ligands,7-9,12,14,32,36-40 but also on the disposition of the heme peripheral groups and out-of-plane distortions of the heme macrocycle as reflected by activation of out-of-plane heme modes.7-9,41-43 The propionate bending modes typically occur in the region of ~360-380 cm−1 with features observed in the higher range being ascribed to stronger H-bonding of the terminal carboxyl groups with active site residues.28,29,44 Similarly, the frequencies of the so-called “vinyl bending” modes, which occur in the 400-440 cm−1 region, like their ν(C=C) counterparts mentioned above, are normally considered to be sensitive the orientation of the vinyl groups with respect to the planes of the pyrrole rings to which they are attached. Specifically, modes observed in the lower half of this frequency region are typically associated with an in-plane vinyl group orientation, while modes observed in the higher range are linked with vinyl groups experiencing out-of-plane distortions.27-29,32 Documentation of the dispositions of both types of heme peripheral groups as well as the activation of out-of-plane modes of the heme macrocyle is important inasmuch as convincing arguments have been presented for the significant impact of all of these structural features on heme protein functional properties, including heme reduction potential and affinities for endogenous and exogenous ligands.10,11,29,45,46
In the present study the spectrum of the substrate-free (SF) sample (Figure 3, trace A) exhibits a single feature associated with the propionate bending mode (at 376 cm−1) and a prominent vinyl bending mode at 424cm−1 along with a weak shoulder at 414 cm−1, noting that evidence for two ν(C=C) vinyl stretching modes were seen at 1629 and 1621 cm−1 in Figure 2. However, it is emphasized that the assignment of these low frequency modes to “vinyl-bending” motions is an oversimplification and that the modes are not as pure as those observed for the C=C vinyl stretching. This point has been convincingly demonstrated previously by the fact that replacement of the pyrrole -CH3 substituents with -CD3 groups leads to large shifts in these “vinyl bending” modes,31,47,48 indicating that these modes are more complex and are better described as “pyrrole deformation” modes (albeit, pyrrole deformation modes that are apparently somewhat sensitive to vinyl group orientation).14,29-32,47,48 Furthermore, these low frequency “vinyl bending” modes are complex and there is little information available regarding their enhancement factors; i.e., the absence of two observed features in this region would not imply that two modes are directly overlapped. Significantly, addition of any of the four substrates (Figure 3, traces B through E) causes no significant changes in the frequencies of the 376 cm−1 mode, indicating that hydrogen bonding to the propionate groups is not altered upon binding of any of these substrates. Inspection of the spectra in the vinyl bending mode region indicates that the prominent mode at 424 cm−1 is not altered; while there appears to be some difference in the weak lower frequency modes (414 vs 405 cm−1), it is deemed inadvisable to attach significance to this owing to their low intensity. These observations of relative insensitivity of the bands associated with peripheral substituent disposition to substrate binding are not surprising. In the case of CYP21,49 such insensitivity was seen for binding of PROG and 17-OH PROG, while for CYP19, on the other hand, changes were seen in propionate and vinyl bending modes upon binding of androstenedione and its structural derivatives.50 Though substrate binding to CYP17 causes minimal changes in bending modes of the peripheral substituents, there is clear indication of activation of out-of-plane (OOP) modes at 318, ~490 cm−1 and 816 cm−1, assigned to the γ7, γ12 and γ11 modes (also see Figure S2).30
Figure 3.

The low frequency RR data of ferric ND:CYP17 in substrate-free state (A) and with the following substrates: PROG (B), 17-OH PROG (C), PREG (E) and 17-OH PREG (F). The spectra were normalized to the ν8 mode observed at around 345 cm−1.
Another type of important information gained from the low frequency region of the rR spectra is the status of the endogenous and exogenous axial ligands. In the case of cytochromes P450, the ν(Fe-S) mode is effectively enhanced only in the case of the HS ferric form, using excitation near 350 nm.29,32,49-52 As shown in Figure 4, this mode is observed at 347 cm−1 for all substrates used, indicating that substrate structure does not affect the trans-axial linkage with the cysteine thiolate. Note that the apparent diminished intensities of the ν(Fe-S) modes in traces B and D are a result of normalization with the ν7 mode which contains contributions from the substantial remnants of the LS forms in these two samples. The main point to be made here is that any changes in the modes associated with exogenous axial ligands, such as the CO fragment being studied here, are attributable to distal side interactions. Finally, it is noted that the 347 cm−1 value observed for the ν(Fe-S) mode is at the low end of values obtained for other cytochromes P450, which range between ~353 down to 347 cm−1.29,32,49-52 As will be seen, this lower ν(Fe-S) has an impact on the ν(Fe-C) and ν(C-O) modes of the trans-axial Fe-C-O fragment (vide infra).
Figure 4.

The low frequency RR data of ferric ND:CYP17 with the following substrates: PROG (A), 17-OH PROG (B), PREG (C) and 17-OH PREG (D). The spectra were normalized to the ν7 mode observed at 675 cm−1. Excitation laser line is 356.7 nm.
B. Ferrous CO adducts of ND:CYP17 and its interaction with substrates
The ferrous CO adducts of cytochrome P450 are usually quite stable as compared to the oxygenated forms and have often been exploited to probe interactions of exogenous axial ligands with the distal heme pocket residues. The changes in frequencies and intensities of modes associated with the Fe-C-O fragment reflect the strength of the heme linkage with the proximal endogenous ligand and reliably report on the steric and electronic influences presented by the distal pocket environment, such as electrostatic or H-bonding interactions with distal pocket amino acid residues, water molecules or substrates. The key vibrational modes of interest are the ν(Fe-C) stretching modes, usually observed in the region of 460-490 cm−1, and the ν(C-O) stretching modes seen between 1920 and 1970 cm−1. The δ(Fe-C-O) bending modes, very often quite weak and difficult to detect, are seen in the range from 550-570 cm−1. A well-documented negative linear correlation between the ν(C-O) and ν(Fe-C) stretching frequencies arises from back donation of the Fe(II) dπ electrons to π* orbitals of diatomic ligands, resulting in an increase of the Fe-C bond strength while weakening that of the C-O bond.7-9,12
Referring to the high frequency rR spectra of the CO-bound ferrous samples (Figure S3), the ν4 oxidation state marker band of all ferrous CO samples is located at 1370 cm−1, while the spin state marker bands, the ν3 and ν2 modes, are seen at 1497 cm−1 and at 1585 cm−1, respectively; all of these are characteristic for low spin 6-coordinate CO adducts in accordance with previously published data on other cytochromes P450.14,26,29,32,36,37 Figure 5 shows the low and high frequency rR spectra in the regions where the bands associated with the Fe-C-O fragment are seen. Before proceeding to analysis of the behavior of the modes associated with the Fe-C-O fragments, it is important to point out that binding of substrates to the ferrous CO form of Nanodisc-incorporated CYP17 has no impact on the heme core modes nor the orientation of vinyl groups, based on invariant frequencies; though it possibly causes a slight lowering (1 or 2 cm−1) of the propionate bending modes, indicative of a slightly weaker H-bond interaction of propionate groups with the nearby amino acid residues, these small changes are of borderline significance. So, in agreement with the data for the substrate-bound ferric forms, the structural differences among the 4 substrates have little or no impact on the heme structure or its interactions with active site residues.
Figure 5.

The low frequency (left panel) and high frequency (right panel) RR data of ferrous CO adducts of substrate-free ND:CYP17 (A) and with the following substrates: PROG (B), 17-OH PROG (C), PREG (D) and 17-OH PREG (E). The low frequency spectra were normalized to the ν7 mode observed at 673 cm−1 and the high frequency data were normalized to the ν4 mode (not shown on the picture).
Turning attention to the modes of the Fe-C-O fragment, in the spectrum of the SF form (Figure 5, trace A, left panel) in the region near 450-500 cm−1, appears a rather broad envelope with components having maxima near 472 and 485 cm−1. This low frequency spectrum of the SF form shows evidence for the presence of two Fe-C-O conformers, an interpretation that is confirmed by the observation of two ν(CO) stretching modes in the high frequency region (Figure 5, trace A, right panel). Based on relative intensities, the ν(C-O) mode at 1946 cm−1 corresponds to the 485 cm−1 ν(Fe-CO) mode and the 1957 cm−1 feature is correlated with the 472 cm−1 ν(Fe-CO) mode. Obviously, this behavior is consistent with the well-documented inverse frequency relationship.7-9,12 The only remaining point to be noted about the general features of the spectral data is that the weak bands appearing in the 1840-1870 cm−1 region are well fit to combination modes involving the ν(Fe-C) and ν4 modes. Thus, in the case of the SF sample, two combination modes are seen near 1839 and 1852 cm−1 corresponding to (472 +1370 cm−1 and 485 +1370 cm−1), while the combination modes for the other forms occur at frequencies about 15-25 cm−1 higher; e.g., 498 + 1370 cm−1 corresponds to 1867 cm−1 and 491 + 1370 cm−1 corresponds to the broadened combination band centered at 1858 cm−1. It seems important to emphasize the point that the inherent intensities of the ν(C-O) modes are quite weak, being comparable to those of the observed combination bands.
Returning attention to the fundamental modes associated with the Fe-C-O fragments, upon substrate binding (Figure 5) quite substantial shifts in the internal modes of the Fe-C-O fragment occur. The samples with PROG and PREG (traces B and D) exhibit ν(C-O) modes at 1932 and 1940 cm−1, respectively, and an identical frequency of 498 cm−1 for the ν(Fe-C) modes. As can be seen by inspection of the inverse-correlation plot shown in Figure 6, these points for the PROG and PREG substrates fall along the line generated by the data set for the CYP17 samples, but at the same time, there is a clear indication that unexpected variations occur inasmuch as these two substrates show identical ν(Fe-C) modes but ν(C-O) frequencies that differ by 8 cm−1. It is important to note that such large shifts of the internal modes of the Fe-C-O fragment upon substrate binding are characteristic for enzymes with relatively small and tightly organized heme pockets; e.g., substrate associated shifts to higher frequency of the ν(Fe-CO) mode of 20 25 cm−1 magnitude were previously observed in bacterial cytochrome P450s such as cytochrome P450cam (CYP101).26,37 On the other hand, in the case of mammalian drug-metabolizing cytochromes P450, which have larger and relatively flexible heme pockets,1,2 addition of substrates typically causes minimal changes in the ν(Fe-CO) frequency (<5 cm−1).32,52,53 The δ(Fe-C-O) bending modes for substrate-bound samples are upshifted 3 cm−1 relative to the bending mode seen in substrate-free sample, consistent with the higher frequencies of the Fe-CO stretching modes.
Figure 6.

The experimental spectra of the ferrous 12C16O (A) and the 13C16O (B) adducts of 17-OH PREG-bound ND:CYP17 and their difference trace. The thin black lines represent fitted 50/50 % Gaussian/Lorenzian functions and their bandwidths are shown in parentheses.
Turning attention to the two substrates that possess a hydroxyl group in a position to interact with the Fe-C-O fragment, it is noted that the spectral data obtained for the 17-OH PROG-bound sample reveals only one ν(Fe-C) and one ν(C-O) mode, appearing at 491 and 1938 cm−1, data that are consistent with a single Fe-C-O conformer. It is further noted that the Fe-CO associated modes in the 17-OH PROG-bound spectra are relatively narrow, an observation that likely reflects a highly directional H-bonding interaction between Fe-CO fragment and the 17-OH PROG substrate, restricting the degree of fluctuation of the fragment within the pocket. Similar behavior of the vibrations of the Fe-C-O fragment was previously seen in the ERY-bound Nanodisc-incorporated CYP3A4,14 where the H-bonding from the substrate was suggested by crystallographic data.54 While the spectral data acquired for the 17-OH PROG-bound CYP17 are thus consistent with the existing interpretational framework derived from many previous studies,7-9,12,14,36,51-53 the spectra obtained for the sample containing 17-OH PREG, shown in trace E, is surprisingly complex, providing evidence for two conformational isomers. In the low frequency region features are seen at 481 and 505 cm−1, with indications of a weak shoulder near 522 cm−1. On the other hand very carefully conducted rR studies of the region containing the ν(C-O) stretching modes near 1900-1950 cm−1 shows only two features attributable to ν(C-O), occurring at 1953 and 1928 cm−1; based on relative intensities, the higher frequency mode correlates with the 481 cm−1 feature with the other forming the 505/1928 cm−1 pair. Given the fact that only two ν(C-O) modes were observed and the relative weakness of the shoulder near 522 cm−1, spectra were also acquired using 13C16O in an effort to clarify the three-component envelope. Figure 6, shows rR spectra of the 12C16O and 13C16O substituted ferrous CO adducts in the low frequency region and their difference pattern. The analysis of the difference trace is not straightforward because the isotopic shifts are smaller than the bandwidths and as result the peaks seen in difference trace represent frequencies of the bands’ wings instead of true frequencies of Fe-C stretches. Therefore, in order to extract the frequencies of these unresolved Raman bands, the absolute spectra were deconvoluted in the region of 430-610 cm−1 with 50/50 % Gaussian/Lorenzian functions. The ν(Fe-C) envelope of 12C16O spectrum was best fitted with three bands at 481 cm−1, 505 cm−1 and 522 cm−1; attention was given mainly to the ν(Fe-CO) stretching modes, since the region containing the weak δ(Fe-C-O) bending modes, as well as several weak heme modes, was not sufficiently enhanced to properly treat by deconvolution. A similar procedure was applied to the 13C16O spectrum, where it was indicated that the Fe-C associated modes were shifted by 4 cm−1 to lower frequency (i.e., to 477 cm−1 and 501 cm−1), while the band at 522 cm−1 did not shift. It is noted that a heme mode is observed at this frequency in the spectra obtained for many LS ferric forms and the other CO adducts studied here (deconvolution data not shown).
In order to rule out the possibility that the complicated spectrum of the 17-OH PREG sample is overly photosensitive or converts to the P420 form, separate studies were conducted to confirm such reactions were not occurring, as described in Supporting Information (Figures S4 and S5). One initial reaction to this unexpected spectral complexity observed when using the 17-OH PREG substrate was concern regarding the chemical integrity of this particular batch of substrate. However, identical results were obtained when using a second batch of substrate, newly purchased from a different supplier; more convincingly, quite extensive efforts were made to document the purity of the material used, including mass spectral analysis and 1H and 13C-NMR, with all of these results supporting its high purity, as is thoroughly described in Supporting Information (Part 3, Figures S6, S7, S8 and S9).
Even though binding of 17-OH PREG to CYP17 gives rise to a mixture of two Fe-C-O conformers, it is interesting to note that all of these data observed here for CYP17 still yield a reasonably well behaved inverse linear correlation, as shown in Figure 6. This figure shows a negative correlation between ν(Fe-C) and ν(CO) for the Nanodisc-incorporated CYP17 samples investigated here (red triangles and red line), along with similar plots for other previously reported bacterial and mammalian cytochromes P450 (green squares and line) and for mammalian forms of nitric oxide synthase (NOS), another thiolate-ligated heme protein (blue diamonds and line). The actual values for all of the points plotted in Figure 6 are listed in Table S1 of Supporting Information.
Several comments are warranted when considering the plots shown in Figure 6. First, it is generally accepted that the slopes of such correlations are mainly determined by distal pocket polarity with positive electrostatic potentials, often associated with H-bonding, shifting the points to the upper left, reflecting enhanced Fe dπ → π(CO) back-bonding.12 On the other hand, the position of such plots along the vertical axis is influenced by the relative strength of the donor ability of the trans-axial heme ligand; e.g., the line for histidine-ligated heme is shifted vertically to higher regions than the lines shown in Figure 6.
The thiolate in P450cam (CYP101) has three backbone hydrogen bonds from NH groups of nearby amides, yielding an Fe-S bond that exhibits a ν(Fe-S) mode near ~353 cm−1.51 The point in the lower right of the P450cam line represents the substrate-free state and the points to the higher left side are associated with the substrate-bound states. The CYP101 substrates bind close to the heme iron, expelling water molecules from the heme pocket and allowing the CO ligand to directly interact with H-bond donor groups, which involve the D251 and T252 residues and possibly their associated water molecules. These residues are postulated to play a central role in proton shuttle required for O-O cleavage. The NOS points fall on a separate ν(Fe-C)/ν(CO) line that is much higher than that for cytochrome P450cam, owing to weakening of the Fe-S linkage (Table S1). The heme-bound thiolate of NOS has only two H-bonds from backbone NH groups, but more importantly, it is involved in one H-bond from a tryptophan sidechain residue. This stronger H-bond was shown to weaken the thiolate donor strength, as reflected in a lower ν(Fe-S) mode (~340 cm−1) relative to CYP101. The result of this weakened trans-axial bond leads to increases in the opposing Fe-C bond strength (and ν(Fe-C) frequency), raising the back-bonding correlation line above that of cytochrome P450cam.
Considering all of the data acquired herein for CYP17, two aspects of the derived correlation need to be addressed. First, it is noted that the points generally fall above those obtained for the CYP101, an observation that is entirely consistent with the weakened Fe-S bond with its ν(Fe-S) mode near 347 cm−1 as documented in this work (Figure 4), versus the value of 353 cm−1 reported for CYP101.51 A secondary, and unexpected observation was that the slope of the derived line obtained for CYP17 is significantly greater than those for the other two sets of data. One cause of this might be that the number of data points is insufficient to generate a valid correlation; however, the number of points for the CYP101 and NOS plots is not substantially larger, yet both yield comparable slopes. Alternatively, it seems plausible that the active site structural organization for steroidogenic cytochromes P450 might be relatively more rigid, as reflected by the strict requirements for a high degree of stereospecificity attending the chemical conversions which they orchestrate.1-3 Thus, the presence of specific functional groups on the substrate periphery might introduce highly directional steric or electrostatic interactions that lead to a greater slope and/or deviations of some points (substrates) from the typical plots that are determined by more remote or delocalized electrostatic interactions.
Returning attention to the spectra shown in Figure 5, an especially intriguing observation is the dramatically different behavior exhibited for the sample containing 17-OH PREG. Like, 17-OH-PROG it introduces into the heme distal pocket the highly polar R-O-H functionality, yet the spectral differences are quite remarkable with the 17-OH PREG substrate giving rise to two Fe-C-O conformers. One conformer (point 9) gives spectral parameters similar to those seen for the two substrate-free forms (points 8 and 10) and is comparably broad, most likely reflecting a conformer that is weakly H-bonded to distal pocket residues or loosely associated water molecules. A second conformer (point 14) is positioned nearer to the points observed for the other three substrates, but apparently experiencing a stronger electropositive field that the other three; i.e., lying higher to the left along the line.12
C. Functional Implications
The rR spectral data acquired here reveal key information regarding the substrate/protein interplay associated with the substrate binding event that can affect the active site structure and reactivity. Careful analysis of the data acquired for the ferric forms show that binding of substrate causes variable degrees of conversion from the LS to HS state, with the non-hydroxylated PROG and PREG yielding almost complete HS form, while the two hydroxylated substrates (17-OHPROG and 17-OH-PREG) give only about a 60% conversion to HS. This incomplete conversion is attributable to either a direct interaction of the 17-OH fragment with the heme iron or possibly retention of an active site water molecule that can interact with the heme iron. For either explanation, the presence of these hydroxylated substrates is expected to lower the efficiency of electron transfer from the associated reductase,55 although binding of the reductase to the enzyme may enhance the spin-state conversion; experiments to probe the structural effects of reductase binding are planned for the near future. Another important result from the present study is that the acquired rR spectra of the ferric HS forms, as well as the ferrous CO adducts, convincingly show that the heme structure and its interactions with the active site protein residues remain constant for all four substrates; i.e., the heme group is held in a relatively fixed orientation with respect to the associated protein, remaining unaffected by any differences in the molecular structure of these four substrates.
An important corollary to the above finding is that any differences encountered in enzymatic processing of these four physiologically important substrates are most reasonably attributed to subtle alterations in the intermolecular interactions of given substrate with the associated heme axial ligand; within the natural enzymatic cycle this includes the bound dioxygen ligand, its reduced peroxo- or hydroperoxo- forms, or any species arising from an O-O bond cleavage process, such as the putative Compound I intermediate. In the present work, the exogenous heme ligand is CO and, indeed, the largest changes in the rR spectra acquired for the four different substrates occur for the internal modes of the Fe-C-O fragment. The most intriguing observation is the appearance of two Fe-C-O conformers for the sample containing 17-OH PREG, because it might have been expected that the presence of the 17-OH fragment also would give rise to two conformers for the sample containing 17-OH PROG. However, consistent with this rather surprising observation is some very recent work by Scott and coworkers on crystals of CYP17 bound with either 17-OH PROG or 17-OH PREG.56 Specifically, while the details of this work are not yet published, what is known to us is that all 4 enzyme molecules of the asymmetric unit cell for the 17-OH PROG sample have the substrate in a single conformation (designated “up”),56 whereas for the 17-OH PREG sample, three of the molecules have the substrate in the “up” position, but the remaining one has 17-OH PREG in a different position (designated “down”).56 Despite the lack of further details, the salient point for the rR results reported here is that these crystallographic results imply that, in solution, the 17-OH PREG may permit two arrangements of the substrate/CO pair, a suggestion that is clearly consistent with our current rR data for the CO adducts. The important functional implication of this is that the subtle difference between the −OH and -C=O substituents at the remote 3β-position of the 17-OH PREG and 17-OH PREG, respectively, can significantly alter the interaction of the 17-OH functionality with the Fe-C-O fragment. While this difference in substrate disposition may lead only to disorder for the weakly H-bonding Fe-C-O fragment, its effects could be enhanced with more strongly H-bonding fragments, including Fe-O-O. Thus, this behavior is satisfyingly consistent with the recent rR results obtained by our groups for the dioxygen adducts of these four substrates with CYP17. As noted in the Introduction, those data are most reasonably interpreted to demonstrate a significant difference in the interactions of these two hydroxylated substrates of heme-bound O2, with the 17-OH PROG being H-bonded to the terminal oxygen of the Fe-O-O fragment, while the 17-OH PREG forms an H-bond with the proximal oxygen atom.17
Supplementary Material
Figure 7.
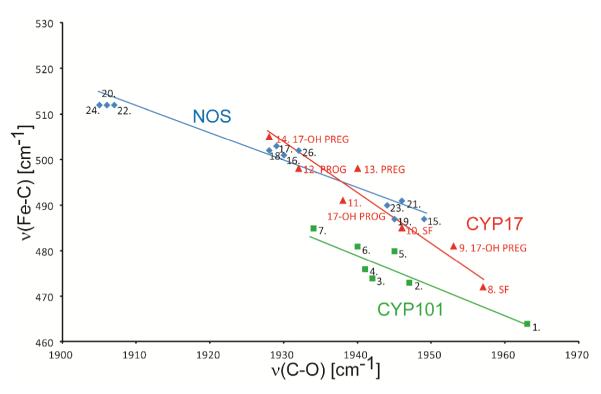
The diagrams showing an inverse correlation for CYP’s and NOS’s. The numbers represent proteins listed in Table S1. The green squares show points for CYP101 (green line), the red triangles show points for CYP17 (red line) and the blue diamonds for mammalian NOS’s (blue line).
Acknowledgments
This work was supported by National Institutes of Health grants GM31756 and GM33775 to S.G.S. and GM96117 to J.R.K.
Abbreviations
- P450
Cytochrome P450
- CYP
cytochrome P450
- CYP17
Cytochrome P450c17
- ND:CYP17
Nanodisc incorporated CYP17
- NOS
nitric oxide synthase
- rR
resonance Raman
- PROG
progesterone
- PREG
pregnenolone
- 17-OH PROG
17 α -hydroxyprogesterone
- 17-OH PREG
17 α –ydroxypregnenolone
- AD
androstenedione
- DHEA
dehydroepiandrosterone
- ERY
erythromycin
- HS5c
high spin five-cordinated
- LS6c
low spin six-coordinated
- SF
substrate-free
Footnotes
Supporting Information. Included in the supporting information are Figures depicting the rR spectra of ferric and ferrous CO adducts over wider frequency ranges, experimental details of rR procedures, ferrous-CO photodissociation studies, validation of purity of 17-OH PREG (ESI-MS, GC-MS, 1H NMR, 13C NMR) and a table with Fe-CO data for CYPs and NOS’s. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Sigel A, Sigel H, Sigel RKO. In: Metal Ions in Life Sciences. Sigel A, Sigel H, Sigel RKO, editors. Vol. 3. John Wiley & Sons, Ltd.; Chichester: 2007. [Google Scholar]
- (2).Ortiz de Montellano PR. In: Cytochrome P450: Structure, Mechanism and Biochemistry. 3rd Ed Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- (3).Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chung BC, Picado-Leonard J, Haniu M, Bienkowski M, Hall PF, Shively JE, Miller WL. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc. Natl. Acad. Sci. U. S. A. 1987;84:407–411. doi: 10.1073/pnas.84.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Attard G, Reid AH, Olmos D, de Bono JS. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009;69:4937–4940. doi: 10.1158/0008-5472.CAN-08-4531. [DOI] [PubMed] [Google Scholar]
- (6).DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482:116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Spiro TG. In: Biological Applications of Raman Spectroscopy. Spiro TG, editor. John Wiley & Sons; New York: 1998. [Google Scholar]
- (8).Kadish KM, Smith KM, Guilard R. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 7. Academic Press; 2000. pp. 225–291. [Google Scholar]
- (9).Kitagawa T, Mizutani Y. Resonance Raman spectra of highly oxidized metalloporphyrins and heme proteins. Coord. Chem. Rev. 1994;135/136:685–735. [Google Scholar]
- (10).Lee KB, Jun E, La Mar GN, Rezzano IN, Pandey RK, Smith KM, Walker FA, Buttlaire DH. Influence of heme vinyl- and carboxylate-protein contacts on structure and redox properties of bovine cytochrome b5. J. Am. Chem. Soc. 1991;113:3576–3583. [Google Scholar]
- (11).Reid LS, Lim AR, Mauk AG. Role of heme vinyl groups in cytochrome b5 electron transfer. J. Am. Chem. Soc. 1986;108:8197–8201. [Google Scholar]
- (12).Spiro TG, Soldatova AV, Balakrishnan G. CO, NO and O2 as vibrational probes of heme protein interactions. Coord. Chem. Rev. 2013;257:511–527. doi: 10.1016/j.ccr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS letters. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mak PJ, Denisov IG, Grinkova YV, Sligar SG, Kincaid JR. Defining CYP3A4 Structural Responses to Substrate Binding. Raman Spectroscopic Studies of a Nanodisc-Incorporated Mammalian Cytochrome P450. J. Am. Chem. Soc. 2011;133:1357–1366. doi: 10.1021/ja105869p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Denisov IG, Sligar SG. Cytochromes P450 in Nanodiscs. Biochim. Biophys. Acta. 2012;1814:223–229. doi: 10.1016/j.bbapap.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nath A, Grinkova YV, Sligar SG, Atkins WM. Ligand Binding to Cytochrome P450 3A4 in Phospholipid Bilayer Nanodiscs: The Effect of Model Membranes. J Biol. Chem. 2007;282:28309–28320. doi: 10.1074/jbc.M703568200. [DOI] [PubMed] [Google Scholar]
- (17).Gregory M, Mak PJ, Sligar SG, Kincaid JR. Differential Hydrogen Bonding in Human CYP17 Dictates Hydroxylation versus Lyase Chemistry. Angew. Chem. Int. Ed. 2013;52:5342–5345. doi: 10.1002/anie.201300760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Imai T, Globerman H, Gertner JM, Kagawa N, Waterman MR. Expression and Purification of Functional Human 17α-Hydroxylase/17,20-Lyase (P450c17) in Escherichia coli. J. Biol. Chem. 1993;268:19681–19689. [PubMed] [Google Scholar]
- (19).Omura T, Sato R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J. Biol. Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- (20).Luthra A, Gregory M, Grinkova YV, Denisov IG, Sligar SG. Nanodiscs in the studies of membrane-bound cytochrome P450 enzymes. Methods Mol. Biol. 2013;987:115–127. doi: 10.1007/978-1-62703-321-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McKenna CE, Gutheil WG, Song W. A method for preparing analytically pure sodium dithionite. Dithionite quality and observed nitrogenase-specific activities. Biochim. Biophys. Acta. 1991;1075:109–117. doi: 10.1016/0304-4165(91)90082-r. [DOI] [PubMed] [Google Scholar]
- (22).O’Keeffe DH, Ebel RE, Peterson JA. Purification of Bacterial Cytochrome P-450. Meth. Enzymol. 1978;52:151–157. doi: 10.1016/s0076-6879(78)52017-x. [DOI] [PubMed] [Google Scholar]
- (23).Gunsalus IC, Wagner GC. Bacterial P-450cam Methylene Monooxygenase Components: Cytochrome m, Putidaredoxin, and Putidaredoxin Reductase. Meth. Enzymol. 1978;52:166–188. doi: 10.1016/s0076-6879(78)52019-3. [DOI] [PubMed] [Google Scholar]
- (24).Line Shriver DF, Dunn JBR. The Backscattering Geometry for Raman Spectroscopy of Colored Materials. Appl. Spectrosc. 1974;28:319–323. [Google Scholar]
- (25).Champion PM, Gunsalus IC, Wagner GC. Resonance Raman investigations of cytochrome P450cam from Pseudomonas putida. J. Am. Chem. Soc. 1978;100:3743–3751. [Google Scholar]
- (26).Wells AV, Li P, Champion PM, Martinis SA, Sligar SG. Resonance Raman investigations of Escherichia coli-expressed Pseudomonas putida cytochrome P450 and P420. Biochemistry. 1992;31:4384–4393. doi: 10.1021/bi00133a002. [DOI] [PubMed] [Google Scholar]
- (27).Marzocchi MP, Smulevich G. Relationship between heme vinyl conformation and the protein matrix in peroxidases. J. Raman. Spectrosc. 2003;34:725–736. [Google Scholar]
- (28).Peterson ES, Friedman JM, Chien EYT, Sligar SG. Functional implications of the proximal hydrogen-bonding network in myoglobin: a resonance Raman and kinetic study of Leu89, Ser92, His97, and F-helix swap mutants. Biochemistry. 1998;37:12301–12319. doi: 10.1021/bi980752u. [DOI] [PubMed] [Google Scholar]
- (29).Chen Z, Ost TWB, Schelvis JPM. Phe393 Mutants of Cytochrome P450 BM3 with Modified Heme Redox Potentials Have Altered Heme Vinyl and Propionate Conformations. Biochemistry. 2004;43:1798–1808. doi: 10.1021/bi034920g. [DOI] [PubMed] [Google Scholar]
- (30).Hu S, Smith KM, Spiro TG. Assignment of protoheme resonance Raman spectrum by heme labeling in myoglobin. J. Am. Chem. Soc. 1996;118:12638–12646. [Google Scholar]
- (31).Mak PJ, Kaluka D, Manyumwa EM, Zhang H, Deng T, Kincaid JR. Defining Resonance Raman Spectral Response to Substrate Binding by Cytochrome P450. Biopolymers. 2008;89:1045–1053. doi: 10.1002/bip.21058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mak PJ, Im S-C, Zhang H, Waskell LA, Kincaid JR. Resonance Raman Studies of Cytochrome P450 2B4 in Its Interactions with Substrates and Redox Partners. Biochemistry. 2008;47:3950–3963. doi: 10.1021/bi800034b. [DOI] [PubMed] [Google Scholar]
- (33).Sligar SG, Gunsalus IC. A Thermodynamic Model of Regulation: Modulation of Redox Equilibria in Camphor Monoxygenase. Proc. Natl. Acad. Sci. U. S. A. 1976;73:1078–1082. doi: 10.1073/pnas.73.4.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fisher M, Sligar S. Control of Heme Protein Redox Potential and Reduction Rate: Linear Free Energy Relation Between Potential and Ferric Spin State Equilibrium. J. Am. Chem. Soc. 1985;107:5018–5019. [Google Scholar]
- (35).Mak PJ, Zhu Q, Kincaid JR. Using resonance Raman data to estimate the spin state populations of Cytochromes P450. J. Raman Spectrosc. 2013 doi: 10.1002/jrs.4401. DOI: 10.1002/jrs.4401, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ibrahim M, Xu C, Spiro TG. Differential sensing of protein influences by NO and CO vibrations in heme adducts. J. Am. Chem. Soc. 2006;128:16834–16845. doi: 10.1021/ja064859d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Uno T, Nishimura Y, Makino R, Iizuka T, Ishimura Y, Tsuboi M. The resonance Raman frequencies of the iron-carbon monoxide stretching and bending modes in the carbon monoxide complex of cytochrome P-450cam. J. Biol. Chem. 1985;260:2023–2026. [PubMed] [Google Scholar]
- (38).Bangcharoenpaurpong O, Rizos AK, Champion PM, Jollie D, Sligar SG. Resonance Raman detection of bound dioxygen in cytochrome P-450cam. J. Biol. Chem. 1986;261:8089–8092. [PubMed] [Google Scholar]
- (39).Macdonald IDG, Sligar SG, Christian JF, Unno M, Champion PM. Identification of the Fe-O-O Bending Mode in Oxycytochrome P450cam by Resonance Raman Spectroscopy. J. Am. Chem. Soc. 1999;121:376–380. [Google Scholar]
- (40).Hu S, Schneider A, Kincaid JR. Resonance Raman Studies of Oxycytochrome P-450 cam: The Effect of Substrate Structure on ν(O-O) and ν(Fe-O2) J. Am. Chem. Soc. 1991;113:4815–4822. [Google Scholar]
- (41).Huang Q, Schweitzer-Stenner R. Non-planar heme deformations and excited state displacements in horseradish peroxidase detected by Raman spectroscopy at Soret excitation. J. Ram. Spectrosc. 2005;36:363–375. doi: 10.1021/jp052986a. [DOI] [PubMed] [Google Scholar]
- (42).Jentzen W, Ma J-G, Shelnutt JA. Conservation of the Conformation of the Porphyrin Macrocycle in Hemoproteins. Biophys. J. 1998;74:753–763. doi: 10.1016/S0006-3495(98)74000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shelnutt JA, Song X-Z, Ma J-G, Jia S-L, Jentzen W, Medforth CJ. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 1998;27:31–42. [Google Scholar]
- (44).Cerda-Colon JF, Silfa E, Lopez-Garriga J. Unusual rocking freedom of the heme in the hydrogen sulfide-binding hemoglobin from Lucina pectinata. J. Am. Chem. Soc. 1998;120:9312–9317. doi: 10.1002/(sici)1520-6343(1998)4:5<311::aid-bspy3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- (45).Funk WD, Lo TP, Mauk MR, Brayer GD, MacGillivray RTA, Mauk AG. Mutagenic, electrochemical, and crystallographic investigation of the cytochrome b5 oxidation-reduction equilibrium: involvement of asparagine-57, serine-64, and heme propionate-7. Biochemistry. 1990;29:5500–5508. doi: 10.1021/bi00475a013. [DOI] [PubMed] [Google Scholar]
- (46).Ma J-G, Zhang J, Franco R, Jia S-L, Moura I, Moura JJG, Kroneck PMH, Shelnutt JA. The Structural Origin of Nonplanar Heme Distortions in Tetraheme Ferricytochromes c3. Biochemistry. 1998;37:12431–12442. doi: 10.1021/bi981189i. [DOI] [PubMed] [Google Scholar]
- (47).Mak PJ, Podstawka E, Kincaid JR, Proniewicz LM. Effects of Systematic Peripheral Group Deuteration on the Low Frequency Resonance Raman Spectra of Myoglobin Derivatives. Biopolymers. 2004;75:217–228. doi: 10.1002/bip.20115. [DOI] [PubMed] [Google Scholar]
- (48).Podstawka E, Mak PJ, Kincaid JR, Proniewicz LP. Low Frequency Resonance Raman Spectra of isolated alpha and beta subunits of Hemoglobin and Their Deuterated Analogues. Bioploymers. 2006;83:455–466. doi: 10.1002/bip.20573. [DOI] [PubMed] [Google Scholar]
- (49).Tosha T, Kagawa N, Arase M, Waterman MR, Kitagawa T. Interaction between Substrate and Oxygen Ligand Responsible for Effective O–O Bond Cleavage in Bovine Cytochrome P450 Steroid 21-Hydroxylase Proved by Raman Spectroscopy. J. Biol. Chem. 2008;283:3708–3717. doi: 10.1074/jbc.M707338200. [DOI] [PubMed] [Google Scholar]
- (50).Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman MR, Kitagawa T. Raman Evidence for Specific Substrate-Induced Structural Changes in the Heme Pocket of Human Cytochrome P450 Aromatase during the Three Consecutive Oxygen Activation Steps. Biochemistry. 2006;45:5631–5640. doi: 10.1021/bi060094a. [DOI] [PubMed] [Google Scholar]
- (51).Champion PM, Stallard BR, Wagner GC, Gunsalus IC. Resonance Raman detection of an iron-sulfur bond in cytochrome P 450cam. J. Am. Chem. Soc. 1982;104:5469–5472. [Google Scholar]
- (52).Mak PJ, Yang Y, Im S-C, Waskell LA, Kincaid JR. Experimental Documentation of the Structural Consequences of Hydrogen-Bonding Interactions to the Proximal Cysteine of a Cytochrome P450. Angew. Chem. Int. Ed. 2012;51:10403–10407. doi: 10.1002/anie.201205912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mak PJ, Zhang H, Hollenberg PF, Kincaid JR. Defining the Structural Consequences of Mechanism-Based Inactivation of Mammalian Cytochrome P450 2B4 Using Resonance Raman Spectroscopy. J. Am. Chem. Soc. 2010;132:1494–1495. doi: 10.1021/ja910276s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nagano S, Cupp-Vickery JR, Poulos TL. Crystal Structures of the Ferrous Dioxygen Complex of Wild-type Cytochrome P450eryF and Its Mutants, A245S and A245T: INVESTIGATION OF THE PROTON TRANSFER SYSTEM IN P450eryF. J. Biol. Chem. 2005;280:22102–22107. doi: 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]
- (55).Estrada DF, Laurence JS, Scott EE. Substrate-modulated cytochrome P 450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 2013;288:17008–17018. doi: 10.1074/jbc.M113.468926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Scott Emily. personal communication of results presented at the 18th International Conference on Cytochrome P450; Seattle, WA. June, 2013; Professor. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.