

Abstract
Here we investigate the effect of hypothermia on the expression of apoptosis-regulating protein TIMP-3 after fluid percussion traumatic brain injury (TBI) in rats. We began with 210 adult male Sprague-Dawley rats and randomly assigned them to three groups: TBI with hypothermia treatment (32°C), TBI with normothermia (37°C), and sham-injured controls. TBI was induced by a fluid percussion TBI device. Mild hypothermia (32°C) was achieved by partial immersion in a water bath (0°C) under general anesthesia for 4 h. The rats were killed at 4, 6, 12, 24, 48, and 72 h and 1 week after TBI. The mRNA and protein level of TIMP-3 in both the injured and uninjured hemispheres of the brains from each group were measured using RT-PCR and Western blotting. In the normothermic group, TIMP-3 levels in both the injured and uninjured hemispheres were significantly increased after TBI compared with those of sham-injured animals (p < 0.01). In contrast, post-traumatic hypothermia significantly attenuated this increase. According to the RT-PCR and Western blot analyses, the maximum mRNA levels of TIMP-3 were reduced to 60.60 ± 2.30%, 55.83 ± 1.80%, 66.03 ± 2.10%, and 64.51 ± 1.50%, respectively, of the corresponding values in the normothermic group in the injured and uninjured hemispheres (cortex and hippocampus) of the hypothermia group (p < 0.01), while the respective maximum protein levels of TIMP-3 were reduced to 57.50 ± 1.50, 52.67 ± 2.20, 60.31 ± 2.50 and 54.76 ± 1.40 (p < 0.01). Our data suggest that moderate fluid percussion brain injury significantly upregulates TIMP-3 expression, and that this increase may be suppressed by hypothermia treatment.
Key words: : hippocampus, mild hypothermia, TIMP-3, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a major cause of morbidity and mortality (Thurman et al. 1999). Studies over the last two decades have demonstrated that a significant amount of central nervous system (CNS) damage after TBI occurs as a result of secondary autodestructive insults (Hayes et al., 1992; Faden, 1996; McIntosh, et al. 1998). Secondary injury involves a complex cascade of biochemical events that contributes to delayed tissue damage and cell death (Kermer et al., 1999; Graham et al., 2000).
Tissue inhibitors of metalloproteinases (TIMPs) are natural matrix metalloproteinase (MMP) inhibitors that impede the proteolytic activity of MMPs by forming noncovalent 1:1 stoichiometric complexes (Fassina et al., 2000). After traumatic spinal cord injury, transient upregulation of TIMP-3 has been detected (Buss et al., 2007). Wallace and colleagues reported that marked TIMP-3 expression in neurons in the cortex and caudate of ischemic hemispheres was observed, together with co-localization of TIMP-3 with TUNEL immunostaining (Wallace et al., 2002). On the other hand, our early studies confirmed that mild to moderate hypothermia can significantly reduce mortality and improve behavioral outcomes in a rodent brain injury model (Clifton et al., 1991). Furthermore, moderate hypothermia provided remarkable protection against secondary insults after impact acceleration in a model of diffuse TBI coupled with both moderate and severe periods of hypoxia and hypotension (Yamamoto et al., 1999).
A hypothesis can be drawn from the above studies that hypothermia may provide protection against secondary insults, partly due to its effect of downregulating TIMP-3, which is associated with reductions in pro-apoptotic events. However, the investigations of Buss and Wallace were carried out in models of traumatic spinal cord injury (SCI) and cerebral ischemia (Buss et al., 2007; Wallace et al., 2002), whereas no data are available on the role of TIMP-3 in TBI and its expression resulting from hypothermic treatment post-TBI.
To test this hypothesis, a model simulating the clinical conditions of TBI was chosen for this study (Dixon et al., 1987), and the expression of TIMP-3 after TBI and hypothermia was investigated, and the results were compared with the values seen in sham-injured animals. Levels of TIMP-3 mRNA were measured quantitatively by reverse transcription-polymerase chain reaction (RT-PCR), while TIMP-3 protein expression was detected by Western immunoblot analysis.
Methods
Animals
Rats weighing 320–380 g were randomly divided into three groups: TBI with hypothermia (32°C), TBI with normothermia (37°C), and sham-injured controls. The time points of assays of all three groups were 4, 6, 12, 24, 48, and 72 h, and 1 week (n = 10 for each time point). The animal procedures were approved by the Animal Care and Experiment Committee of the School of Medicine, Shanghai JiaoTong University.
Surgical preparation
The animals were anesthetized with a nitrous oxide/oxygen mixture (70%/30%) containing 2% halothane. Tracheal intubation was performed, and positive-pressure ventilation was initiated. Both the femoral artery and vein on the right side were cannulated with PE50 polyethylene tubing for monitoring blood pressure and for blood gas analysis. After cannulation, the wound was sutured, and the animals were turned to the prone position. The animals were then placed in a stereotactic frame and the scalp was incised sagittally. A 4.8-mm-diameter burr hole in the skull was bored 2 mm to the left of the midline between the bregma and the lambda. Two fixation screws were then placed 1 mm rostral to the bregma and 1 mm caudal to the lambda. A Luer Lock (Becton Dickinson, Mountain View, CA) needle hub was placed in the skull hole and cemented in position with cyanoacrylate adhesive. Dental acrylic was then poured around the needle hub and the two screws for rigidity. A 1.5-mm burr hole was then drilled at the middle of the occipital bone (1.5 mm from the midline and 1.5 mm from the horizontal edge). A guide (a modified no. 20 1.5-in. needle) for the brain temperature probe was affixed to the surface of the skull over the burr hole with cyanoacrylate adhesive and dental acrylic. During the entire experiment, the mean arterial pressure was monitored continuously, and blood gases were measured before injury and 15 min after injury.
Fluid percussion TBI
A fluid percussion device was used to produce TBI as described in detail previously (Dixon et al., 1987). Briefly, this device consists of a acrylic glass cylindrical reservoir 60 cm long and 4.5 cm in diameter, bounded at one end by a rubber-covered acrylic glass piston mounted on O rings. The opposite end is fitted with a transducer housing terminated with a 2.6-mm inside diameter Luer Lock fitting. The entire system is filled with 37°C isotonic saline. Injury is induced by the descent of a metal pendulum striking the piston, thereby injecting a small volume of saline epidurally into the closed cranial cavity and producing brief displacement and deformation of neuronal tissue. The resulting pressure pulse is measured in atmospheres by an extracranial transducer (Statham PA 85-100; Gould, Oxnard, CA) and recorded on a storage oscilloscope (Tektronix 5111; Tektronix, Beaverton, OR). The animals were injured at a moderate magnitude of injury (2.1 ± 0.2 atm), as described in detail previously (Dixon et al., 1987).
Temperature measurement and hypothermia
Frontal cortex brain temperature was monitored with a digital electronic thermometer (Model DP 80; Omega Engineering, Inc., Stamford, CT) with a 0.15-mm-diameter temperature probe (Model HYP-033-1-T-G-60-SMP-M; Omega Engineering, Inc.) inserted 4.0 mm ventral to the surface of the skull. The probe was removed before fluid percussion injury and replaced immediately after injury. Rectal temperatures were measured with an electronic thermometer (Model 43 TE; YSI, Inc., Yellow Springs, OH) and temperature probe (Series 400; YSI, Inc.). Brain temperature was 37°C before the hypothermic treatment. Hypothermia was achieved as described in a previous study (Jiang et al., 1991). Briefly, a brain temperature of 32°C was achieved by partially immersing the body of the anesthetized rat in an ice-cold water bath (0°C). The skin and fur of all animals were protected from direct contact with the water by placing the animal in a plastic bag with the head exposed before immersion in the water bath. The animals were removed from the water bath when their brain temperature had fallen to within 2°C of the target temperature. It took approximately 15 min to reach the target brain temperature, which was maintained for 4 h under general anesthesia at room temperature by intermittent application of ice packs when needed. Gradual rewarming to normothermic levels (37°C) was done over a 90-min period as described previously (Koizumi and Povlishock, 1998) to avoid rapid rewarming that may have influenced secondary injury processes.
RT-PCR
The time course of mRNA for TIMP-3 was measured by RT-PCR at 4, 6, 12, 24, 48, and 72 h and 1 week post-injury, and comparisons were made between the values of the different groups at each time point. Five rats were studied at each time point. Quantitative real-time RT-PCR with the Lightcycler® System (Roche Diagnostics Corp., CITY?, STATE?) was used to measure mRNA for TIMP-3. Tissue harvested from the hippocampus was homogenized in Trizol reagent (Invitrogen, Carlsbad, CA) for extraction of total RNA according to the manufacturer's protocol, and 2 μg of RNA was used in the following procedures. In a sterile RNase-free microcentrifuge tube, add 20 μm 1 μL of the oligo(dT)15 primer in a total volume of 15 μL in water. The tube was heated to 70°C for 5 min to melt the secondary structure within the template, followed by immediate cooling on ice to prevent the secondary structure from reforming. The tube was then spun briefly and the solution at the bottom was collected. Then we added the following components to the annealed primer/template in this order: 5 μL 5× M-MLV Reaction Buffer, 1.25 μL 10 mM dNTPs, 25 U RNasin Rnase Inhibitor, and 200 U M-MLV RT Rnase H. These were added to the reagent to yield 25 μL total reaction volume. They were mixed by gently shaking the tube, and incubated for 60 min at 42°C before the reaction was terminated at −20°C.
The following primer pairs were used: rat TIMP-3, Forward: 5′-GTGGGAAAGAAGCTGGTGAAG-3′, reverse: 5′-GCACATGGGGCATCTTACTG-3′, and expression of the GAPDH gene acted as the housekeeping gene control. Separate PCR reactions (25 μL) were conducted for each transcript and comprised cDNA (2.0 μL), 12.5 μL 2× SYBR Premix Ex Taq (TaKaRa, Ltd., CITY?, Japan) and each of 0.5 μL 10 μM gene-specific forward and reverse primers. PCR conditions were optimized to 95°C (30 sec), followed by 40 cycles (45 sec each) at 95°C, 60°C (5 sec), and 72°C (30 sec), then the reaction was completed at 37°C for 30 sec. The relative expression levels for each gene of interest were calculated by normalizing the quantified cDNA transcript level (cycle threshold, Ct) to that of GAPDH.
Western blotting
Western blots were used to measure TIMP-3 protein of each animal in each group at all time points (the same as for RT-PCR) post-TBI. Brain tissue was homogenized by disruption through a syringe, and it was then mixed with protein extraction buffer (1 M Tris-HCl [pH 6.8] 0.625 mL, 10% SDS 2 mL, 50% glycerol 2 mL, with water added to yield 10.00 mL). Total protein concentration was determined using the BCA protein assay kit (Bio-Rad Laboratories, Hercules, CA). Then 0.01% bromophenol blue and 10% β-mercaptoethanol were added to the protein sample, and it was boiled for 5 min. Total protein of 100 μg was separated on a 12–15% SDS-polyacrylamide gel by electrophoresis. The proteins were transferred to PVDF immunoblotting membranes (Bio-Rad Laboratories) and run at 120 V for 1 h at 4°C. The membranes were blocked with 5% nonfat milk in PBS with 0.1% Tween-20, rinsed, and then incubated overnight at 4°C with rabbit anti-TIMP-3 antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-human β-actin (1:5000; Abcam, Cambridge, MA). After rinsing, the membranes were incubated with secondary antibody IRDye 800CW goat anti-rabbit fluorescent-labeled secondary antibody (1:2000, LI-COR Biosciences, Lincoln, NE) and IRDye 800CW goat anti-mouse fluorescent-labeled secondary antibody (1:5000, LI-COR Biosciences). The TIMP-3 protein was detected immunologically using the LI-COR imaging system, which allows for the detection of sub-picogram quantities of protein. After a 3-min incubation in the detection reagent, the blot was immediately exposed to Kodak Diagnostic film (Eastman Kodak Co., Rochester, NY). Molecular weight and TIMP-3 standard were included on each blot. Standards for TIMP-3 were a kind gift from P. Alexander (Triple Point Biologics, Portland, OR). Gels from the independent hemispheres from all animals at each time point were run simultaneously to allow for comparisons between the different groups. The gels were scanned with a transparent scanner (Agfa DuoScan; Agfa Corp., Ridgefield Park, NJ), and the scanned images were quantified with a gel electrophoresis analysis program (Alpha Ease; Alpha Innotech Corp., San Leandro, CA). The relative levels of protein expression were calculated by normalizing to that of β-actin.
Statistical analysis
Data are presented as mean ± SD. A one-way analysis of variance (ANOVA) was used to compare the physiological variables measured between groups. Between-group comparisons of TIMP-3 data were conducted using a two-way ANOVA. Post-hoc comparisons were made using the Tukey test. Differences were considered significant if p < 0.05.
Results
Physiological data
Physiological parameters were assessed for each group at 15 min before and 15 min after TBI or sham procedures. All physiological variables with the exception of temperature were within normal ranges for the TBI normothermic and TBI hypothermic groups as well as the sham group (p < 0.01).
RT-PCR
The TIMP-3 mRNA as measured by quantitative RT-PCR remained at a constant level within the tested time period in the sham group as shown in Figure 1(the data for TIMP-3 RT-PCR represents values normalized to GAPDH, while GAPDH was maintained as a relative constant throughout the study [Thellin et al., 1999; Jiang et al., 2009; Aswal et al., 2008]). At 4 h post-TBI, the amount of TIMP-3 mRNA (in both the ipsilateral and contralateral cortex and ipsilateral and contralateral hippocampus) in the normothermic group was significantly increased compared to the sham group (p < 0.01). It continued to increase with time after TBI, and reached the maximum value at around 12 h (in the ipsilateral cortex and hippocampus) and 48 h (in the contralateral cortex and hippocampus) after TBI. A smaller peak in the amount of TIMP-3 mRNA was observed at around 48 h post-TBI in the ipsilateral side. TIMP-3 mRNA expression in the hypothermic group was also increased compared with that in the sham group at 4 h after TBI (p < 0.01). It also continued to increase in a similar manner as that of the normothermic group, with the maximum values reached at the same time point in both the ipsilateral cortex and hippocampus and the contralateral cortex and hippocampus. However, this increase was significantly attenuated in this group compared with the normothermic group. The maximum mRNA levels of TIMP-3 were significantly reduced in the hypothermic group to 60.60 ± 2.30%, 55.83 ± 1.80%, 66.03 ± 2.10%, and 64.51 ± 1.50%, respectively, of the corresponding values in the normothermic group in the injured (ipsilateral) and uninjured (contralateral) hemispheres of the cortex, and the injured (ipsilateral) and uninjured (contralateral) hemispheres of the hippocampus. However, compared with the sham group, TIMP-3 mRNA expression was still significantly higher in the hypothermic group at all time points except for 1 week (p < 0.01). At 1 week after TBI, the TIMP-3 mRNA levels of all groups dropped to 0.013 ??, with no significant differences in the ipsilateral cortex and hippocampus or the contralateral cortex and hippocampus. For the sham-injured control group, the mRNA levels of TIMP-3 remained at a relatively constant low value in the ipsilateral cortex (0.010 ± 0.001) and hippocampus (0.009 ± 0.001), and the contralateral cortex (0.011 ± 0.002) and hippocampus (0.010 ±0.001) (p > 0.05).
FIG. 1.
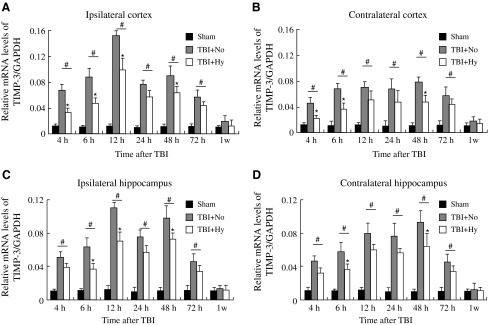
Time course of TBI-induced expression of TIMP-3 mRNA. RT-PCR was used to measure relative levels of TIMP-3 mRNA in the ipsilateral cortex (A) and contralateral cortex (B), and the ipsilateral (C) and contralateral (D) hippocampus of the rats in the three study groups. The relative expression levels for each gene of interest were calculated by normalizing the quantified cDNA transcript level (cycle threshold, Ct) to that of GAPDH (Sham, sham-injured; TBI + No, TBI and normothermia; TBI + Hy, TBI and hypothermia). Data represent mean ± SD (n = 5; #p < 0.01 hypothermia and normothermia groups versus sham-operated group; *p < 0.01 hypothermia group versus normothermia group).
Thus it can be seen that after TBI, levels of expression of TIMP-3 mRNA were higher than those in the sham-injured group, and that hypothermia significantly inhibited the expression of TIMP-3 mRNA both in the injured and the uninjured hemispheres.
Western blot
Western blot analysis was carried out to further assess the expression of TIMP-3 in the protein level. At all time points, the expected 22-kd band was seen, representing the standard TIMP-3 expression (Fig. 2).
FIG. 2.

Time course of TBI-induced expression of TIMP-3 proteins. Western blots of TIMP-3 in the ipsilateral cortex (A) and contralateral cortex (B), and the ipsilateral (C) and contralateral (D) hippocampus of the rats in the three study groups. The relative levels of protein expression were calculated by normalizing to that of β-actin (Sham, sham-injured; NO and TBI + No, TBI and normothermia; Hy and TBI + Hy, TBI and hypothermia). Data represent mean ± SD (n = 5; #p < 0.01 for the hypothermia and normothermia groups versus the sham-operated group; *p < 0.01 for the hypothermia versus the normothermia group).
TIMP-3 expression was seen in the sham-injured animals, indicating its presence in normal tissues. At 4 h post-TBI, the amount of TIMP-3 in both the normothermic and hypothermic groups was increased significantly over that seen in the sham-injured group (p < 0.01). With increasing time, the TIMP-3 protein levels in the normothermic and hypothermic groups continued to increase, with the maximum levels reached at around 24 h and 72 h post-TBI in the ipsilateral and contralateral hemispheres, respectively (Fig. 2). Similarly to the RT-PCR results, a second peak in the amount of TIMP-3 protein was seen at around 72 h post-TBI on the ipsilateral side. However, in the cortex of the injured (ipsilateral) hemisphere, the increases in TIMP-3 protein in the hypothermic group were less than those seen in the normothermic group at all time points (p < 0.01). The maximum protein levels of TIMP-3 in the hypothermic group were 57.50 ± 1.50%, 52.67 ± 2.20%, 60.31 ± 2.50%, and 54.76 ± 1.40%, respectively, of the values in the normothermic group in the injured (ipsilateral) and uninjured (contralateral) cortex, and the injured (ipsilateral) and uninjured (contralateral) hippocampus. However, compared with the sham group, the TIMP-3 protein expression was still significantly higher in the hypothermic group at all time points except for 1 week in the uninjured (contralateral) hemisphere (p < 0.01). At 1 week post-TBI, the TIMP-3 protein level in the injured hemispheres dropped to 0.012 ??, and there were no significant differences in the contralateral cortices and hippocampi. For the sham-injured control group, the protein levels of TIMP-3 remained at a relatively constant low value in the ipsilateral cortex (0.009 ± 0.001) and hippocampus (0.009 ±0.002), and the contralateral cortex (0.011 ± 0.001) and hippocampus (0.010 ± 0.002) (p > 0.05).
Discussion
It was shown in this study that the TIMP-3 levels in both the injured and uninjured hemispheres increased significantly after fluid percussion brain injury, while mild hypothermia induced soon after injury significantly attenuated this increase, both in the gene and the protein levels. These findings suggest that the protection against secondary insults provided by hypothermia may partially be due to its effect in downregulating TIMP-3 in both injured and uninjured tissues.
The extracellular matrix-bound TIMP-3 has several functions, including the facilitation of apoptosis (Leco et al., 1994; Baker et al., 1999; Brew et al., 2000; Deng et al., 2006; Chetty et al., 2008). Although the mechanism by which TIMP-3 promotes cell death has not been fully elucidated, prior studies suggest a link between death receptor-mediated apoptosis and MMP inhibition by TIMP-3 (Smith et al., 1997; Bond et al., 2000, 2002). Tissue inhibitors of metalloproteinases (TIMPs) are important regulators of matrix MMP and adamalysin metalloproteinase activity. MMPs are essential for remodeling the extracellular matrix, tissue morphogenesis, and wound healing (Werb, 1997). However, excessive proteolytic activity of MMPs can be detrimental, leading to numerous pathologic conditions, including disruption of the blood–brain barrier (Yong et al., 2001), apoptosis (Gu et al., 2002), and inflammation (Mun-Bryce and Rosenberg, 1998). In agreement with the findings detailed here, TIMP-3 and MMP-9 have been found to be upregulated after TBI and inhibited by hypothermia (Truettner et al., 2005). These observations imply a physiological role for the balance of TIMP-3 and MMP activity at the neuronal surface in regulating death receptor sensitivity.
It has been proposed that TIMP-3 inhibits MMPs and A Disintegrin And Metalloproteinase (ADAMs) proteins that are involved in the shedding of death receptors (DRs) and death-inducing ligands (DILs) from the cell surface (Amour et al., 1998, 2000). Programmed cell death occurs via the release of caspase-activating factors from mitochondria, and by stimulating cell surface receptors that may act through mitochondrial activation (Ashkenazi and Dixit, 1998). Previous studies have suggested that TIMP-3 acts through cell surface receptors. When cell surface DRs are engaged by DILs in association with the activation of the caspase cascade, cell death usually occurs, according to the findings of Ashkenazi and Dixit (Ashkenazi and Dixit, 1998). Some DRs may be induced by TBI, such as Fas (CD95/APO-1) and tumor necrosis factor-α (TNF-α) receptor (Qiu et al., 2002; Hang et al., 2005). Moreover, the inhibition of TNF-α-receptor cleavage has also been reported to be caused by TIMP-3, and this also facilitates cell death (Smith et al., 1997).
To the best of our knowledge, few studies have been carried out to examine the relationship between TIMP-3 and post-TBI hypothermia. In this study, we found that the expression of TIMP-3 increased significantly in both the hypothermia and normothermia TBI groups when assayed at 4 h post-TBI. A significant decrease in the expression of TIMP-3 was observed in the hypothermia group compared with the normothermia group, as confirmed both by the RT-PCR results and Western blot analysis. Thus it can be seen that TBI leads to significant enhancements in the expression of TIMP-3 mRNA and protein compared to sham-injured animals up to 1 week post-TBI, and that hypothermia significantly inhibits the expression of TIMP-3 mRNA and protein in both the injured and uninjured hemispheres over the same period.
Our study also showed that the TIMP-3 protein and mRNA expression profiles were different not only in the different hemispheres, but also in different regions of the same hemisphere. Two expression peaks of TIMP-3 mRNA and protein were observed in the ipsilateral cortex and hippocampus. The finding that the first TIMP-3 mRNA and protein peaks in the ipsilateral cortex were higher than those seen in the ipsilateral hippocampus may due to the fact that the direct mechanical injury to the cortex was more serious than that to the ipsilateral hippocampus, and similar results were found by Buss and colleagues. In that study, they showed that traumatic spinal cord injury induces transient upregulation of TIMP-3 (Buss et al., 2007). We infer that the higher value of the first peak may due to this direct damage, while the lower level of the second peak may be due to the secondary damage caused by autodestructive insults such as ischemia. However, only one peak in TIMP-3 mRNA was observed in the contralateral cortex and hippocampus. The TIMP-3 mRNA peaked at 48 h in both the cortex and hippocampus of that side. The higher TIMP-3 values seen in the contralateral hippocampus than in the contralateral cortex may also be due to secondary damage (p < 0.05; mRNA T = 48 h; protein, T = 72 h), or perhaps the hippocampus was more susceptible to secondary damage from ischemia (Chen et al., 2007; Yamashita et al., 2007). The increases seen in the early period (peaks in mRNA and protein at 12 h and 24 h, respectively) in the ipsilateral hemisphere may due to direct mechanical damage, or perhaps the distance between the ipsilateral hippocampus and the damaged site (ipsilateral cortex) attenuated the increase, such that the increase in the ipsilateral hippocampus was significantly lower than that seen in the ipsilateral cortex (p < 0.01). Although further studies will be necessary to reveal the underlying mechanism behind the activation of TIMP-3 after hypothermically-treated TBI, the results of this study provide convincing evidence of increases in TIMP-3 levels in both the injured and uninjured hemispheres. This is also consistent with the finding of TIMP-3 gene expression post-brain injury in Wobbler mice (Jaworski et al., 2000; Rathke-Hartlieb et al., 2000).
Several factors may explain why hypothermia influences TIMP-3 expression after TBI. It has been reported that therapeutic hypothermia targets various pathologic mechanisms, including temperature-dependent reductions in the cerebral metabolic rate of oxygen, decreases in free radical production (Hayashi et al., 1997), limitations of blood–brain barrier disruption (Jiang et al., 1991; Smith and Hall, 1996; Lotocki et al., 2006), and reductions in brain edema (Marion and White, 1996). Others have reported attenuation of ionic disruption (Yamamoto et al., 1999), decreased release of excitatory amino acids, increased expression of inflammatory response genes (Busto et al., 1989; Mitani et al., 1991; Truettner et al., 2005), shunting of an increased fraction of glucose metabolism (Kaibara et al., 1999), and inhibition of excessive calcium neuronal entry and intracellular calcium overload (Mitani et al., 1991). Although our findings suggest that hypothermic protection can downregulate TIMP-3 expression, more thorough investigations are needed of the mechanisms behind the regulation of TIMP-3 levels in the brain. In conclusion, the study detailed here not only opens up the possibility of using hypothermia and other agents to interfere with the action of TIMP-3, but also may herald the creation of novel treatments to reduce apoptosis.
Acknowledgments
This work was supported by grants from the National Key Basic Research Project (no. 2005CB522604), National Science and Nature Grant (no. 30571908), the Science and Technology Committee of Shanghai (no. 07JC14038), and the Program for Shanghai Outstanding Medical Academic Leader.
Author Disclosure Statement
No competing financial interests exist.
References
- Amour A., Knight C.G., Webster A., Slocombe P.M., Stephens P.E., Knauper V., Docherty A.J., and Murphy G. (2000). The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 473, 275–279 [DOI] [PubMed] [Google Scholar]
- Amour A., Slocombe P.M., Webster A., Butler M., Knight C.G., Smith B.J., Stephens P.E., Shelley C., Hutton M., Knauper V., Docherty A.J., Murphy G. (1998). TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39–44 [DOI] [PubMed] [Google Scholar]
- Ashkenazi A., and Dixit V.M. (1998). Death receptors: signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- Aswal A.P., Raghav S., De S., Thakur M., Goswami S.L., and Datta T.K. (2008). Expression stability of two housekeeping genes (18S rRNA and G3PDH) during in vitro maturation of follicular oocytes in buffalo (Bubalus bubalis). Anim. Reprod. Sci. 103, 164–171 [DOI] [PubMed] [Google Scholar]
- Baker A.H., George S.J., Zaltsman A.B., Murphy G., and Newby A.C. (1999). Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br. J. Cancer 79, 1347–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M., Murphy G., Bennett M.R., Amour A., Knauper V., Newby A.C., and Baker A.H. (2000). Localization of the death domain of tissue inhibitor of metalloproteinase-3 to the N terminus. Metalloproteinase inhibition is associated with proapoptotic activity. J. Biol. Chem. 275, 41358–41363 [DOI] [PubMed] [Google Scholar]
- Bond M., Murphy G., Bennett M.R., Newby A.C., and Baker A.H. (2002). Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J. Biol. Chem. 277, 13787–13795 [DOI] [PubMed] [Google Scholar]
- Brew K., Dinakarpandian D., and Nagase H. (2000). Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim. Biophys. Acta. 1477, 267–283 [DOI] [PubMed] [Google Scholar]
- Buss A., Pech K., Kakulas B.A., Martin D., Schoenen J., Noth J., and Brook G.A. (2007). Matrix metalloproteinases and their inhibitors in human traumatic spinal cord injury. BMC Neurol. 26, 7–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busto R., Globus M.Y.T., Dietrich W.D., Martinez E., Valdés I., and Ginsberg M.D. (1989). Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke 20, 904–910 [DOI] [PubMed] [Google Scholar]
- Chen Z.Y., Asavaritikrai P., Prchal J.T., and Noguchi C.T. (2007). Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J. Biol. Chem. 31, 25875–25883 [DOI] [PubMed] [Google Scholar]
- Chetty C., Lakka S.S., Bhoopathi P., Kunigal S., Geiss R., and Rao J.S. (2008). Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Res. 15, 4736–4745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton G.L., Jiang J.Y., Lyeth B.G., Jenkins L., and Hayes R.L. (1991). Marked protection by moderate hypothermia after experimental TBI. J. Cereb. Blood Flow. Metab. 11, 114–121 [DOI] [PubMed] [Google Scholar]
- Deng X., Bhagat S., Dong Z., Mullins C., Chinni S.R., and Cher M. (2006). Tissue inhibitor of metalloproteinase-3 induces apoptosis in prostate cancer cells and confers increased sensitivity to paclitaxel. Eur. J. Cancer 2006, 3267.—3273 [DOI] [PubMed] [Google Scholar]
- Dixon C.E., Lyeth B.G., Povlishock J.T., Findling R.L, Hamm R.J., Marmarou A., Young H.F., and Hayes R.L. (1987). A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119 [DOI] [PubMed] [Google Scholar]
- Faden A.I. (1996). Pharmacologic treatment of acute traumatic brain injury. J.A.M.A. 276, 569–570 [PubMed] [Google Scholar]
- Fassina G., Ferrari N., Brigati C., Benell R., Santi L., Noonan D.M., and Albini A. (2000). Tissue inhibitors of metalloproteases: regulation and biological activities. Clin. Exp. Metastasis 18, 111–120 [DOI] [PubMed] [Google Scholar]
- Graham D.I., McIntosh T.K., Maxwell W.L., and Nicoll J.A. (2000). Recent advances in neurotrauma. J. Neuropathol. Exp. Neurol. 59, 641–651 [DOI] [PubMed] [Google Scholar]
- Gu Z., Kaul M., Yan B., Kridel S.J., Cui J., Strongin A., Smith J.W., Liddington R.C., and Lipton S.A. (2002). Signaling pathway to neuronal cell death S-nitrosylation of matrix metalloproteinases. Science 297, 1186–1190 [DOI] [PubMed] [Google Scholar]
- Hang C.H., Shi J.X., Li J.S., Li W.Q., and Wu W. (2005). Expressions of intestinal NF-kappaB, TNF-alpha, and IL-6 following traumatic brain injury in rats. J. Surg. Res. 123, 188–193 [DOI] [PubMed] [Google Scholar]
- Hayashi N., Kinosita K., and Utagawa A. (1997). Prevention of cerebral thermo-pooling, free radical reactions, and protection of A10 nervous system by control of brain tissue temperature in severely brain injured patients, in: Neurochemistry. New York: Plenum Press, pps. 97–103 [Google Scholar]
- Hayes R.L., Jenkins L.W., and Lyeth B.G. (1992). Neurotransmitter mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J. Neurotrauma 9, S173–S187 [PubMed] [Google Scholar]
- Jiang C., Meng L., Zhu W., Shahzad M., Yang X., and Lu S. (2009). Housekeeping gene stability in pristane-induced arthritis and antigen-induced pulmonary inflammation of rats. Inflamm. Res. 2009April9 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Jiang J.Y., Lyeth B.G., Clifton G.L., Jenkins L.W., Harem R.J., and Hayes R.L. (1991). Relationship between body and brain temperature in traumatically brain-injured rodents. J. Neurosurg. 74, 492–496 [DOI] [PubMed] [Google Scholar]
- Kermer P., Klocker N., and Baèhr M. (1999). Neuronal death after brain injury. Models, mechanisms, and therapeutic strategies in vivo. Cell Tissue Res. 298, 383–395 [DOI] [PubMed] [Google Scholar]
- Kaibara T., Sutherland G.R., Colbourne F., and Tyson R.L. (1999). Hypothermia. Depression of tricarboxylic acid cycle flux and evidence for pentose phosphate shunt upregulation. J. Neurosurg. 90, 339–347 [DOI] [PubMed] [Google Scholar]
- Koizumi H., and Povlishock J.T. (1998). Posttraumatic hypothermia in the treatment of axonal damage in an animal model of traumatic axonal injury. J. Neurosurg. 89, 303–309 [DOI] [PubMed] [Google Scholar]
- Leco K.J., Khokha R., Pavloff N., Hawkes S.P., and Edwards D.R. (1994). Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J. Biol. Chem. 269, 9352–9360 [PubMed] [Google Scholar]
- Lotocki G., de Rivero Vaccari J.P., Perez E.R., Alonso O.F., Curbelo K., Keane R.W., and Dietrich W.D. (2006). Therapeutic hypothermia modulates TNFR1 signaling in the traumatized brain via early transient activation of the JNK pathway and suppression of XIAP cleavage. Eur. J. Neurosci. 24, 2283–2290 [DOI] [PubMed] [Google Scholar]
- Mitani A., Kadoya F., and Kataoka K. (1991). Temperature dependence of hypoxia induced calcium accumulation in gerbil hippocampal slices. Brain Res. 562, 159–163 [DOI] [PubMed] [Google Scholar]
- Mitani A., and Kataoka K. (1991). Critical levels of extracellular glutamate mediating gerbil hippocampal delayed neuronal death during hypothermia: Brain microdialysis study. Neuroscience 42, 661–770 [DOI] [PubMed] [Google Scholar]
- Marion D.W., and White M.J. (1996). Treatment of experimental brain injury with moderate hypothermia and 21-aminosteroids. J. Neurotrauma 13, 139–141 [DOI] [PubMed] [Google Scholar]
- McIntosh T.K., Saatman K.E., Raghupathi R., Graham D.I., Smith D.H., Lee V.M., and Trojanowski J.Q. (1998). The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol. Appl. Neurobiol. 24, 251–267 [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S., and Rosenberg G.A. (1998). Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am. J. Physiol. 274, R1203–R1211 [DOI] [PubMed] [Google Scholar]
- Qiu J., Whalen M.J., Lowenstein P., Fiskum G., Fahy B., Darwish R., Aarabi B., Yuan J., and Moskowitz M.A. (2002). Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J. Neurosci. 22, 3504–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.R., Kung H., Durum S.K., Colburn N.H., and Sun Y. (1997). TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine 9, 770–780 [DOI] [PubMed] [Google Scholar]
- Smith S.L., and Hall E.D. (1996). Mild pre- and posttraumatic hypothermia attenuates blood-brain barrier damage following controlled cortical impact injury in the rat. J. Neurotrauma. 13, 1–9 [DOI] [PubMed] [Google Scholar]
- Thellin O., Zorzi W., Lakaye B., De Borman B., Coumans B., Hennen G., Grisar T., Igout A., and Heinen E. (1999). Housekeeping genes as internal standards: use and limits. J. Biotechnol. 75, 291–295 [DOI] [PubMed] [Google Scholar]
- Thurman D.J., Alverson C., Dunn K.A., Guerrero J., and Sniezek J.E. (1999). Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 14, 602–615 [DOI] [PubMed] [Google Scholar]
- Truettner J.S., Alonso O.F., and Dalton Dietrich W.D. (2005). Influence of therapeutic hypothermia on matrix metalloproteinase activity after traumatic brain injury in rats. J. Cereb. Blood Flow. Metab. 25, 1505–1516 [DOI] [PubMed] [Google Scholar]
- Truettner J.S., Alonso O.F., and Dietrich W.D. (2005). The effect of therapeutic hypothermia on the expression of inflammatory response genes following moderate traumatic brain injury in the rat. Brain Res. Mol. Brain Res. 138, 124–134 [DOI] [PubMed] [Google Scholar]
- Wallace J.A., Alexander S., Estrada E.Y., Hines C., Cunningham L.A., and Rosenberg G.A. (2002). Tissue inhibitor of metalloproteinase-3 is associated with neuronal death in reperfusion injury. J. Cereb. Blood Flow Metab. 22, 1303–1310 [DOI] [PubMed] [Google Scholar]
- Werb Z. (1997). ECM and cell surface proteolysis: regulating cellular ecology. Cell 91, 439–442 [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Marmarou C.R., Stiefel M.F., Beaumont A., and Marmarou A. (1999). Neuroprotective effect of hypothermia on neuronal injury in diffuse traumatic brain injury coupled with hypoxia and hypotension. J. Neurotrauma 16, 487–500 [DOI] [PubMed] [Google Scholar]
- Yamashita A., Kunimatsu T., Yamamoto T., and Yoshida K. (2007). Hypothermic, but not normothermic, ischemia causes a drastic increase in cyclooxygenase-2 immunoreactive granule cells in rat dentate gyrus after 4 hours of ischemic reperfusion. Arch. Histol. Cytol. 70, 197–205 [DOI] [PubMed] [Google Scholar]
- Yong V.W., Power C., Forsyth P., and Edwards D.R. (2001). Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]