

Abstract
Alzheimer's disease (AD) is a heterogeneous neurodegenerative disorder and the most prevalent form of dementia worldwide. AD is characterized pathologically by amyloid-β plaques, neurofibrillary tangles and neuronal loss, and clinically by a progressive loss of cognitive abilities. At present, the fundamental molecular mechanisms underlying the disease are unclear and no treatment for AD is known. Epidemiological evidence continues to mount linking vascular diseases, such as hypertension and diabetes, and hypercholesterolaemia with an increased risk for developing AD. A growing amount of evidence suggests a mechanistic link between cholesterol metabolism in the brain and the formation of amyloid plaques in AD development. Cholesterol and statins clearly modulate β-amyloid precursor protein (βAPP) processing in cell culture and animal models. Statins not only reduce endogenous cholesterol synthesis but also exert other various pleiotrophic effects, such as the reduction in protein isoprenylation. Through these effects statins modulate a variety of cellular functions involving both cholesterol (and membrane rafts) and isoprenylation. Although clearly other factors, such as vascular inflammation, oxidative stress and genetic factors, are intimately linked with the progression of AD, this review focuses on the present research findings describing the effect of cholesterol, membrane rafts and isoprenylation in regulating βAPP processing and in particular γ-secretase complex assembly and function and AD progression, along with consideration for the potential role statins may play in modulating these events.
Keywords: cholesterol, secretases, Alzheimer's disease, statins, isoprenoids, APP metabolism, Abeta, Tau, membrane rafts
Introduction
Alzheimer's disease (AD) is a devastating neurologi-as the number of people worldwide over the age of cal disorder that has become increasingly common 65 increases. Understanding the complex etiological and pathological underpinnings of the disease represent our best hope for developing therapies capable of treating and/or reversing this devastating disease. Although the several pathological hallmarks associated with AD, such as amyloid plaques and neurofibrillary tangles are well known, no single theory appears to completely explain and/or account for all of the complex pathological changes that ultimately lead to neurodegeneration and cognitive dysfunction. For the past decade, the prevailing theory underlying AD has been the ‘amyloid cascade hypothesis’, in which the major causative molecule responsible for the disease is amyloid (A).Therefore, much of the research community has focused on elucidating the mechanisms underlying the processing and production of this peptide.
βAPP processing and Aβ generation
It is well established that the abnormal generation and deposition of amyloid β peptides (Aβ) is a pathologic hallmark of AD. Aβ is generated by two sequential proteolytic cleavage steps from the β-amyloid precursor protein (βAPP) [Reviewed in 1–3]. βAPP is initially cleaved by β-secretase, which has been identified as a β-site APP-cleaving transmembrane aspartic protease (BACE) [4], which has been suggested to occur in the endocytic pathway. This cleavage is followed by the subsequent intramembrane proteolysis of the membrane-bound C-terminal fragment (CTF, C99) catalyzed by the γ-secretase complex, which has been suggested to occur at the plasma membrane, although the exact cellular location of either of the cleavage events is not clearly understood.
Importantly, the Aβ peptides produced by γ-secretase cleavage are heterogeneous in size, ranging from 38 to 43 amino acids, with the highly amyloid-genic 42 amino acid peptide suggested to be the most pathogenic [5]. These Aβ peptides can exist as monomers, oligomers or as amyloid fibrils [5]. Recent evidence suggests that the oligomeric form of these Aβ peptides may be the key form responsible for the cognitive problems of people with AD [5, 6]. Interestingly, the exact function of βAPP processed Aβ peptides remains unknown. In an alternative pathway βAPP is cleaved by α-secretase, a member of the ADAMs (a disintegrin and metallo-protease) family of enzymes, within the Aβ domain and the remaining CTF (C83) is also cleaved by γ-secretase to release the non-amyloidgenic p3 peptide. Not all Aβ is released extracellularly, as recent evidence suggests that intraneuronal Aβ is also toxic and appears to play a significant role in the perturbation of cognitive function [7–9].
The activation of the γ-secretase complex activity requires the formation of a stable high-molecular-weight multiprotein complex, which includes presenilin, nicastrin (NCT), anterior pharynx-defective-1 (APH-1) and PS-enhancer-2 (PEN-2) [Reviewed in 10]. These four transmembrane proteins are presumed to be indispensable for γ-secretase activity, because their co-expression enables reconstitution of the γ-secretase activity [10–14], while the absence of even one results in the absence of γ-secretase activity and defects in the expression and/or maturation of the remaining partners [10, 11]. NCT, an integral membrane protein, has been reported to bind tightly to βAPP-C99 and recruit it into the γ-secretase complex [15]. Conformation changes in and/or N-linked glycosylation of NCT appears to play a prominent role in modulating γ-secretase activity [16–19]. Various APH-1 variants are believed to stabilize presenilin within the γ-secretase complex [20], while PEN-2 appears necessary for the endoproteolysis of presenilin, a step essential for the activity of the γ-secretase complex [21, 22]. In addition to the processing of βAPP, the γ-secretase complex facilitates the regulated intramembrane proteolysis of other select type I membrane proteins, such as Notch, CD44 and E-cadherin, which play diverse physiological roles in multiple cell types and tissue [1].
Cholesterol
There is growing evidence that cholesterol is linked to the development of AD [Reviewed in 23, 24]. Clearly there exists a relationship between AD and hypercho-lesterolaemia, in addition to coronary artery disease and hypertension [25–27]. Various experimental and clinical findings also strongly suggest that brain vascular and hemodynamic alternations may play an important role in the progression of AD [28–30]. In addition, the regulation of cholesterol homeostasis appears to also be perturbed, as the 4 allele of ApoE, a major apolipoprotein in the brain, has been identified as important risk factors for AD [31–34].
The central nervous system (CNS) contains about 25% of the total body cholesterol, the highest content of any organ, the majority of which is found in neuronal plasma membranes and the myelin sheaths of axons [35]. Cholesterol is a major regulator of membrane lipid organization and fluidity, and as such, mammals have developed complex and sophisticated homeostatic mechanisms that function to maintain cellular cholesterol levels in membranes within a narrow range. Alterations and/or disturbances in these homeostatic mechanisms, either by genetic, environmental, diet-induced or natural aging can lead to altered cell function and disease [36]
In AD, the γ-secretase complex catalyzes the cleavage of βAPP within the hydrophobic lipid bilayer suggesting that perturbations in cellular cholesterol levels, trafficking and/or organization that disorganize the structure of the protein–lipid bilayer may accelerate or contribute to the generation of Aβ. Animals fed cholesterol rich diets exhibit increased brain Aβ levels that can be reduced upon return of the animals to normal chow diets [37, 38]. In vivo studies have demonstrated that high cholesterol diets can increase Aβ levels in rabbits and in an AD mouse model [39]. While in vitro studies indicate that increased cellular cholesterol levels result in the increased production of Aβ peptides [40, 41]. In addition, cholesterol depletion also inhibits Aβ generation in hippocampal neurons [41] and increases γ-secretase activity in cultured cells [42, 43]. Although these studies reveal that cellular cholesterol levels can modulate βAPP processing, the exact mechanism by which this occurs remains unclear. To understand the relationship between cellular cholesterol levels and cellular γAPP processing and Aβ production, we must discuss the distribution of membrane cholesterol and membrane rafts.
Membrane rafts
Membrane rafts (previously referred to as lipid rafts) are dynamic highly ordered membrane microdomains enriched in cholesterol, sphingolipids and saturated phospholipids distinct from the surrounding bilayer of mostly unsaturated phospholipids. Proteins can be selectively included or excluded from these microdomains [Reviewed in 44–47]. Although there remains considerable uncertainty about the abundance, size, duration and exact composition of membrane rafts (some of which we believe is attributable to cell type variation), membrane rafts are believed to be around 50 nm in diameter (10–100 nm range), with each individual raft potentially carrying around 20 protein molecules [described in 48, 49]. Theoretically, a cell may have around 1,000,000 membrane rafts covering more than half of its membrane surface. In this fashion, it is unlikely that a protein in one raft would encounter its interaction partner or substrate in the same individual raft. This function underlies the theory that the small size of individual membrane rafts may serve to segregate and hold membrane signaling proteins in the ‘off’ state.
Once the cell is activated, membrane rafts are believed to function as a concentrating platform for a variety of signal transduction molecules [44–47]. During activation, many rafts would cluster, forming a larger platform, thus allowing functional proteins to concentrate and interact, likely with cytoplasmic signaling factors, such as small G proteins, that are recruited to the cytoplasmic face of rafts in response to clustering, thereby facilitating the initiation of signaling events. Due to the highly ordered nature of lipid rafts, glycosyl-phosphatidylinositol (GPI) anchored and doubly acylated proteins tend to cluster in these microdomains. Additionally, other proteins have shown the ability to move in and out of membrane rafts in response to ligand binding or oligomerization. Importantly, this clustering is believed to be cholesterol dependent [48, 49]. In this regard, membrane rafts are believed to play a central role in regulating several cellular processes, such as membrane sorting, trafficking and signal transduction [45, 47].
Several lines of evidence suggest the involvement of membrane rafts in β- and γ-cleavage of βAPP [Reviewed in 48, Refer to Fig. 1]. It has been reported that the proteins relevant to Aβ generation, including presenilin, NCT, APH-1, PEN-2 and a small portion of βAPP, localize in membrane rafts [49–59]. In addition to the γ-secretase components, it has also been reported that β-secretase localizes in membrane rafts and that cholesterol depletion abrogates this localization [49, 50]. Ehehalt et al. [49] have reported that βAPP exists in two pools, one associated with membrane rafts, in which β-cleavage occurs, and another outside of membrane rafts, where α-cleavage occurs, suggesting that raft localization favors the generation of A. Recent reports have suggested that γ-secretase activity is predominantly localized in membrane rafts and cholesterol can directly regulate the γ-secretase activity [59]. Collectively, these findings suggest that the raft or non-raft membrane distribution of βAPP may in part determine its processing fate. It has been suggested that during embryonic development, the γ-secretase complex may be primarily located in non-raft membranes facilitating the proteolysis of diverse substrates important for development, while the translocation of β-secretase to membrane rafts in adults facilitates the processing of adult-specific substrates, including βAPP CTFs, while limiting the processing of other potential substrates [60]. Although the experimental evidence remains unclear, under such a scenario, high cellular cholesterol levels and/or lipid bilayer alterations that increase the number or potentially the size of membrane rafts would disrupt the segregation ability of the raft/non-raft membrane and unintentionally allow the interaction between βAPP and the γ-secretase complex in an individual raft, thus favoring Aβ production. Low cellular cholesterol levels or disruption of lipid rafts could be envisaged to decrease Aβ production and favor the non-pathogenic cleavage by α-secretase. In further support of the importance of membrane rafts in AD progression, it has been recently reported that following dimeric Aβ accumulation in membrane rafts, both ApoE and phopshorylated tau, were observed to increase in membrane rafts in the aged Tg2576 mouse model of AD [61].
1.
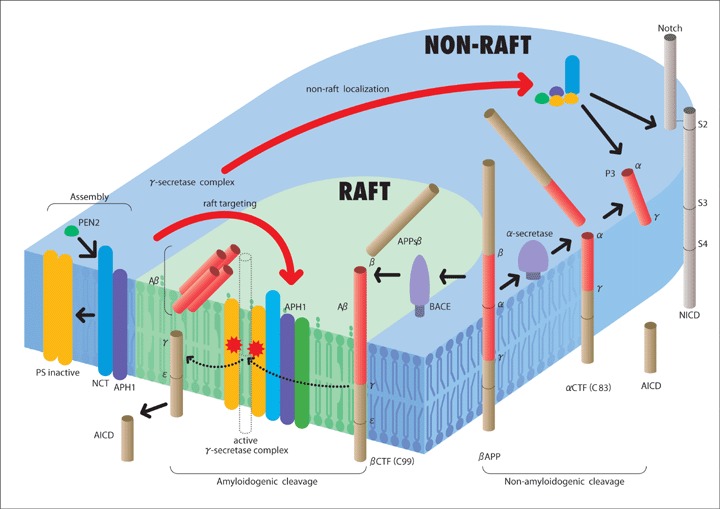
Schematic diagram of lipid mediated dynamic association of γ-secretase complex with its substrates. Assembly of presenilin (PS) with nicastrin (NCT) and APH-1 stabilizes PS, and the association of PEN2 activates PS to fragmented form [11]. Active γ-secretase complex is targeted to membrane rafts by lipid modifications, such as myristoylation, palmitoylation or double isoprenylation. Amyloid precursor protein (βAPP) is mostly cleaved by α-secretase in non-raft domain to generate APPsα and αCTF, but some part of βAPP in the membrane raft is cleaved at the beta site by β-secretase, which resides in cholesterol rich membrane raft, to generate APPsβ and βCTF. After the α- or β-cleavage, NCT serves as a receptor for the γ-secretase substrates. Accordingly, the βCTF (C99) is cleaved at gamma site by γ-secretase in the raft domain to generate pathogenic form of Aβ peptides. C-terminal peptide is further cleaved at ζ or site by γ-secretase to generate amyloid intracellular domain (AICD) peptide. In the non-raft domain, αCTF (C83) is further cleaved by γ-secretase to generate P3 and AICD peptides. Other γ-secretase substrates, such as the CTFs derived from Notch1, Jagged2, deleted in colorectal cancer (DCC), and N-cadherin, are cleaved in non-raft domain [61].
Diet, genetic factors and environmental factors may well contribute to, in association with membrane lipid changes associated with aging [Reviewed in 62], the perturbation of membrane raft structure and function and thus ultimately imbalance this important segregation of proteolytic complex, γ-secretase and/or α-secretase and βAPP titling the balance toward pathogenic Aβ production. This scenario could be envisaged to play out over years and be intimately intertwined with the aging process, in a fashion similar to cardiovascular disease. Due to similarities between AD and cardiovascular disease, and the well-established benefit of reducing cholesterol levels in patients with cardiovascular disease, similar therapeutic strategies have received significant consideration for the treatment of AD [31].
Statins
Several epidemiological studies have suggested that the use of 3-hydroxy-3-methylglutaryl-CoA-reduc-tase inhibitors (‘statins’) to treat hypercholestero-laemia may reduce the risk of dementia [63–66]. Statins efficiently inhibit the rate-limiting enzyme, HMG CoA reductase, in the cholesterol biosynthetic pathway responsible for the production of non-sterol isoprenoids, notably farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), ubiquinone and dolichol, in addition to cholesterol [67]. Statins also effect intracellular cholesterol distribution, gene expression and proteasome activity [68]. Although there have been initial reports that treatment with cholesterol synthesis inhibitors can lower the Aβ levels in guinea pig and in the AD mouse [40, 68], recent findings suggest that the lowering of Aβ levels by statins may not be so clear [69, 70]. Statins are believed to exert their effects by manipulating cholesterol and isoprenoid levels intracellularly leading to altered βAPP processing and membrane signaling. Statin treatment has been shown to decrease the association of the active form of the γ-secretase complex with membrane rafts [59]. Whether this effect is due to statin's reduction in cholesterol biosynthesis or a reduction in isoprenylation, or the more likely scenario, in which it is a complex combination of both, remains to be clearly elucidated [Reviewed in 71, 72]. It has also been suggested that statin-treatment-induced inhibition of isoprenylation results in a reduction in membrane raft clustering involved in βAPP processing and Aβ generation [73].
The clinical relevance of these findings is questionable, as it has been shown that at clinical dosages, only low statin concentrations are detected in the cerebrospinal fluid, and at such concentrations, although cholesterol biosynthesis may be reduced, it appears highly unlikely that isoprenoid synthesis is inhibited in the CNS [74–76]. Therefore, there may be other mechanisms underlying the clinical benefit of statin therapy [77]. It remains highly plausible that many of the positive benefits of statin treatment may in fact be associated with modifications and modulations of both the structure and functioning of membrane rafts, not only in the CNS, but possibly predominantly in the periphery, leading to statin's anti-oxidant and anti-inflammatory properties.
Isoprenoids and Ras superfamily of GTPases
Isoprenylation, the after translational attachment of FPP or GGPP to a protein, is an important cellular regulated process of protein localization and function [Reviewed in 78, 79]. It is well established that one of the pleiotrophic benefits of statin therapy is the reduction in protein isoprenylation. Isoprenylation appears to play a crucial role in regulating the subcellular localization of proteins, including targeting these proteins to plasma membranes, possibly specifically to membrane lipid microdomains and intracellular membranes as well as modulating protein interactions. Whether a direct relationship, if any, exists between isoprenylation and cellular or membrane cholesterol levels remains unknown. Isoprenylation affects a large variety of cellular processes and cellular functions, including vesicular transport, cytoskeletal alterations and cell signaling [Reviewed in 79].
A large number of proteins are isoprenylated, including the subunits of trimeric G proteins, protein kinases and the more than 150 members of the Ras GTPase superfamily, including Rac, Ral, Rap and Rho. Members of the Ras superfamily of GTPases are integral components of complex signaling net-works and control diverse cellular activities including intracellular vesicle transport, cell adhesion, endocytosis, cytoskeletal organization, receptor signaling, cell cycle progression and gene expression [73, 80–82]. Experimental evidence suggests that GTPases may play a significant role in AD pathogenesis [82, 83]. Ras has been implicated in the Aβ signaling cascade [80]. Neuronal Ras, Rac and RhoA appear to function in neuronal and synaptic plasticity [84]. Rab1B and Rab6 appear to function in the intra-cellular trafficking and processing of βAPP [85]. Other GTPases have also been implicated in AD progression [86, 87]. Due to the fact that isoprenylation, in part, regulates both the functionality and the sub-cellular localization of these GTPases, it is likely that statin-induced inhibition of isoprenylation, as some experimental evidence suggests, would have complex ramifications on βAPP trafficking and processing, Aβ formation and neuronal and synaptic plasticity [88]. We have shown that statin treatment decreases the association of the active form of the γ-secretase complex with lipid rafts, and that this effect can be partially rescued through the addition of GGPP, suggesting that GGPP may in part regulate active γ-secretase complex association with membrane rafts [59]. Consistent with this finding, and highlighting the importance of isoprenylation in AD progression, it has been suggested that two pools of Aβ exist and appear to function independently of each other, with the intracellular pool regulated by isoprenoids and the secreted pool regulated by cellular cholesterol levels [73].
ACAT inhibitors
In addition to interest in statin therapies for the treatment of AD, recent reports suggest that inhibitors of acylcoenzyme A: cholesterol acyltransferase (ACAT) may hold therapeutic promise [89]. In vitro and in vivo findings demonstrated that treatment with an ACAT inhibitor reduced the production of Aβ and reduced the accumulation of amyloid plaques and insoluble amyloid in the CNS of mice [90]. Cholesterol distribution in membranes, in particular membrane rafts, appears to be more important for Aβ processing than cellular cholesterol levels. The mechanism underlying this observed effect have not been clearly defined, yet inhibition of cholesterol esterification may result in a greater or enhanced mobilization of the cellular cholesterol efflux machinery thereby decreasing membrane cholesterol levels.
Conclusions
Clearly AD is a complex disease caused by various genetic and environmental factors. Although there has recently been a number of exciting findings in the AD field, the relationship between cholesterol, iso-prenylation and the processing of βAPP is far from understood. It remains controversial what is the primary causative agent or factor versus what are secondary effects/responses. Consistent with the view of many researchers, it remains important for the AD research community to approach the ever-increasing number of new research findings with an open mind, and continue to search for a unifying theory that can account for all of the biochemical and pathological complexities associated with AD.
References
- 1.Haass C. Take five-BACE and the gamma-secretase quartet conduct Alzheimer's amyloid beta-peptide generation. EMBO J. 2004;23:483–8. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Deciphering the genesis and fate of amy-loid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest. 2002;110:1375–81. doi: 10.1172/JCI16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haass C, Steiner H. Alzheimer disease gamma-secretase: a complex story of GxGD-type presenilin pro-teases. Trends Cell Biol. 2002;12:556–62. doi: 10.1016/s0962-8924(02)02394-2. [DOI] [PubMed] [Google Scholar]
- 4.Vassar R. The beta-secretase, BACE: a prime drug target for Alzheimer's disease. J Mol Neurosci. 2001;17:157–70. doi: 10.1385/JMN:17:2:157. [DOI] [PubMed] [Google Scholar]
- 5.Klein WL, Stine WBJ, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–80. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi YE, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nature Neuroscience. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 7.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuropro-tective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–6. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–88. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 10.DeStrooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 11.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi T, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–41. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- 12.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–8. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 13.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA. 2003;100:6382–7. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi I, Urano Y, Fukuda R, Isoo N, Kodama T, Hamakubo T, Tomita T, Iwatsubo T. Selective reconstitution and recovery of functional gamma-secretase complex on budded baculovirus particles. J Biol Chem. 2004;279:38040–6. doi: 10.1074/jbc.M405597200. [DOI] [PubMed] [Google Scholar]
- 15.Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005;122:435–47. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Yang DS, Tandon A, Chen F, Yu G, Yu H, Arawaka S, Hasegawa H, Duthie M, Schmidt SD, Ramabhadran TV, Nixon RA, Mathews PM, Gandy SE, Mount HT, St George-Hyslop P, Fraser PE. Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J Biol Chem. 2002;277:28135–42. doi: 10.1074/jbc.M110871200. [DOI] [PubMed] [Google Scholar]
- 17.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Complex N-linked glycosylated nicastrin associates with active gamma-secretase and undergoes tight cellular regulation. J Biol Chem. 2002;277:35113–7. doi: 10.1074/jbc.M204446200. [DOI] [PubMed] [Google Scholar]
- 18.Shirotani K, Edbauer D, Capell A, Schmitz J, Steiner H, Haass C. Gamma-secretase activity is associated with a conformational change of nicastrin. J Biol Chem. 2003;278:16474–7. doi: 10.1074/jbc.C300095200. [DOI] [PubMed] [Google Scholar]
- 19.Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits ML, Xu H, Thinakaran G. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J Biol Chem. 2002;277:19236–40. doi: 10.1074/jbc.C200148200. [DOI] [PubMed] [Google Scholar]
- 20.Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H. Identification of distinct gamma-secretase complexes with different APH-1 variants. J. Biol. Chem. 2004;279:41340–5. doi: 10.1074/jbc.M405768200. [DOI] [PubMed] [Google Scholar]
- 21.Bergman A, Hansson EM, Pursglove SE, Farmery MR, Lannfelt L, Lendahl U, Lundkvist J, Näslund J. Pen-2 is sequestered in the endoplasmic reticulum and subjected to ubiquitylation and proteasome-mediated degradation in the absence of presenilin. J Biol Chem. 2004;279:16744–53. doi: 10.1074/jbc.M313999200. [DOI] [PubMed] [Google Scholar]
- 22.Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, Haass C. PEN-2 is an integral component of the gamma-secretase complex required for coordinated expression of presenilin and nicastrin. J Biol Chem. 2002;277:39062–5. doi: 10.1074/jbc.C200469200. [DOI] [PubMed] [Google Scholar]
- 23.Simons M, Keller P, Dichgans J, Schulz JB. Cholesterol and Alzheimer's disease: is there a link? Neurology. 2001;57:1089–93. doi: 10.1212/wnl.57.6.1089. [DOI] [PubMed] [Google Scholar]
- 24.Wolozin B. Cholesterol and the biology of Alzheimer's disease. Neuron. 2004;41:7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- 25.Masters CL, Multhaup G, Simms G, Pottigiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–63. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soneira CF, Scott TM. Severe cardiovascular disease and Alzheimer's disease: senile plaque formation in cortical areas. Clin Anat. 1996;9:118–27. doi: 10.1002/(SICI)1098-2353(1996)9:2<118::AID-CA4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 27.Kalback W W, Esh C, Castano EM, Rahman A, Kokjohn T, Luehrs DC, Sue L, Cisneros R, Gerber F, Richardson C, Bohrmann B, Walker DG, Beach TG, Roher AE. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer's disease. Neurol Res. 2004;24:525–39. doi: 10.1179/016164104225017668. [DOI] [PubMed] [Google Scholar]
- 28.Skoog I, Gustafson D. Hypertension and related factors in the etiology of Alzheimer's disease. Ann NY Acad Sci. 2002;977:29–36. doi: 10.1111/j.1749-6632.2002.tb04796.x. [DOI] [PubMed] [Google Scholar]
- 29.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Oden A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–5. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez RG, Fischman AJ, Guimaraes AR, Carr CA, Stern CE, Halpern EF, Growdon JH, Rosen BR. Functional MR in the evaluation of dementia: correlation of abnormal dynamic cerebral blood volume measurements with changes in cerebral metabolism on positron emission tomography with fludeoxyglucose F 18. Am J Neuroradiol. 1995;16:1763–70. [PMC free article] [PubMed] [Google Scholar]
- 31.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 32.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F, Iakovidou V, Maddalena A, Lutjohann D, Ghebremedhin E, Hegi T, Pasch T, Traxler M, Bruhl A, Benussi L, Binetti G, Braak H, Nitsch RM, Hock C. Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol. 2003;60:29–35. doi: 10.1001/archneur.60.1.29. [DOI] [PubMed] [Google Scholar]
- 34.Papassotiropoulos A, Wollmer MA, Tsolaki M, Brunner F, Molyva D, Lutjohann D, Nitsch RM, Hock C. A cluster of cholesterol-related genes confers susceptibility for Alzheimer's disease. J Clin Psychiatry. 2005;66:940–7. [PubMed] [Google Scholar]
- 35.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–21. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 37.Sparks DL, Scheff SW, Hunsaker JC, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol. 1994;126:88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- 38.Sparks DL. Intraneuronal beta-amyloid immunoreactivity in the CNS. Neurobiol Aging. 1999;17:291–9. doi: 10.1016/0197-4580(95)02067-5. [DOI] [PubMed] [Google Scholar]
- 39.Refolo LM, Malester MB, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolaemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–31. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 40.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, Von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–61. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simons M, Keller P, De Strooper D, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM. Proc Natl Acad Sci USA. 2001;98:5815–20. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–40. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 44.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 45.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 46.Ikonen E. Roles of lipid rafts in membrane transport. Curr Opin Cell Biol. 2001;13:470–7. doi: 10.1016/s0955-0674(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 47.Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;11:724–38. doi: 10.1034/j.1600-0854.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 48.Cordy JM, Hooper NM, Turner AJ. The involvement of lipid rafts in Alzheimer's disease. Mol Mem Bio. 2006;23:111–22. doi: 10.1080/09687860500496417. [DOI] [PubMed] [Google Scholar]
- 49.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol. 2001;11:1288–93. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- 51.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2003;100:11735–40. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 53.Wada S, Morishima-Kawashima M, Qi Y, Misono H, Shimada Y, Ohno-Iwashita Y, Ihara Y. Gamma-secretase activity is present in rafts but is not cholesterol-dependent. Biochemistry. 2003;42:13977–86. doi: 10.1021/bi034904j. [DOI] [PubMed] [Google Scholar]
- 54.Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–54. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parkin ET, Hussain I, Karran EH, Turner AJ, Hooper NM. Characterization of detergent-insoluble complexes containing the familial Alzheimer's disease-associated presenilins. J Neurochem. 1999;72:1534–43. doi: 10.1046/j.1471-4159.1999.721534.x. [DOI] [PubMed] [Google Scholar]
- 56.Bouillot C, Prochiantz A, Rougon G, Allinquant B. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J Biol Chem. 1996;271:7640–4. doi: 10.1074/jbc.271.13.7640. [DOI] [PubMed] [Google Scholar]
- 57.Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–4. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 58.Murphy MP, Das P, Nyborg AC, Rochette MJ, Dodson MW, Loosbrock NM, Souder TM, McLendon C, Merit SL, Piper SC, Jansen KR, Golde TE. Overexpression of nicastrin increases Abeta production. FASEB J. 2003;17:1138–40. doi: 10.1096/fj.02-1050fje. [DOI] [PubMed] [Google Scholar]
- 59.Urano Y, Hayashi I, Isoo N, Reid PC, Shibasaki Y, Noguchi N, Tomita T, Iwatsubo T, Hamakubo T, Kodama T. Association of active gamma-secretase complex with lipid rafts. J Lipid Res. 2005;46:904–12. doi: 10.1194/jlr.M400333-JLR200. [DOI] [PubMed] [Google Scholar]
- 60.Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawarabayashi T, Shoji M, Younkin LH, Wen-Leng L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafs followed by apolipoportein E and Phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neuroscience. 2004;24:3801–9. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wood WG, Schroeder F, Igbayboa U, Avdulov NA, Chochina SV. Brain membrane cholesterol domains, aging, and amyloid beta-peptides. Neurobiol Aging. 2002;23:685–94. doi: 10.1016/s0197-4580(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 63.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 64.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 65.Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, Wolfson C, McDowell I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–7. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- 66.Kumar AP, Reynolds WF. Statins downregulate myeloperoxidase gene expression in macrophages. Biochem Biophys Res Commun. 2005;331:442–51. doi: 10.1016/j.bbrc.2005.03.204. [DOI] [PubMed] [Google Scholar]
- 67.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–17. [PubMed] [Google Scholar]
- 68.Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2000;8:890–9. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 69.Hoglund K, Wiklund O, Vanderstichele H, Eikenberg O, Vanmechelen E, Blennow K. Plasma levels of beta-amyloid(1-40), beta-amyloid(1-42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolaemia after treatment with statins. Arch Neurol. 2004;61:333–7. doi: 10.1001/archneur.61.3.333. [DOI] [PubMed] [Google Scholar]
- 70.Fassbender K, Stroick M, Bertsch T, Ragoschke A, Kuehl S, Walter S, Walter J, Brechtel K, Muehlhauser F, Von Bergmann K, Lutjohann D. Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology. 2002;59:1257–8. doi: 10.1212/wnl.59.8.1257. [DOI] [PubMed] [Google Scholar]
- 71.Wierzbicki AS, Poston R, Ferro A. The lipid and non-lipid effects of statins. Pharmacol Ther. 2003;99:95–112. doi: 10.1016/s0163-7258(03)00055-x. [DOI] [PubMed] [Google Scholar]
- 72.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an iso-prenoid-dependent mechanism. J Biol Chem. 2005;280:18755–70. doi: 10.1074/jbc.M413895200. [DOI] [PubMed] [Google Scholar]
- 74.Botti RE, Triscari J, Pan HY, Zayat J. Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin Neuropharmacol. 1991;14:256–61. doi: 10.1097/00002826-199106000-00010. [DOI] [PubMed] [Google Scholar]
- 75.Haley RW, Dietschy JM. Is there a connection between the concentration of cholesterol circulating in plasma and the rate of neuritic plaque formation in Alzheimer disease. Arch Neurol. 2000;57:1410–2. doi: 10.1001/archneur.57.10.1410. [DOI] [PubMed] [Google Scholar]
- 76.Scheffler IE. Metabolic pathways inside mitochondria. In: Scheffler IE, editor. Mitochondria. New York: Wiley-Liss; 1999. pp. 246–72. [Google Scholar]
- 77.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–9. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 78.Cox AD, Der CJ. Farnesyltransferase inhibitors and cancer treatment: targeting simply Ras. Biochim Biophys Acta. 1997;1333:F51–F71. doi: 10.1016/s0304-419x(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 79.Cole S, Vassar R. Isoprenoids and Alzheimer's disease: A complex relationship. Neuro Dis. 2006;22:209–22. doi: 10.1016/j.nbd.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 80.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 81.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–55. [PMC free article] [PubMed] [Google Scholar]
- 82.Ridley AJ. Rho proteins: linking signaling with membrane trafficking. Traffic. 2001;2:303–10. doi: 10.1034/j.1600-0854.2001.002005303.x. [DOI] [PubMed] [Google Scholar]
- 83.Shimohama S, Kamiya S, Taniguchi T, Sumida Y, Fujimoto S. Differential involvement of small G proteins in Alzheimer's disease. Int J Mol Med. 1999;3:597–600. doi: 10.3892/ijmm.3.6.597. [DOI] [PubMed] [Google Scholar]
- 84.Lee M, You HJ, Cho SH, Woo CH, Yoo MH, Joe EH, Kim JH. Implication of the small GTPase Rac1 in the generation of reactive oxygen species in response to beta-amyloid in C6 astroglioma cells. Biochem J. 2002;366:937–43. doi: 10.1042/BJ20020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scheper W, Zwart R, Baas F. Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Brain Res Mol Brain Res. 2004;122:17–23. doi: 10.1016/j.molbrainres.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 86.Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999;8:1263–9. doi: 10.1093/hmg/8.7.1263. [DOI] [PubMed] [Google Scholar]
- 87.Kametani F, Usami M, Tanaka K, Kume H, Mori H. Mutant presenilin (A260V) affects Rab8 in PC12D cell. Neurochem Int. 2004;44:313–20. doi: 10.1016/s0197-0186(03)00176-1. [DOI] [PubMed] [Google Scholar]
- 88.Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 2005;280:34202–9. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- 89.Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM. Acyl-coenzyme A: cholesterol acyl-transferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;3:905–12. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 90.Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, Kovacs DM. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron. 2004;44:227–38. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]