

Abstract
Human islet transplantation could represent an attractive alternative to insulin injections for the treatment of diabetes type 1. However, such an approach requires a better understanding of the molecular and cellular switches controlling β-cell function in general as well as after transplantation into the liver. Although much research has been done into the suitability of stem or progenitor cells to generate a limitless supply of human β-cells, a reproducible and efficient protocol for the differentiation of such cells into stably insulin-secreting β-cells suitable for transplantation has yet to be reported. Fueled by recent findings showing that mature β-cells are able to regenerate, many efforts have been undertaken to expand this cell pool. Unfortunately, also these approaches had problems to yield sufficiently differentiated human islet cells. The aim of this review is to summarize recent findings describing some of the molecular and cellular key players of islet biology. A more complete understanding of their orchestration and the use of new methods such as real time confocal imaging for the assessment of islet quality may yield the necessary advancements for more successful human islet transplantation.
Keywords: islet transplantations, vascularization, diabetes type 1, cell therapy, islet of Langerhans, real time confocal imaging
Human cell therapy
One of the most exciting fields in translational medicine are cell-based therapies. Their potential can be seen in blood transfusions and bone marrow transplantations, which are the two most convincing examples for successful applications of cell-based therapies.
Recent progress in fast growing scientific disciplines such as molecular biology, stem cell biology, biomaterials, immunology, clinical research and transplantation biology has expanded the potential applications of cell therapies.
Since most diseases are not caused by the deficiency of a single molecule but develop due to changes in the interactions of a variety of cell components, such cell-based therapies may prove to be more successful than classical therapies by providing a more dynamic therapeutic approach capable of responding to the individual pathophysiological conditions [1]. In this regard, one of the most promising cell-based therapeutic applications are the innovative approaches for tissue regeneration or replacement, using cells to rebuild or replace damaged organs and tissues [2].
The basic problem for every cell-based therapy is cell sourcing since cells have to be expanded into large enough quantities while maintaining uniformity in function. Cell expansion as well as uniformity requirement are dependent on the proliferation capacity of either stem/progenitor or already differentiated cells. The use of human embryonic versus adult stem cells is still under scientific/ethical debate [3]. The extent of adult stem cell plasticity which could provide access to novel sources of (trans-) differentiated cells has been questioned by recent reports demonstrating stem cell fusion with tissuespecific differentiated cells rather than true stem cell transdifferentiation with normal diploid chromosomal numbers [4–6]. Due to safety concerns the utilization of immortalized cells is still not a real alternative [7]. Currently cell-based approaches focus on adult tissue- specific stem cell expansion but in the future this might possibly change depending on the ongoing discussion addressing the use of human embryonic stem (ES) cells.
Besides autologous also non-autologous human cells may be taken into consideration in the formulation of a cell-based therapy. However, non-autologous cells must be protected from the natural immunologic rejection process of the host thereby limiting this approach. Microencapsulation of the transplanted material [8], development of better immunosuppressant therapies or tolerance induction are fields of research awaiting progress before such allotransplantats will enter clinical routine. Also the use of autologous cells has its own problems since it requires obtaining the patient's own cells, expand them in vitro to large quantities over a more or less long period of time and then transplant them in a site-specific manner. Therefore, each treatment is an individualized and non-scalable process with substantial logistical and regulatory problems including maintenance of the uniform quality of cells, avoidance of introduced pathogens during cell processing, and potential retrievability after implantation [1].
A final obstacle that has to be tackled for cellbased therapies is the maintenance of cell viability during long-term implantation. Long-term function of the transplanted cells is only possible upon successful integration after transplantation and adequate nutrient and oxygen delivery to the cellular implant.
Human islet transplantation
Although cell-based therapy has substantial technological, regulatory and ethical barriers, the potential to develop new treatment modalities for a large number of clinical disorders is expanding rapidly.
Type 1 diabetic patients will probably be one of the major beneficiaries from the advancement of regenerative medicine through cellular therapies. Type 1 diabetes mellitus results from the immune destruction of insulin producing β-cells, located in the pancreatic islets of Langerhans [9].
Diabetes prevalence (type 1 and 2) has increased from a world estimate of 135 million in 1995 to 180 million currently, and is predicted to rise to 300 million by the year 2025 [10].
Although insulin therapy has saved the lives of diabetic patients, diabetics still are in danger to develop chronic diabetes-related complications such as renal failure, myocardial infarction, blindness or vascular problems, that appear years after the onset of diabetes and contribute to shortened lifespan [11, 12]. Such complications not only diminish the quality of life of patients but also represent a burden for health care systems [13, 14]. Although several clinical trials have shown that a strict glycemic control can slow and even prevent the progression of diabetic complications, such an intensive insulin therapy on the other hand increases the incidence of hypoglycaemic episodes and is suitable only for selected patients [15, 16].
As an alternative, pancreas and islet transplantation have been shown to be efficient in replacing the function of the impaired islets and, thus induce continuous normoglycaemia. The freedom from daily insulin injections and blood glucose monitoring is reported to improve the successfully transplanted recipient's sense of well being, independence, and promote a perception of normality [17].
While pancreas transplantation requires major surgery, islet transplantation has the advantage of being less invasive. Since both procedures require lifelong immunosuppression, only patients with severe late-stage complications or those already undergoing kidney transplantation and immunosuppression are candidates for such therapies [18].
Astonishingly, attempts to treat type 1 diabetes mellitus through transplantation predate insulin therapy by more than 30 years. Minkowski was the first in 1891 to describe the subcutaneous implantation of pancreatic tissue autografts from pancreatectomized dogs. He laid the foundation for the first attempts to transplant isolated islets in a rodent model [19], which were followed by the first successful islet infusion using the intra-hepatic approach [20], which is nowadays the classical transplantation site for human islets. Since 1993, pancreatic islet therapy has become subject to regulation by the U.S. Food and Drug Administration as biologic product [21]. Based on the exceptionally successful study of Shapiro et al. at the University of Alberta in Edmonton, Canada, in which seven patients out of seven were insulin-free at the end of one year after islet transplantation [22], an exponential increase in clinical islet transplantation followed [23].
However, follow-up investigations have revealed a progressive attrition of the grafted islets with less than 10% of patients remaining insulin-free after 5 years despite most patients having more than one transplant [24].
The most ambitious goal of allogenic islet transplantation is to establish long-term normoglycaemia and phasic insulin secretion and thereby achieve prolonged insulin independence in the diabetic patient; a goal already achieved in islet autotransplantation for treatment of chronic painful pancreatitis in non-diabetic patients (Fig. 1).
1.
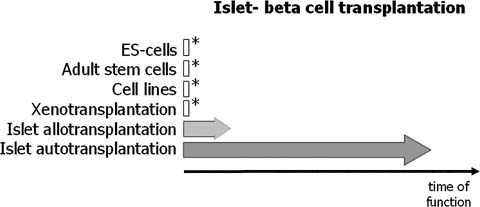
Schematic representation of the progress/success in islet or β-cell transplantation. Asterisks symbolize the experimental state of the approach. The approach using embryonal stem cells still awaits ethical decisions. Similar to the approach aiming at the establishment of cell lines capable to substitute islet β-cells also embryonal stem cells bear the danger of oncologic transformations due to the many necessary in vitro population doublings. Another possibility is the use of xeno-islets, such as procine islets, for transplantation. Although having the advantage of being a sustainable islet source, many obstacles such as the antigenicity (due to the β-galactosyl antigen which is present in most non-human primates) and infection with zoonoses (such as the pig endogenous retroviruses) still remain [23]. The biological proof of principle showing a long-lasting and stable insulin production of islets transplanted into the liver has already been shown in autotransplantations [29]. Although the current immunosuppressive therapies have improved the outcome of islet allotransplantations, most patients once they reach 5 years post-transplantation do not sustain insulin independence [24].
Chronic pancreatitis is a continuing inflammatory disease characterized by irreversible morphological change and typically gives rise to pain and in some cases permanent impairment of endocrine and exocrine function. Pancreatic stellate cells are crucially involved in the development of fibrosis, a hallmark of chronic pancreatitis and may represent suitable targets for gene therapy modulating cellular interactions in the pancreas [25]. Autoimmune pancreatitis accounts for 4.6–6% of all forms of chronic pancreatitis and possession of the HLA DRB1*0405–DQB1*0401 genotype confers a risk for its development. Autoimmune pancreatitis was proposed to be triggered by Helicobacter pylori through several different and probably cooperating mechanisms, including molecular mimicry between human and bacterial ntigens [26, 27]. As described recently, human carbonic anhydrase II and _–carbonic anhydrase of Helicobacter pylori, an enzyme, which is fundamental for the survival and proliferation of the bacterium n the gastric environment, display a significant homology. Moreover, the homologous segments contain the binding motif of the HLA molecule DRB1*0405, thereby strengthening the hypothesis that gastric Helicobacter pylori infection can trigger autoimmune pancreatitis in genetically predisposed persons [28].
Treatment of chronic pancreatitis is directed toward the major problems of pain control and malabsorption. Diabetes inevitably accompanies chronic pancreatitis once sufficient pancreatic tissue has been destroyed. Surgery is generally performed upon discovery of a pseudocyst or localized ductal obstruction. In the most extreme cases, total or neartotal pancreatic resection is performed for relief of unrelenting pain [29]. Such patients have the option to receive an intraportal infusion of their own islets without the need for immunosuppressive drugs. In spite of the problems also autologous transplanted islets have to deal with in their new surrounding, pancreatic islet autotransplantation has prevented the onset of diabetes in pancreatectomized patients for up to two decades [29]. Therefore, the biological proof of principle for a long-lasting stable glucose control by islets transplanted into the liver has been established. This success is equally surprising as well as inspiring for the more difficult task of islet allotransplantation.
Understanding how autotransplanted islets can sustain their homeostasis and function in the liver even for decades might help us to find answers for still open questions regarding the molecular and cellular basis necessary for a successful islet allotransplantation. A better understanding of proliferation and differentiation processes taking place in progenitor and fully differentiated β-cells, both in the embryo as well as in the adult, will be essential for all future strategies aiming at achieving an unlimited source of functional β-cells to cure type 1 diabetes.
In the United States of America, approximately one million individuals have type 1 diabetes mellitus, and 16 million more are estimated to have type 2 diabetes. Since both situations result at least in part from loss of islet function, both could benefit from an optimized islet transplantation solution. In spite of big efforts being made, only about 6000 human braindead donors are suitable for organ donation each year. In addition, even the most up-to-date islet isolation facility achieves an isolated islet yield of sufficient quality and quantity to transplant only every second isolation [30]. Taking also into account that almost all islet recipients currently require islets from 2–4 donors, then only less than 1000 recipients per year (out of the 17 million in need) could benefit from an islet transplantation [17].
In addition, the quality of isolated islets is still poorly defined and the functional potential of transplanted islets are difficult to predict. Current standard assays for islet product release criteria are still unable to predict the outcome after clinical islet transplantation [31]. Therefore, one of the challenges in islet transplantation is to identify and understand the changes taking place in islets after isolation, culture and transport. Description of such changes in living islet cells may offer insights, which cannot be gained by use of fixed cell techniques. Confocal real time analysis of vital islets is a new method well suited for a fast and accurate assessment of islet quality [32]. An example of such a real time confocal analysis is shown in Figure 2. Four fluorescent dyes, tetramethylrhodamine methyl ester perchlorate (TMRM), Alexa Fluor labeled wheat germ agglutinin (WGA), cell permeant acetoxymethylester (Rhod-2), propidium iodide (PI) and fluorescein isothiocyanate (FITC)- labeled annexin V were used to assess either, time-dependent changes in mitochondrial membrane potentials (Fig. 2A), localization of oligosaccharides on the cell surfaces (Fig. 2A and B), calcium (Fig. 2B), cell death and apoptosis (Fig. 2C). Confocal microscopy was performed with a microlens-enhanced Nipkow disk-based confocal system mounted on an inverse microscope. Especially in light of the many other fluorescent dyes which can be used as sub-cellular markers, a combination of these with a confocal system allowing live cell imaging will be of great value for a better and faster cell quality assessment after isolation, culture and transport of human islet cells. Besides these advantages, live confocal imaging may yield new insights into the biology of human islets and thereby help us answering the still open question in islet transplantation: How can the current disparity between supply and demand be tackled? Only when new sources of pancreatic islets or β-cells are found, cell replacement as a treatment for diabetes will become widely available [16]. Despite their in vivo self-replication potential [33], differentiated β-cells cannot be expanded efficiently in vitro[34]. Therefore alternatives are being explored to generate cell-replacement therapies for diabetics.
2A.
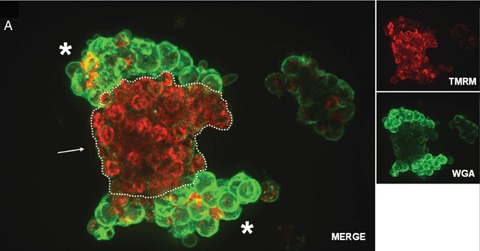
Confocal real time microscopy of an entrapped human islet stained with tetramethylrhodamine methyl ester perchlorate (TMRM; red) and Alexa Fluor labeled wheat germ agglutinin (WGA; green). Vital mitochondria with an intact potential appear in red due to the TMRM staining. While islet (dotted line) mitochondria show a significantly higher vitality (see arrow), compared with the surrounding exocrine cells, the glycoconjugates, stained viaWGA, at the membrane surfaces are present in a significantly higher amount in the exocrine cells (see asterisks) compared with the islet cells.
2B.
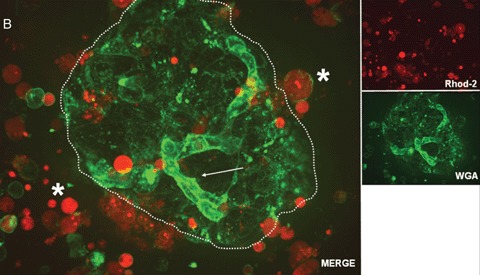
Confocal real time microscopy of a human islet surrounded by stressed single cells in suspension after islet isolation procedure. The cell surfaces are stained with WGA.While endothelial cells (see arrow) show a strong staining of the cell surface after addition of WGA, islet cells (dotted circle) are only weakly stained. Note the strong positivity for cell permeant acetoxymethylester (Rhod-2) in the stressed single cells and the high cell vitality documented by the nearly absent staining for Rhod-2 positive cells in the islet.
2C.
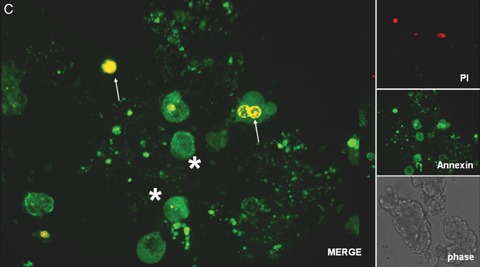
Confocal real time microscopy of a human islet isolation stained with propidium iodide (PI; red; see arrows) and the FITC labeled apoptosis marker annexin V (green; see asterisks). Confocal microscopy was performed with a microlens-enhanced Nipkow disk-based confocal system mounted on an inverse microscope. All pictures were acquired with a 40 x water immersion objective.
In search of an “endless pool” of β-cells: embryonic stem cells, adult mesenchymal stem cells, progenitor cells, cell lines and xenotransplantations
The shortage of primary human islets has led to several efforts to identify and create high numbers of β-cells that can be used for regulated insulin therapy. Approaches to generate β-cells from embryonic or adult stem cells or by transdifferentiation from other cell types such as liver cells or exocrine/ductal pancreatic cells are still far away from reaching the stage of clinical application [35–38]. This also holds true for xenotransplantations, especially those using porcine islets as a source. Concerns about transmission of infectious agents and the severe xenobiotic immune response are major hurdles which still have to be taken [39]. Currently, fueled by the publication of Dor and co-workers showing that mature β-cells are able to regenerate and proliferate, many efforts have been undertaken to expand the pool of mature human islet cells. Unfortunately, also these approaches had problems to yield sufficiently differentiated human islet cells. β-cell function is normally lost as a consequence of cell expansion [21].
Another possible strategy to generate human pancreatic β-cells in sufficient quantity is to transform human primary β-cells with immortalizing genes and excise these potentially tumorigenic genes after propagation before the final recovery of primary β-cell function.
Recently such an approach yielded a reversibly immortalized human β-cell line [40]. The immortalized cell line was generated by transformation of human islet cells with retroviral vectors expressing SV40 large T-antigen (SV40T) and human telomerase reverse transcriptase (TERT). Both genes were flanked by loxP sites for later Cre recombinasebased excision. One cell line obtained, thereafter termed NAKT-15, expressed insulin and four β-cell specific transcription factors. Interestingly, insulin secretion from these cells was shown to be regulated by glucose in a dose-dependent manner comparable to human islets. In addition, also tolbutamide, glucagon like peptide 1 (GLP-1), or fatty acids, all known potentiators of the glucose response in β-cells, increased the glucose response in NAKT-15 cells. Implantation of approximately 3 million NAKT-15 cells, which is roughly equivalent to 2000 islets, under the kidney capsule of streptozotocin-induced diabetic severe combined immunodeficiency mice resulted in perfect control of blood glucose within 2 weeks. Normoglycaemia was achieved in these mice for a period of 30 weeks with no evidence of hypo- or hyperglycaemic episodes.
Nevertheless, several β-cell enriched transcription factors, hormone processing enzymes, secretory granule proteins as well as insulin were not as abundant as compared with human islet cells. Therefore, it can be estimated that about one billion NAKT-15 cells will be needed for the therapy of one human diabetic patient. Such an approach bears the danger of somatic mutations due to the high numbers of population doublings required to achieve the needed amount of cells. In addition, the response of such cells to immunosuppressive regimens is still an open question. Due to these and other reasons it also remains to be seen how regulatory agencies such as the U.S. Food and Drug Administration will respond to such growth promoting strategies involving genetic engineering of cell lines with oncogenes [40,41].
cell proliferation and differentiation
Studies showing that in the postnatal period as well as in adult life newly formed pancreatic β-cells originate from pre-existing β-cells have led to an increased emphasis on understanding and promoting the replication of adult β-cells [33,42]. A tight equilibrium between cell proliferation and differentiation is essential for the homeostasis of a tissue, an organ and an organism it resides in. On the one hand, several factors are known to either promote or inhibit proliferation or differentiation of pancreatic β-cells. On the other hand, some factors such as those of the Growth Hormone family, IGF-I and IGF-II, promote both proliferation as well as differentiation (Fig. 3) [43]. The number of studies analyzing the mechanisms that govern β-cell proliferation and growth in vivo as well as in vitro has increased exponentially during the last decade. These studies exploring the role of cell-cell and cell-matrix contacts in combination with growth and differentiation factors are the basis for future strategies aimed at expanding human β-cells in vitro and/or in vivo after transplantation.
3.
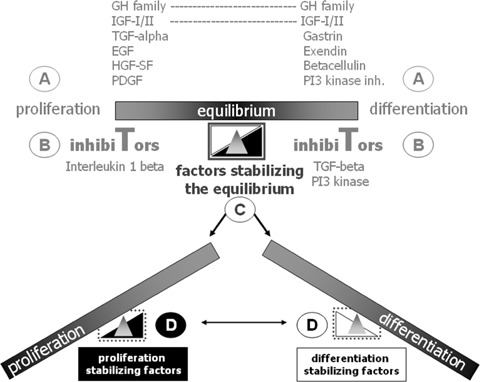
Schematic model proposing four different molecular control levels of pancreatic β-cell proliferation and differentiation. For a more detailed description of the extrinsic factors influencing β-cell proliferation and differentiation see Soria, 2001 [43]. Pancreatic β-cell growth and differentiation requires precise control of the molecular mechanisms leading to either entry or exit of a replicative or differentiated state. A, B, C and D symbolize different control levels. Developmental state, age, metabolic factors, injuries or diseases such as diabetes affect both proliferation as well as differentiation of β-cells. Local signals coming either from surrounding cells or the extracellular matrix balance the proliferation and differentiation status of β-cells and thereby also their function. As impressively documented by islet autotransplantation, β-cells can handle with “big changes” i.e. even the transplantation into another organ such as the liver. The questions are how big the dependence of the β-cells is concerning an intact cytoarchitecture of the transplanted islet and how much help coming from the neighbouring cells, is needed (see also Figure 4)? Answering these questions is fundamental for further cell therapeutic approaches aiming at an expansion of the available pool of human β-cells.
By maintaining the 3-D configuration of human islets in fibrin gels, β-cells were shown to expand in response to hepatocyte growth factor. In addition, the physiologic glucose responsiveness both in vitro as well as in vivo after transplantation into nude mice was preserved [44]. This and several other studies demonstrate the importance of the above mentioned interactions with components of the extracellular matrix in combination with soluble factors for β-cell physiology in general as well as for the ex vivo expansion of human pancreatic cells [45–47]. Recently a protocol for the in vitro differentiation of mouse ES cells into insulin-producing cells was published. In a first step embryoid bodies are created which then in a second step differentiate into progenitor cells of ecto-, meso- and endodermal lineage. In a final step, differentiation of the early progenitor cells into pancreatic lineages is induced via growth and extracellular-matrix factors, such as laminin, nicotinamide and insulin [46].
Nevertheless, we still lack a sufficient understanding of the regulatory mechanisms, signalling pathways and molecules that orchestrate β-cell physiology maintaining the cells in a perfect equilibrium between growth and differentiation. Two things are essential for such equilibrium. First of all, cells residing in such equilibrium must still be able to react dynamically to the changing demands of the organism, i.e.depending on the situation either expansion or differentiation rules their basic physiology. Secondly, once the equilibrium is reached a stabilization of this balance is important for a cell such as the β-cell. Such a balance is exemplified by the square in figure 3. Minor changes caused i.e.by infections, diseases, aging or nutritional changes would thereby be counterbalanced. The contrary is the case with other “physiological” challenges for example during development or pancreatic regeneration in which a β-cell or a precursor cell would theoretically benefit from the absence of factors stabilizing the current balance, thereby enabling a more rapid and sensitive reaction in order to respond correctly to local changes (Fig. 3). It is therefore important to differentiate between the embryonic, adult or regeneration/transplantation situations when issues of islet proliferation and differentiation are discussed. Apart from these three situations a fourth in vitro/ex vivo situation dealing with issues of β-cell engineering should be discriminated as regulatory mechanisms might totally differ from the in vivo situation (Fig. 4).
4.
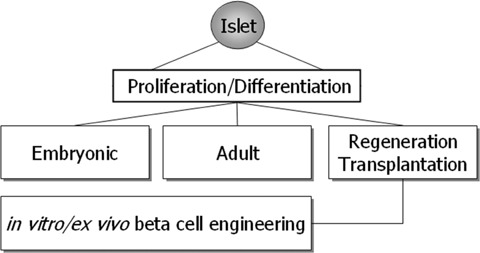
Human islet β-cell proliferation and differentiation takes place either under embryonic or adult conditions. Besides these two physiological conditions a third one addressing regeneration or even transplantation of islets into the liver can be distinguished. In all three conditions, a correct balance between proliferation and differentiation is pivotal in order to ensure proper β-cell function. This balance is facing again a different situation when β-cells are maintained and propagated in vitro/ex vivo with the goal to gain an unlimited source of transplantable β-cells for human islet transplantation. It is this fourth situation that will benefit from a better understanding of the three other ones. Besides this, knowledge gained from the in vitro experiments with β-cells will round up our knowledge of in vivoβ-cell physiology.
The β-cell mass plays an important role in determining the amount of insulin that is secreted in order to maintain the body's glucose levels within a narrow range. Therefore, the regulation of β-cell mass is crucial, especially taking into consideration its dynamic nature depending on the demand of insulin needed. Besides functional adaptations, such as changes in threshold for glucose induced insulin secretion during pregnancy [48], the β-cell mass itself is the major factor in the amount of insulin that can be secreted [49]. As shown in a morphometric study using autopsied pancreata from non-insulin-dependent diabetes mellitus patients (NIDDM) and non-diabetic patients, in the diabetic as well as the non-diabetic patient, the β-cell mass was approximately 40% increased in the obese as compared with the lean patients, indicating a compensatory growth of the β-cell mass [50]. In rodents, the postnatal period is characterized by a massive increase in β-cell mass, which is primarily due to the replication of terminally differentiated β-cells [51]. The question is, how the terminally differentiated β-cells transit between a quiescent and a replicative state during such a period. A better understanding of the regulation of β-cell growth and differentiation will help us to develop the necessary tools and strategies for a successful therapy of diabetes.
The three D-cylins (cylins D1, D2 and D3) are known key regulators of cell proliferation. Their primary roles are to sense the cells’ readiness to replicate and to couple extracellular signals to the biochemical machinery of the cells thereby inducing progression through G1 phase of the mammalian cell division cycle [52]. After the mitogenic stimulus Dcyclins associate with partner cyclin-dependent kinases CDK4 and CDK6 thereby driving cells into Sphase [53]. From the three D-cyclins, primarily cyclin D2 is required for β-cell replication and proper expansion of β-cell mass during the murine postnatal development of the pancreas. While replication of exocrine and ductal cells is unperturbed in cyclin D2 deficient mice, the absence of endocrine cell replication in these mice results in a 3-fold decrease in β-cell mass [42]. Whereas in the majority of other cell types up-regulation of the remaining D-cyclins can compensate for the loss of any particular D-cyclin [54], this does not seem to be the case in pancreatic β-cells. The inability of β-cells to up-regulate other Dcyclins in early postnatal development indicates that the mitotic signaling mediating the high rates of β-cell replication specifically depends on cyclin D2 [42]. Such a rigid connection of the signalling pathway to the cell cycle machinery in β-cells may offer an opportunity for selective intervention during the course of islet transplantation in order to either propagate β-cells in vitro or give them the proper mitotic stimulus before transplanting them into the liver. Before such new strategies can be tackled, a better understanding of the cell cycle progression and regulation of β-cells is needed. Especially, as it is known that forcing endocrine cells to re-enter the cell cycle may trigger programmed cell death [55]. In addition, a recent study showed that dependent on the genetic background, other mechanisms could compensate for the absence of cyclin D2. Surprisingly cyclin D2−/- mice on a C57BL/6 sv129 genetic background have normal early postnatal β-cell proliferation, which ceases by 3 months of life, causing eventual diabetes.
This demonstration of intact β-cell proliferation in 16-day-old cyclin D2−/- mice but its lack in cyclin D1+/- D2−/- mice show that, in C57BL/6 sv129 genetic background mice, cyclin D1 contributes to β-cell expansion. Cyclin D3 does not appear to influence islet function, as superimposed disruption of one cyclin D3 allele does not worsen the diabetic phenotype of cyclin D2−/- mice [56].
Interestingly, the analogue of glucagon-like peptide 1, exendin-4, which is also known to augment pancreatic islet mass and β-cell proliferation, was shown to increase cyclin D1 expression when administered to the pancreatic β-cell line INS-1 [57]. In addition, the same study described an exendin-4 induced phosphorylation of Raf-1 and extracellularsignal-regulated kinase (ERK). Recently ERK1/2 were reported to control phosphorylation of the cAMP-responsive element-binding protein (CREB) thereby playing a key role in glucose-mediated pancreatic β-cell survival [58]. Notably, Raf-1 kinase inhibitory protein (RKIP), which has been shown to inhibit the ERK-signalling pathway [59] was shown to be present in most pancreatic β-cells as well as in a subset of pancreatic polypeptide-expressing cells [60]. Besides showing a down-regulation of RKIP in human insulinomas, the authors used antisense technology in the transformed hamster β-cell line HIT-T15 to document an inhibitory role of RKIP in β-cell proliferation. While it is very probable that β-cell growth is influenced by RKIP via the ERK-signalling pathway, additional interactions of RKIP with other signalling pathways cannot be ruled out.
Signal transducer and activator of transcription 5 (STAT5) activation was shown to play a key role in growth hormone (GH) and prolactin-mediated transcriptional induction of cyclin D2 in rat pancreatic β-cells. The authors concluded that STAT5 activation is sufficient to drive proliferation of the β-cells and that cyclin D2 may be a critical target gene for STAT5 in this process [61].
After their interaction with cyclins, cyclin-dependent kinases Cdk4 and Cdk6 translate the mitotic stimulus by phosphorylation and subsequent inactivation of the retinoblastoma gene products pRb and pRb-related proteins [53]. Upon phosphorylation, pRb releases tethered transcription factor E2F and irreversibly commits the cell to progress through the cell cycle [62]. Mice lacking Cdk4 expression develop insulin-deficient diabetes due to a reduction in the number of pancreatic β-cells. While loss of Cdk4 has no detrimental effects on the development of the exocrine pancreas, Cdk4 appears to specifically affect the development of endocrine β-cells. The observation that islets of newborn Cdk4-deficient mice are similar to those of the control littermates indicates that loss of Cdk4 affects postnatal proliferation of β-cells. Consequently, mice expressing a mutant Cdk4 that cannot bind the cell-cycle inhibitor p16INK4a display pancreatic hyperplasia due to abnormal proliferation of pancreatic β-cells, thereby establishing the importance of Cdk4 as an essential regulator of β-cell proliferation [63]. An association of cyclin D1 and Cdk4 in neonatal rat pancreatic β-cells in vitro after mitogenic stimulation further emphasizes the role of cyclin D1 and Cdk4 in the control of β-cell proliferation [64].
Besides the Cyclin D2-Cdk4 complex formation, other factors such as the cyclin kinase inhibitors (CKIs) play a role in the decision whether a β-cell remains quiescent or starts to divide. The CKIs are divided into two families, the Ink4-family members, which specifically inhibit cyclin E-Cdk4/6 activity and the CIP/KIP family, which includes p21Cip1, p27Kip1 and p57Kip2. Addressing the question which role CKIs play in the proliferation of murine β-cells during embryogenesis and the postnatal period Georgia and Bhushan recently reported that newly differentiated β-cells accumulate p27Kip1 during embryogenesis and that disabling p27Kip1 allowed newly differentiated β-cells, which are normally quiescent during embryogenesis, to re-enter the cell cycle and proliferate, thereby generating an excess of β-cells at birth. Interestingly, the postnatal expansion of β-cells was unaffected in the p27Kip1-deficient mice, suggesting that the main function of p27Kip1 is to maintain the quiescent state of newly differentiated β-cells during embryogenesis. Notably, when assessing the role of p27Kip1 in β-cell regeneration the authors showed that β-cells retained the ability to re-enter the cell cycle at a far greater frequency in p27Kip1 deficient, streptozotocin-induced diabetic mice [65]. Given the limited amount of islets available for transplantation, key regulators in β-cell division such as p27Kip1 might play a central role in future strategies to enhance islet regeneration or proliferation.
In early pancreas development, the balance between expansion and differentiation of Pdx1 — expressing multipotent progenitor cells is critical for proper pancreatic development [66]. As recently reported, the cyclin kinase inhibitor p57Kip2 regulates cell cycle exit of Pdx-1-expressing progenitor cells during the early stages of pancreas formation. Interestingly, p57Kip2 was shown to be a direct target of transcriptional repression by the Notch effector Hes1, which previously has been shown to prevent differentiation by suppressing the expression of pancreatic differentiation factors such as Ngn3 [67]. Inactivation of Hes1 resulted in up-regulation of p57Kip2 expression, cell cycle arrest, precocious differentiation and depletion of the progenitor cells [68]. These results are in line with previous studies showing that Notch signalling viaHes1 plays a central role in the development of the pancreas by selecting progenitor cells to either differentiate or self-renew [69,70]. Therefore, Hes1activation in pancreatic progenitors can suppress the expression of both Ngn3 and p57Kip2 to simultaneously prevent cell cycle exit and differentiation. These findings are of great importance for the successful development of future methods for the expansion and manipulation of pancreatic precursor cell proliferation and differentiation in vitro. A better knowledge of the regulation of the pathways described above will result in an improved understanding of the control of β-cell growth and function.
Adult pancreatic β-cells have to cope with the stress caused by the isolation procedure (see Fig. 2B and C), transport, injection into the portal vein and the engraftment into the sinusoids of the liver. Numerous challenges which surprisingly enough β-cells are not only able to survive but in addition they reassure their original physiological role in the liver for decades as shown in islet autotransplantation.
Revascularization of transplanted islets
There is still a need for more intensive research designed to characterize and improve the environment into which islets are being transplanted. A better knowledge of the local cellular and molecular changes taking place after islet transplantation into the liver will yield the necessary basis for a more successful islet implantation. The influence of the surrounding tissue on islet physiology can be demonstrated for the release of glucagon: while islets transplanted into the peritoneal cavity release glucagon normally during hypgoglycaemia [71], this is not the case when the islets are transplanted intrahepatically [72]. It will be interesting to see, whether such differences in islet function might be attributed e.g. to differences in the vascular basement membrane composition dependent on the implantation site.
Up to now, most of the attention and discussions in the field of islet transplantation focused on β-cells and their precursors/progenitors or stem cells [21, 35, 36,73,74]. Research into the contribution of microvascular endothelial cells of pancreatic islets to the outcome of islet transplantation has been largely neglected in these discussions.
Nevertheless, a major issue of islet cells survival after transplantation is the establishment of new vascular connections. After the islets have been isolated from their familiar surrounding in the pancreas and transplanted into a new microenvironment in the liver they are in need of new blood supply and oxygenation. In this new situation they have to cope with a markedly decreased vascular density and pO2 in their environment [75].
How do islets manage the problem of revascularization? In their physiological state, islets have a dense glomerular-like angioarchitecture [76]. After isolation, the islet vasculature is disrupted and in the immediate post-transplantation period, oxygen and nutrient supply occurs only by diffusion from blood vessels in the surrounding hepatic tissue. Depending on the experimental settings used, it takes several days until the transplanted islets are revascularized [75,77]. It is surprising that the hypoxic islets survive at all up to this time and it is still questionable whether the newly formed blood vessels have the capacity to sufficiently meet the demands of the metabolically highly active islet cells. Even after revascularization a decreased vascular density in murine pancreatic islets was reported [78].
In a nude mouse model it was shown that murine as well as human islets when transplanted under the kidney capsule had a markedly decreased vascular density and pO2 compared with endogenous islets [79]. A decreased vascular engraftment may be one of the reasons why high numbers of islets are needed to obtain insulin independence. Consequently, improvements in islet revascularization should decrease this number.
A prerequisite for such improvements are detailed histological analysis of the transplanted, intraportal islets. In a non-human primate islet transplantation model using the Edmonton Protocol immunosuppressed rhesus macaques underwent total pancreatectomy and then had allogeneic isolated islets infused into their portal vein. From their results the authors concluded that islets transplanted into the portal vein lodged like a thrombus in the distal branches of the liver. Islet capillary formation was not detectable 5 days after intrahepatic transplantation, but was present by 30 days. The vascular cells likely originate from the recipient as the capillaries persist even during the later stages of rejection when very few islet cells remain. A very important finding of this study addressed the question whether the portal vein was the best site to infuse the allogeneic islets since it has been known to contain substances from the gut such as the immunosuppressive agents that may prove toxic to the islets. As the authors could show, after the initial capillary formation, endothelial cells grow around the islets rendering them essentially extraluminal relative to the portal vein [77].
Therefore the discovery of new methods of inducing an improved vascularization of transplanted islets or islet-like material is needed. Besides supplying the cells with oxygen and nutrients and removing the often cell toxic metabolites endothelial cells play a more active role than formerly assumed. As shown in pancreas and liver, blood vessels induce the development of both organs even before the blood vessels are functioning [80,81]. In pancreas development signals from blood vessel endothelium induce islet formation [81]. Thereafter islets signal in a paracrine fashion via VEGF-A to the endothelium which then forms the dense vascular network which is known to be required for proper endocrine function and islet size [82]. The future will show to what extent other “helper” cells are needed to guarantee a reproducible and long-lasting propagation and function of human β-cells (Fig. 5A). As recently reported, the attracted VEGFR2-expressing endothelial cells form a basement membrane next to the β-cells that contains different ECM components such as laminins, collagen IV, and fibronectin. This basement membrane is in at least two ways important for β-cell physiology: (i) in contrast to the exocrine pancreatic cells, β-cells do not form a basement membrane (Fig. 5B); (ii) the basement membrane components produced by the endothelial cells promote β-cell proliferation and insulin gene expression [83]. This might explain why islets maintain a capillary network that is five times denser than that of the exocrine pancreatic tissue and therefore exceeds the general tissue requirement for vascular supply [84].
5.
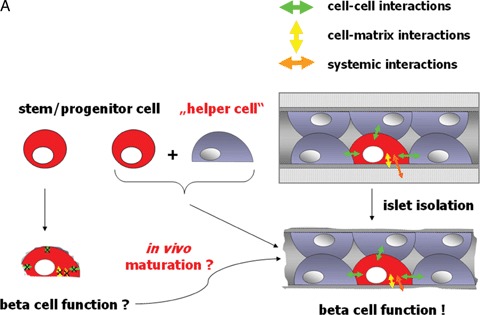
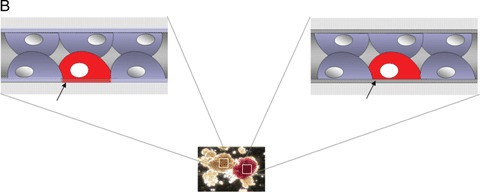
(A) Under normal conditions β-cell function is influenced by cell-cell, cell-matrix and systemic interactions. Even after islet transplantation β-cell survival as well as function are maintained and stable for decades (as seen in autotransplantation of islets). Even after transplantation into the liver, islet cyto- architecture is maintained. Therefore, we hypothesize that β-cells either generated in vitro out of stem cells or ex vivo through expansion of progenitor cells might enface the problem of not being fully functional due to the lack of proper cell-cell or cell-matrix interactions. (B) While epithelial cells in the exocrine compartment (shown in brown colour at the left) produce their own basement membrane (see arrow), β-cells in the Islets of Langerhans (appear in red due to dithizone staining) do not (see arrow) [83]. Their basement membrane is produced from the underlying endothelial cells, a fact underlining the above-mentioned hypothesis.
Therefore, when talking about islet transplantation also endothelial cells should be taken into consideration. Especially as it is known that the enzymatic digestion procedure used to isolate islets partially removes intra-islet endothelial cells thereby contributing to transplant failure [85, 86].
It is still an open question how attracted vascular endothelial cells form capillary structures within the transplanted islets. In a primate model it was shown that islet de novo capillary formation seems to be established between 5 and 30 days after transplantation. Before this time period, islets have to cope not only with a reduced pO2 but possibly also with changes associated with the basement membrane components of the endothelial cells which are known signals promoting β-cell function [17]. Future studies will show whether the addition of matrix components may partially substitute for endothelial cells and thereby bridge the time until new capillaries and vascular basement membranes are formed in the transplanted islets.
It also remains to be shown which cellular changes are induced in islet cells during such hypoxic conditions and what the consequences may be. The two most likely outcomes are the reduction of the survival rate of the transplanted islets and a decreased insulin secretion [87]. Therefore, improving vascularization of transplanted islets should compensate for the vascular dysfunctions and thereby augment islet cell survival and function. By this way the amount of islets needed to cure the diabetic patient should be lowered thereby increasing the number of patients that can be transplanted. As shown with murine islets transplanted under the kidney capsule, VEGF-A overexpression of the transplanted islets leads to improvements in insulin secretion and blood glucose regulation in the recipient mice [88].
Will “new” molecules answer “old” questions?
Tetranectin
Human, as well as murine islets contain about 60% intensely stained Tetranectin (TN) positive β-cells. Interestingly, individual TN positive cells are present within the exocrine acini of human and murine pancreatic tissue [89, 90]. Whether these cells might account for putative precursors of β-cells remains to be investigated. In the search of potential stem cells capable to differentiate into insulin secreting β-cells we should keep in mind, that besides possessing this ability, the cells need to cope with another challenge: the engraftment into the liver. Tetranectin, also shown to be a differentiation marker for myogenesis [91], plays a role in the regulation of pericellular proteolysis [92]. The ability of TN to bind plasminogen indicates, that TN may play a role in the regulation of pericellular proteolysis, and proteolytic activation of latent forms of metalloproteinases and growth factor. It was suggested that TN forms a link between the extracellular matrix and plasminogen by linking it to sulphated polysaccharides thereby enabling local tissue remodelling. One could speculate that TN may be involved in events leading to the proteolysis of matrix proteins, as activated plasminogen is believed to play a central role in the degradation of the extracellular matrix [93]. Together with its ability to bind plasminogen-like hepatocyte growth factor and tissue-type activator TN may play a pivotal role in the survival of islets after islet transplantation. As we could show, TN positive cells can be isolated and maintained in culture after human islet isolation, thereby providing the possibility to clarify its role and function in vivo as well as after islet transplantation [89].
Dickkopf-3
In order to compensate for cell loss, many tissues such as the colon undergo lifelong cellular turnover. Dickkopf-3 (Dkk-3) was shown to be strongly expressed at the base of colon crypts, a region known to contain proliferating epithelial precursor cells. The authors concluded that Dkk-3 might promote non-canonical signalling in the area of the gastrointestinal stem cells, and that these non-canonical signals are crucial in maintaining a proliferating, but undifferentiated stem cell pool [94]. In many other organs such as the pancreas, the identity of cells that give rise to newly differentiated cells is still not well understood. Until recently the relative contribution of adult stem cells and differentiated cells in the pancreas was still a matter of debate.
Interestingly, performing a bioinformatic analysis of gene expression data in EST (expressed sequence tags) libraries of human pancreata (NCBI Server) we identified highest gene expression values for Dkk-3 in human pancreatic tissue. This finding led us to perform a detailed study of the expression and localization of Dkk-3 in the adult human pancreas.
Using a quantitative RT-PCR approach comparing gene expression in the adult human islet fraction versus the exocrine fraction we were able to detect significantly higher expression levels of Dkk-3 in the islet compared with the exocrine fraction. By double labeling confocal immunofluorescence with antibodies directed against Dkk-3 and insulin we identified the β-cells of the islets of Langerhans as the cell type containing Dkk-3. Interestingly, Dkk-3 was present only in a subset of β-cells [95]. β-cell regeneration in the adult human pancreas is still not completely understood. Since not all β-cells contain Dkk-3, this finding may be of help in further distinguishing and characterizing subpopulations of β-cells in the adult human pancreas. A further characterization of the β-cell pool is of great interest as it has been shown that pre-existing β-cells, rather than pluripotent stem cells, are the major source of new β-cells during adult life and after pancreatomy in mice [96]. We, therefore, believe Dkk-3 may be of great use for the further study of islet regeneration, and for the identification of putative adult pancreatic β-cells capable of self-duplication. The possible impact of Dkk-3 on β-cell maturation and function is another important question still awaiting an answer.
Conclusion
A better understanding of the molecular and cellular orchestration of β-cell physiology under normal conditions as well as after transplantation into the liver will have an impact on future therapeutic applications such as islet transplantation. The ambitious goal of a cell therapy for the treatment of type 1 diabetes can only be achieved on such a better understanding. Given the plethora of different factors, regulatory mechanisms and signalling pathways balancing β-cell growth and differentiation a systematic description considering basic data such as the developmental condition or experimental setting should provide useful in gaining a more comprehensive view of β-cell physiology.
Acknowledgments
The authors thank Univ. Doz. Dr. Troppmair, Mag. Pirkebner, Mag. Deutschmann and BMA Draxl for fruitful discussions and critical reading of the manuscript.
References
- 1.Humes HD. Cell therapy: leveraging nature's therapeutic potential. J Am Soc Nephrol. 2003;14:2211–3. doi: 10.1097/01.asn.0000082692.79381.96. [DOI] [PubMed] [Google Scholar]
- 2.Dove A. Cell-based therapies go live. Nat Biotechnol. 2002;20:339–43. doi: 10.1038/nbt0402-339. [DOI] [PubMed] [Google Scholar]
- 3.Brower V. Human ES cells: Can you build a business around them? Nat Biotechnol. 1999;17:139–42. doi: 10.1038/6140. [DOI] [PubMed] [Google Scholar]
- 4.Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–5. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 5.Vassilopoulos G, Russell DW. Cell fusion: An alternative to stem cell plasticity and its therapeutic implications. Curr Opin Genet Dev. 2003;13:480–5. doi: 10.1016/s0959-437x(03)00110-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–901. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- 7.Hacein-Bey-Abina S, Von KC, Schmidt M, Le DF, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–6. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 8.Orive G, Hernandez RM, Gascon AR, Calafiore R, Chang TM, De VP, Hortelano G, Hunkeler D, Lacik I, Shapiro AM, Pedraz JL. Cell encapsulation: promise and progress. Nat Med. 2003;9:104–7. doi: 10.1038/nm0103-104. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson MA, Eisenbarth GS. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–9. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 10.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998;21:1414–31. doi: 10.2337/diacare.21.9.1414. [DOI] [PubMed] [Google Scholar]
- 11.Ritz E, Schomig M. The diabetic patient with renal failure. Nefrologia. 2000;20:16–24. [PubMed] [Google Scholar]
- 12.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 13.Diamond J. The double puzzle of diabetes. Nature. 2003;423:599–602. doi: 10.1038/423599a. [DOI] [PubMed] [Google Scholar]
- 14.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 15.Gautier JF, Beressi JP, Leblanc H, Vexiau P, Passa P. Are the implications of the Diabetes Control And Complications Trial (DCCT) feasible in daily clinical practice? Diabetes Metab. 1996;22:415–9. [PubMed] [Google Scholar]
- 16.Meivar-Levy I, Ferber S. New organs from our own tissues: liver-to-pancreas transdifferentiation. Trends Endocrinol Metab. 2003;14:460–6. doi: 10.1016/j.tem.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Hirshberg B, Rother KI, Gon III BJ, Venstrom J, Harlan DM. State of the art: Islet transplantation for the cure of type 1 diabetes mellitus. Rev Endocr Metab Disord. 2003;4:381–9. doi: 10.1023/a:1027358230402. [DOI] [PubMed] [Google Scholar]
- 18.Robertson RP, Davis C, Larsen J, Stratta R, Sutherland DE. Pancreas and islet transplantation for patients with diabetes. Diabetes Care. 2000;23:112–6. doi: 10.2337/diacare.23.1.112. [DOI] [PubMed] [Google Scholar]
- 19.Ballinger WF, Lacy PE. Transplantation of intact pancreatic islets in rats. Surgery. 1972;72:175–86. [PubMed] [Google Scholar]
- 20.Kemp CB, Knight MJ, Scharp DW, Lacy PE, Ballinger WF. Transplantation of isolated pancreatic islets into the portal vein of diabetic rats. Nature. 1973;244:447. doi: 10.1038/244447a0. [DOI] [PubMed] [Google Scholar]
- 21.Kandeel F, Smith CV, Todorov I, Mullen Y. Advances in islet cell biology: From stem cell differentiation to clinical transplantation: Conference report. Pancreas. 2003;27:e63–78. doi: 10.1097/00006676-200310000-00022. [DOI] [PubMed] [Google Scholar]
- 22.Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–8. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 23.Merani S, Shapiro AM. Current status of pancreatic islet transplantation. Clin Sci. 2006;110:611–25. doi: 10.1042/CS20050342. [DOI] [PubMed] [Google Scholar]
- 24.Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–9. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 25.Brock P, Sparmann G, Ritter T, Jaster R, Liebe S, Emmrich J. Adenovirus-mediated gene transfer of interleukin-4 into pancreatic stellate cells promotes interleukin-10 expression. J Cell Mol Med. 2006;10:884–95. doi: 10.1111/j.1582-4934.2006.tb00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kountouras J, Zavos C, Chatzopoulos D. A concept on the role of Helicobacter pylori infection in autoimmune pancreatitis. J Cell Mol Med. 2005;9:196–207. doi: 10.1111/j.1582-4934.2005.tb00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhatia M. Molecular mimicry in autoimmune pancreatitis: an interesting idea. J Cell Mol Med. 2005;9:745. doi: 10.1111/j.1582-4934.2005.tb00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarneri F, Guarneri C, Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J Cell Mol Med. 2005;9:741–4. doi: 10.1111/j.1582-4934.2005.tb00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson RP, Lanz KJ, Sutherland DE, Kendall DM. Prevention of diabetes for up to 13 years by autoislet transplantation after pancreatectomy for chronic pancreatitis. Diabetes. 2001;50:47–50. doi: 10.2337/diabetes.50.1.47. [DOI] [PubMed] [Google Scholar]
- 30.O'Gorman D, Kin T, Murdoch T, Richer B, Ghee-Wilson D, Ryan EA, Shapiro JA, Lakey JR The standardization of pancreatic donors for islet isolations. Transplantation. 2005;80:801–6. doi: 10.1097/01.tp.0000172216.47547.d5. [DOI] [PubMed] [Google Scholar]
- 31.Barnett MJ, Ghee-Wilson D, Shapiro AM, Lakey JR. Variation in human islet viability based on different membrane integrity stains. Cell Transplant. 2004;13:481–8. doi: 10.3727/000000004783983701. [DOI] [PubMed] [Google Scholar]
- 32.Hermann M, Pirkebner D, Draxl A, Margreiter R, Hengster P. Real-time” assessment of human islet preparations with confocal live cell imaging. Transplant Proc. 2005;37:3409–11. doi: 10.1016/j.transproceed.2005.09.076. [DOI] [PubMed] [Google Scholar]
- 33.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–6. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 34.Halvorsen TL, Beattie GM, Lopez AD, Hayek A, Levine F. Accelerated telomere shortening and senescence in human pancreatic islet cells stimulated to divide in vitro. J Endocrinol. 2000;166:103–9. doi: 10.1677/joe.0.1660103. [DOI] [PubMed] [Google Scholar]
- 35.Colman A. Making new beta cells from stem cells. Semin Cell Dev Biol. 2004;15:337–45. doi: 10.1016/j.semcdb.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YQ, Kritzik M, Sarvetnick N. Identification and expansion of pancreatic stem/progenitor cells. J Cell Mol Med. 2005;9:331–44. doi: 10.1111/j.1582-4934.2005.tb00359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelialto-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–4. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- 38.Zalzman M, Nker-Kitai L, Efrat S. Differentiation of human liver-derived, insulin-producing cells toward the beta-cell phenotype. Diabetes. 2005;54:2568–75. doi: 10.2337/diabetes.54.9.2568. [DOI] [PubMed] [Google Scholar]
- 39.Narang AS, Mahato RI. Biological and biomaterial approaches for improved islet transplantation. Pharmacol Rev. 2006;58:194–243. doi: 10.1124/pr.58.2.6. [DOI] [PubMed] [Google Scholar]
- 40.Narushima M, Kobayashi N, Okitsu T, Tanaka Y, Li SA, Chen Y, Miki A, Tanaka K, Nakaji S, Takei K, Gutierrez AS, Rivas-Carrillo JD, Navarro-Alvarez N, Jun HS, Westerman KA, Noguchi H, Lakey JR, Leboulch P, Tanaka N, Yoon JW. A human beta-cell line for transplantation therapy to control type 1 diabetes. Nat Biotechnol. 2005;23:1274–82. doi: 10.1038/nbt1145. [DOI] [PubMed] [Google Scholar]
- 41.Hohmeier HE, Newgard CB. Islets for all? Nat Biotechnol. 2005;23:1231–2. doi: 10.1038/nbt1005-1231. [DOI] [PubMed] [Google Scholar]
- 42.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–8. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soria B. In-vitro differentiation of pancreatic betacells. Differentiation. 2001;68:205–19. doi: 10.1046/j.1432-0436.2001.680408.x. [DOI] [PubMed] [Google Scholar]
- 44.Beattie GM, Montgomery AM, Lopez AD, Hao E, Perez B, Just ML, Lakey JR, Hart ME, Hayek A. A novel approach to increase human islet cell mass while preserving beta-cell function. Diabetes. 2002;51:3435–9. doi: 10.2337/diabetes.51.12.3435. [DOI] [PubMed] [Google Scholar]
- 45.Beattie GM, Rubin JS, Mally MI, Otonkoski T, Hayek A. Regulation of proliferation and differentiation of human fetal pancreatic islet cells by extracellular matrix, hepatocyte growth factor, and cell-cell contact. Diabetes. 1996;45:1223–8. doi: 10.2337/diab.45.9.1223. [DOI] [PubMed] [Google Scholar]
- 46.Schroeder IS, Rolletschek A, Blyszczuk P, Kania G, Wobus AM. Differentiation of mouse embryonic stem cells to insulin-producing cells. Nat Protoc. 2006;1:495–507. doi: 10.1038/nprot.2006.71. [DOI] [PubMed] [Google Scholar]
- 47.Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, Garcia-Ocana A. Growth factors and beta cell replication. Int J Biochem Cell Biol. 2006;38:931–50. doi: 10.1016/j.biocel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–7. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 49.Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24:297–302. doi: 10.1677/jme.0.0240297. [DOI] [PubMed] [Google Scholar]
- 50.Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–25. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- 51.Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. 2000;11:375–8. doi: 10.1016/s1043-2760(00)00305-2. [DOI] [PubMed] [Google Scholar]
- 52.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–90. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 53.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–76. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–89. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laybutt DR, Weir GC, Kaneto H, Lebet J, Palmiter RD, Sharma A, Bonner-Weir S. Overexpression of c-Myc in beta-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes. 2002;51:1793–804. doi: 10.2337/diabetes.51.6.1793. [DOI] [PubMed] [Google Scholar]
- 56.Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–62. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MJ, Kang JH, Park YG, Ryu GR, Ko SH, Jeong IK, Koh KH, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo YH. Exendin-4 induction of cyclin D1 expression in INS-1 beta-cells: Involvement of cAMP-responsive element. J Endocrinol. 2006;188:623–33. doi: 10.1677/joe.1.06480. [DOI] [PubMed] [Google Scholar]
- 58.Costes S, Broca C, Bertrand G, Lajoix AD, Bataille D, Bockaert J, Dalle S. ERK1/2 control phosphorylation and protein level of cAMP-responsive elementbinding protein: A key role in glucose-mediated pancreatic beta-cell survival. Diabetes. 2006;55:2220–30. doi: 10.2337/db05-1618. [DOI] [PubMed] [Google Scholar]
- 59.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 60.Zhang L, Fu Z, Binkley C, Giordano T, Burant CF, Logsdon CD, Simeone DM. Raf kinase inhibitory protein inhibits beta-cell proliferation. Surgery. 2004;136:708–15. doi: 10.1016/j.surg.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 61.Friedrichsen BN, Richter HE, Hansen JA, Rhodes CJ, Nielsen JH, Billestrup N, Moldrup A. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol Endocrinol. 2003;17:945–58. doi: 10.1210/me.2002-0356. [DOI] [PubMed] [Google Scholar]
- 62.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 63.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 64.Dunlop M, Muggli E, Clark S. Association of cyclindependent kinase-4 and cyclin D1 in neonatal beta cells after mitogenic stimulation by lysophosphatidic acid. Biochem Biophys Res Commun. 1996;218:132–6. doi: 10.1006/bbrc.1996.0023. [DOI] [PubMed] [Google Scholar]
- 65.Georgia S, Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. 2006;55:2950–6. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 66.Gu G, Brown JR, Melton DA. Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mech Dev. 2003;120:35–43. doi: 10.1016/s0925-4773(02)00330-1. [DOI] [PubMed] [Google Scholar]
- 67.Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, German MS. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50:928–36. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- 68.Georgia S, Soliz R, Li M, Zhang P, Bhushan A. p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol. 2006;298:22–31. doi: 10.1016/j.ydbio.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 69.Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de AM, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 70.Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 71.Gupta V, Wahoff DC, Rooney DP, Poitout V, Sutherland DE, Kendall DM, Robertson RP. The defective glucagon response from transplanted intrahepatic pancreatic islets during hypoglycaemia is transplantation site-determined. Diabetes. 1997;46:28–33. doi: 10.2337/diab.46.1.28. [DOI] [PubMed] [Google Scholar]
- 72.Paty BW, Ryan EA, Shapiro AM, Lakey JR, Robertson RP. Intrahepatic islet transplantation in type 1 diabetic patients does not restore hypoglycaemic hormonal counterregulation or symptom recognition after insulin independence. Diabetes. 2002;51:3428–34. doi: 10.2337/diabetes.51.12.3428. [DOI] [PubMed] [Google Scholar]
- 73.Santana A, Ensenat-Waser R, Arribas MI, Reig JA, Roche E. Insulin-producing cells derived from stem cells: Recent progress and future directions. J Cell Mol Med. 2006;10:866–83. doi: 10.1111/j.1582-4934.2006.tb00531.x. [DOI] [PubMed] [Google Scholar]
- 74.Ferber S, Heimberg H, Brownlee M, Colton C. Surrogate beta cells. Diabetologia. 1997;40(Suppl 3):B39–B43. doi: 10.1007/BF03168185. [DOI] [PubMed] [Google Scholar]
- 75.Carlsson PO, Palm F, Mattsson G. Low revascularization of experimentally transplanted human pancreatic islets. J Clin Endocrinol Metab. 2002;87:5418–23. doi: 10.1210/jc.2002-020728. [DOI] [PubMed] [Google Scholar]
- 76.Bonner-Weir S. Morphological evidence for pancreatic polarity of beta-cell within islets of Langerhans. Diabetes. 1988;37:616–21. doi: 10.2337/diab.37.5.616. [DOI] [PubMed] [Google Scholar]
- 77.Hirshberg B, Mog S, Patterson N, Leconte J, Harlan DM. Histopathological study of intrahepatic islets transplanted in the nonhuman primate model using edmonton protocol immunosuppression. J Clin Endocrinol Metab. 2002;87:5424–9. doi: 10.1210/jc.2002-020684. [DOI] [PubMed] [Google Scholar]
- 78.Mattsson G, Jansson L, Carlsson PO. Decreased vascular density in mouse pancreatic islets after transplantation. Diabetes. 2002;51:1362–6. doi: 10.2337/diabetes.51.5.1362. [DOI] [PubMed] [Google Scholar]
- 79.Carlsson PO, Palm F, Mattsson G. Low revascularization of experimentally transplanted human pancreatic islets. J Clin Endocrinol Metab. 2002;87:5418–23. doi: 10.1210/jc.2002-020728. [DOI] [PubMed] [Google Scholar]
- 80.Lammert E, Cleaver O, Melton D. Role of endothelial cells in early pancreas and liver development. Mech Dev. 2003;120:59–64. doi: 10.1016/s0925-4773(02)00332-5. [DOI] [PubMed] [Google Scholar]
- 81.Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294:564–7. doi: 10.1126/science.1064344. [DOI] [PubMed] [Google Scholar]
- 82.Lammert E, Gu G, McLaughlin M, Brown D, Brekken R, Murtaugh LC, Gerber HP, Ferrara N, Melton DA. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2003;13:1070–4. doi: 10.1016/s0960-9822(03)00378-6. [DOI] [PubMed] [Google Scholar]
- 83.Nikolova G, Jabs N, Konstantinova I, Domogatskaya A, Tryggvason K, Sorokin L, Fassler R, Gu G, Gerber HP, Ferrara N, Melton DA, Lammert E. The vascular basement membrane: A niche for insulin gene expression and Beta cell proliferation. Dev Cell. 2006;10:397–405. doi: 10.1016/j.devcel.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 84.Henderson JR, Moss MC. A morphometric study of the endocrine and exocrine capillaries of the pancreas. Q J Exp Physiol. 1985;70:347–56. doi: 10.1113/expphysiol.1985.sp002920. [DOI] [PubMed] [Google Scholar]
- 85.Konstantinova I, Lammert E. Microvascular development: Learning from pancreatic islets. Bioessays. 2004;26:1069–75. doi: 10.1002/bies.20105. [DOI] [PubMed] [Google Scholar]
- 86.Lukinius A, Jansson L, Korsgren O. Ultrastructural evidence for blood microvessels devoid of an endothelial cell lining in transplanted pancreatic islets. Am J Pathol. 1995;146:429–35. [PMC free article] [PubMed] [Google Scholar]
- 87.Jansson L, Carlsson PO. Graft vascular function after transplantation of pancreatic islets. Diabetologia. 2002;45:749–63. doi: 10.1007/s00125-002-0827-4. [DOI] [PubMed] [Google Scholar]
- 88.Zhang N, Richter A, Suriawinata J, Harbaran S, Altomonte J, Cong L, Zhang H, Song K, Meseck M, Bromberg J, Dong H. Elevated vascular endothelial growth factor production in islets improves islet graft vascularization. Diabetes. 2004;53:963–70. doi: 10.2337/diabetes.53.4.963. [DOI] [PubMed] [Google Scholar]
- 89.Hermann M, Pirkebner D, Draxl A, Margreiter R, Hengster P. In the search of potential human islet stem cells: Is tetranectin showing us the way? Transplant Proc. 2005;37:1322–5. doi: 10.1016/j.transproceed.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 90.Christensen L, Johansen N, Jensen BA, Clemmensen I. Immunohistochemical localization of a novel, human plasma protein, tetranectin, in human endocrine tissues. Histochemistry. 1987;87:195–9. doi: 10.1007/BF00492409. [DOI] [PubMed] [Google Scholar]
- 91.Wewer UM, Iba K, Durkin ME, Nielsen FC, Loechel F, Gilpin BJ, Kuang W, Engvall E, Albrechtsen R. Tetranectin is a novel marker for myogenesis during embryonic development, muscle regeneration, and muscle cell differentiation in vitro. Dev Biol. 1998;200:247–59. doi: 10.1006/dbio.1998.8962. [DOI] [PubMed] [Google Scholar]
- 92.Westergaard UB, Andersen MH, Heegaard CW, Fedosov SN, Petersen TE. Tetranectin binds hepatocyte growth factor and tissue-type plasminogen activator. Eur J Biochem. 2003;270:1850–4. doi: 10.1046/j.1432-1033.2003.03549.x. [DOI] [PubMed] [Google Scholar]
- 93.Westergaard UB, Andersen MH, Heegaard CW, Fedosov SN, Petersen TE. Tetranectin binds hepatocyte growth factor and tissue-type plasminogen activator. Eur J Biochem. 2003;270:1850–4. doi: 10.1046/j.1432-1033.2003.03549.x. [DOI] [PubMed] [Google Scholar]
- 94.Byun T, Karimi M, Marsh JL, Milovanovic T, Lin F, Holcombe RF. Expression of secreted Wnt antagonists in gastrointestinal tissues: potential role in stem cell homeostasis. J Clin Pathol. 2005;58:515–9. doi: 10.1136/jcp.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hermann M, Pirkebner D, Draxl A, Berger P, Untergasser G, Margreiter R, Hengster P. Dickkopf-3 is expressed in a subset of adult human pancreatic beta cells. Histochem Cell Biol. 2007;127:513–21. doi: 10.1007/s00418-007-0278-6. [DOI] [PubMed] [Google Scholar]
- 96.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–6. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]